

Psychological Stress-Induced Pathogenesis of Alopecia Areata: Autoimmune and Apoptotic Pathways


Abstract

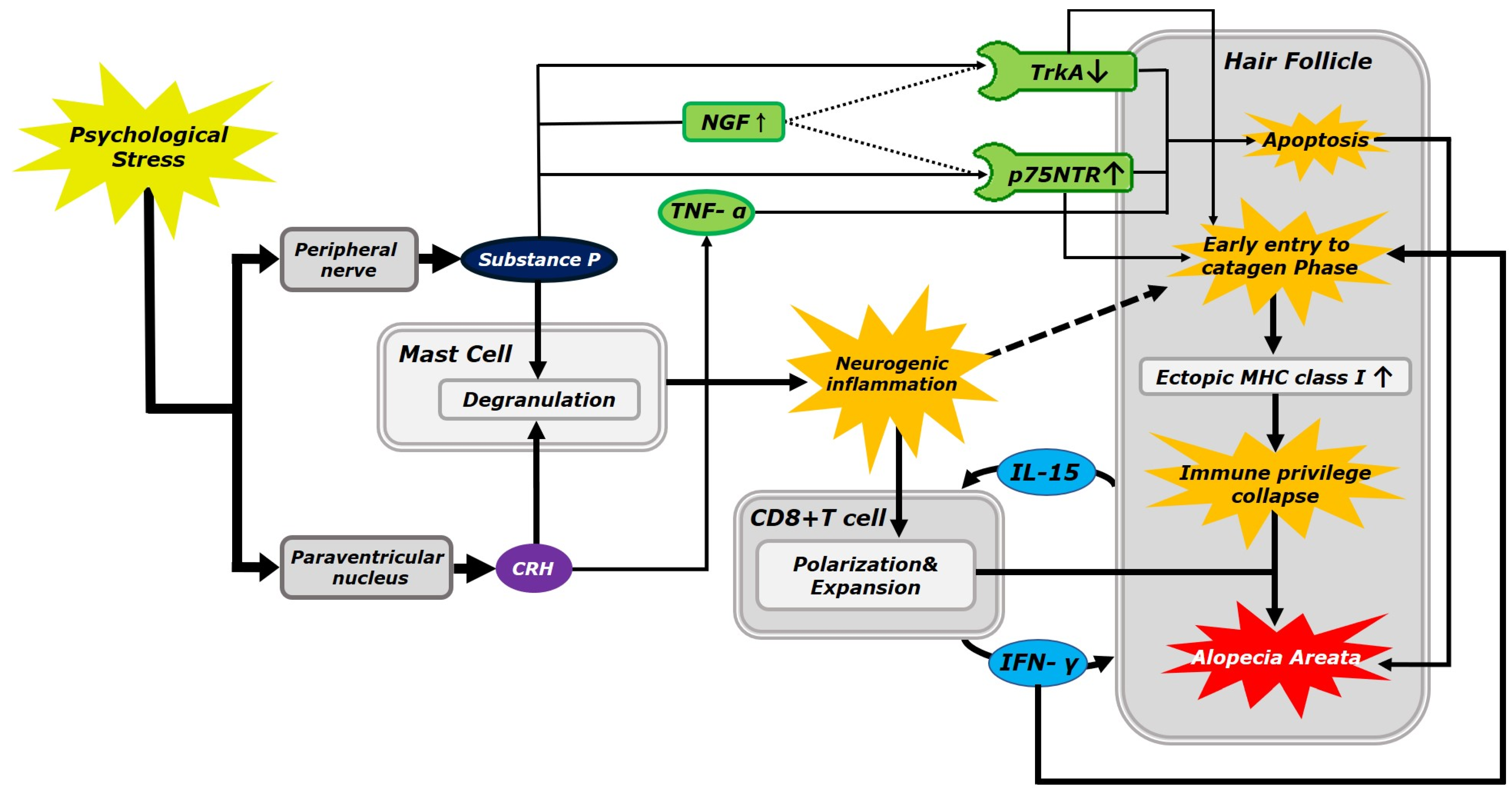
1. Introduction
1.1. Role of Substance P and Corticotropin-Releasing Hormone in the Alopecia Areata
1.2. Neurokinin-1 Receptor Expression in the Human Hair Follicle
1.3. Role of CD8+ T Cells in Alopecia Areata
1.4. Apoptotic Pathway in Alopecia Areata Pathology
1.5. Autoimmune Pathway in Alopecia Areata Pathology
1.6. Improved Therapeutic Strategies Targeting Signaling Pathways for Alopecia Areata
1.7. Potential Therapeutic Treatments for Alopecia Areata
2. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bellocchi, C.; Carandina, A.; Montinaro, B.; Targetti, E.; Furlan, L.; Rodrigues, G.D. The interplay between autonomic nervous system and inflammation across systemic autoimmune diseases. Int. J. Mol. Sci. 2022, 23, 2449. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Fang, F.; Tomasson, G.; Arnberg, F.K.; Mataix-Cols, D.; Fernández de la Cruz, L. Association of stress-related disorders with subsequent autoimmune disease. JAMA 2018, 319, 2388–2400. [Google Scholar] [CrossRef] [PubMed]
- Pratt, C.H.; King, L.E., Jr.; Messenger, A.G.; Christiano, A.M.; Sundberg, J.P. Alopecia areata. Nat. Rev. Dis. Primers 2017, 3, 17011. [Google Scholar] [CrossRef] [PubMed]
- Simakou, T.; Butcher, J.P.; Reid, S.; Henriquez, F.L. Alopecia areata: A multifactorial autoimmune condition. J. Autoimmun. 2019, 98, 74–85. [Google Scholar] [CrossRef] [PubMed]
- AL-Eitan, L.N.; Alghamdi, M.A.; Momani, R.O.A.; Aljamal, H.A.; Elsy, B.; Mohammed, H.M. Genetic Association between Interleukin Genes and Alopecia Areata in Jordanian Patients. Oman. Med. J. 2022, 37, e421. [Google Scholar] [CrossRef]
- Cakirca, G.; Manav, V.; Celik, H.; Saracoglu, G.; Yetkin, E.N. Effects of anxiety and depression symptoms on oxidative stress in patients with alopecia areata. Postepy Dermatol. Alergol. 2020, 37, 412–416. [Google Scholar] [CrossRef]
- Baldini, E.; Odorisio, T.; Sorrenti, S.; Catania, A.; Tartaglia, F.; Carbotta, G. Vitiligo and Autoimmune Thyroid Disorders. Front. Endocrinol. 2017, 8, 290. [Google Scholar] [CrossRef]
- Naik, P.P.; Farrukh, S.N. Association between alopecia areata and thyroid dysfunction. Postgrad. Med. 2021, 133, 895–898. [Google Scholar] [CrossRef]
- McDonagh, A.J.; Messenger, A.G. The pathogenesis of alopecia areata. Dermatol. Clin. 1996, 14, 661–670. [Google Scholar] [CrossRef]
- Rahmani, W.; Sinha, S.; Biernaskie, J. Immune modulation of hair follicle regeneration. NPJ Regen. Med. 2020, 5, 9. [Google Scholar] [CrossRef]
- Peters, E.M.; Botchkarev, V.A.; Botchkareva, N.V.; Tobin, D.J.; Paus, R. Hair-cycle-associated remodeling of the peptidergic innervation of murine skin, and hair growth modulation by neuropeptides. J. Investig. Dermatol. 2001, 116, 236–245. [Google Scholar] [CrossRef]
- Grymowicz, M.; Rudnicka, E.; Podfigurna, A.; Napierala, P.; Smolarczyk, R.; Smolarczyk, K. Hormonal Effects on Hair Follicles. Int. J. Mol. Sci. 2020, 21, 5342. [Google Scholar] [CrossRef]
- Buffoli, B.; Rinaldi, F.; Labanca, M.; Sorbellini, E.; Trink, A.; Guanziroli, E. The human hair: From anatomy to physiology. Int. J. Dermatol. 2014, 53, 331–341. [Google Scholar] [CrossRef]
- McElwee, K.J.; Tobin, D.J.; Bystryn, J.C.; King, L.E., Jr.; Sundberg, J.P. Alopecia areata: An autoimmune disease? Exp. Dermatol. 1999, 8, 371–379. [Google Scholar] [CrossRef]
- Gilhar, A.; Paus, R.; Kalish, R.S. Lymphocytes, neuropeptides, and genes involved in alopecia areata. J. Clin. Investig. 2007, 117, 2019–2027. [Google Scholar] [CrossRef]
- Suchonwanit, P.; Kositkuljorn, C.; Pomsoong, C. Alopecia Areata: An Autoimmune Disease of Multiple Players. Immunotargets Ther. 2021, 10, 299–312. [Google Scholar] [CrossRef]
- Guo, H.; Cheng, Y.; Shapiro, J.; McElwee, K. The role of lymphocytes in the development and treatment of alopecia areata. Expert. Rev. Clin. Immunol. 2015, 11, 1335–1351. [Google Scholar] [CrossRef]
- Daviu, N.; Füzesi, T.; Rosenegger, D.G.; Rasiah, N.P.; Sterley, T.L.; Peringod, G.; Bains, J.S. Paraventricular nucleus CRH neurons encode stress controllability and regulate defensive behavior selection. Nat. Neurosci. 2020, 23, 398–410. [Google Scholar] [CrossRef]
- Rokowska-Waluch, A.; Pawlaczyk, M.; Cybulski, M.; Żurawski, J.; Kaczmarek, M.; Michalak, M.; Mojs, E. Stressful Events and Serum Concentration of Substance P in Acne Patients. Ann. Dermatol. 2016, 28, 464–469. [Google Scholar] [CrossRef]
- Wang, L.; Guo, L.L.; Wang, L.H.; Zhang, G.X.; Shang, J.; Murao, K.; Chen, D.F.; Fan, X.H.; Fu, W.Q. Oxidative stress and substance P mediate psychological stress-induced autophagy and delay of hair growth in mice. Arch. Dermatol. Res. 2015, 307, 171–181. [Google Scholar] [CrossRef]
- Kim, C.; Shin, J.M.; Kim, D.; Park, S.; Hong, D.; Jung, K.E.; Kim, C.D.; Seo, Y.J.; Lee, Y. Role of Substance P in Regulating Micro-Milieu of Inflammation in Alopecia Areata. Ann. Dermatol. 2022, 34, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Katsarou-Katsari, A.; Singh, L.K.; Theoharides, T.C. Alopecia areata and affected skin CRH receptor upregulation induced by acute emotional stress. Dermatology 2001, 203, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Suvas, S. Role of Substance P Neuropeptide in Inflammation, Wound Healing, and Tissue Homeostasis. J. Immunol. 2017, 199, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Rosén, A.; Brodin, K.; Eneroth, P.; Brodin, E. Short-term restraint stress and s.c. saline injection alter the tissue levels of substance P and cholecystokinin in the peri-aqueductal grey and limbic regions of rat brain. Acta. Physiol. Scand. 1992, 146, 341–348. [Google Scholar] [CrossRef]
- Ebner, K.; Rupniak, N.M.; Saria, A.; Singewald, N. Substance P in the medial amygdala: Emotional stress-sensitive release and modulation of anxiety-related behavior in rats. Proc. Natl. Acad. Sci. USA 2004, 101, 4280–4285. [Google Scholar] [CrossRef]
- Peters, E.M.; Liotiri, S.; Bodó, E.; Hagen, E.; Bíró, T.; Arck, P.C.; Paus, R. Probing the effects of stress mediators on the human hair follicle: Substance P holds central position. Am. J. Pathol. 2007, 171, 1872–1886. [Google Scholar] [CrossRef]
- Siebenhaar, F.; Sharov, A.A.; Peters, E.M.; Sharova, T.Y.; Syska, W.; Mardaryev, A.N.; Freyschmidt-Paul, P.; Sundberg, J.P.; Maurer, M.; Botchkarev, V.A. Substance P as an immunomodulatory neuropeptide in a mouse model for autoimmune hair loss (alopecia areata). J. Investig. Dermatol. 2007, 127, 1489–1497. [Google Scholar] [CrossRef]
- Wepler, M.; Preuss, J.M.; Merz, T.; McCook, O.; Radermacher, P.; Tuckermann, J.P.; Vettorazzi, S. Impact of downstream effects of glucocorticoid receptor dysfunction on organ function in critical illness-associated systemic inflammation. Intensive Care Med. Exp. 2020, 8, 37. [Google Scholar] [CrossRef]
- Santibañez, M.; Gysling, K.; Forray, M.I. Desipramine prevents the sustained increase in corticotropin-releasing hormone-like immunoreactivity induced by repeated immobilization stress in the rat central extended amygdala. J. Neurosci. Res. 2006, 84, 1270–1281. [Google Scholar] [CrossRef]
- Osacka, J.; Kiss, A.; Mach, M.; Tillinger, A.; Koprdova, R. Haloperidol and aripiprazole affects CRH system and behaviour of animals exposed to chronic mild stress. Neurochem. Int. 2022, 152, 105224. [Google Scholar] [CrossRef]
- Yamanaka-Takaichi, M.; Mizukami, Y.; Sugawara, K.; Sunami, K.; Teranishi, Y.; Kira, Y.; Paus, R.; Tsuruta, D. Stress and Nasal Allergy: Corticotropin-Releasing Hormone Stimulates Mast Cell Degranulation and Proliferation in Human Nasal Mucosa. Int. J. Mol. Sci. 2021, 22, 2773. [Google Scholar] [CrossRef]
- Westphal, N.J.; Seasholtz, A.F. CRH-BP: The regulation and function of a phylogenetically conserved binding protein. Front. Biosci. 2006, 11, 1878–1891. [Google Scholar] [CrossRef]
- Kim, H.S.; Cho, D.H.; Kim, H.J.; Lee, J.Y.; Cho, B.K.; Park, H.J. Immunoreactivity of corticotropin-releasing hormone, adrenocorticotropic hormone and alpha-melanocyte-stimulating hormone in alopecia areata. Exp. Dermatol. 2006, 15, 515–522. [Google Scholar] [CrossRef]
- Zhang, X.; Yu, M.; Yu, W.; Weinberg, J.; Shapiro, J.; McElwee, K.J. Development of alopecia areata is associated with higher central and peripheral hypothalamic-pituitary-adrenal tone in the skin graft induced C3H/HeJ mouse model. J. Investig. Dermatol. 2009, 129, 1527–1538. [Google Scholar] [CrossRef]
- Singh, L.K.; Pang, X.; Alexacos, N.; Letourneau, R.; Theoharides, T.C. Acute immobilization stress triggers skin mast cell degranulation via corticotropin releasing hormone, neurotensin, and substance P: A link to neurogenic skin disorders. Brain Behav. Immun. 1999, 13, 225–239. [Google Scholar] [CrossRef]
- Asadi, S.; Alysandratos, K.D.; Angelidou, A.; Miniati, A.; Sismanopoulos, N.; Vasiadi, M.; Zhang, B.; Kalogeromitros, D.; Theoharides, T.C. Substance P (SP) induces expression of functional corticotropin-releasing hormone receptor-1 (CRHR-1) in human mast cells. J. Investig. Dermatol. 2012, 132, 324–329. [Google Scholar] [CrossRef]
- Chen, X.Y.; Ru, G.Q.; Ma, Y.Y.; Xie, J.; Chen, W.Y.; Wang, H.J.; Wang, S.B.; Li, L.; Jin, K.T.; He, X.L.; et al. High expression of substance P and its receptor neurokinin-1 receptor in colorectal cancer is associated with tumor progression and prognosis. Onco. Targets. Ther. 2016, 9, 3595–3602. [Google Scholar] [CrossRef]
- Stenn, K.S.; Paus, R. Controls of hair follicle cycling. Physiol. Rev. 2001, 81, 449–494. [Google Scholar] [CrossRef]
- Bertolini, M.; McElwee, K.; Gilhar, A.; Bulfone-Paus, S.; Paus, R. Hair follicle immune privilege and its collapse in alopecia areata. Exp. Dermatol. 2020, 29, 703–725. [Google Scholar] [CrossRef]
- Paus, R.; Bertolini, B. The role of hair follicle immune privilege collapse in alopecia areata: Status and perspectives. J. Investig. Dermatol. Symp. Proc. 2013, 16, S25–S27. [Google Scholar] [CrossRef]
- Fingerlin, T.E.; Zhang, W.; Yang, I.V.; Ainsworth, H.C.; Russell, P.H.; Blumhagen, R.Z.; Schwarz, M.I.; Brown, K.K.; Steele, M.P.; Loyd, J.E.; et al. Genome-wide imputation study identifies novel HLA locus for pulmonary fibrosis and potential role for auto-immunity in fibrotic idiopathic interstitial pneumonia. BMC Genet. 2016, 17, 74. [Google Scholar] [CrossRef] [PubMed]
- Gough, S.C.; Simmonds, M.J. The HLA Region and Autoimmune Disease: Associations and Mechanisms of Action. Curr. Genom. 2007, 8, 453–465. [Google Scholar] [CrossRef]
- Torales, J.; Castaldelli-Maia, J.M.; Ventriglio, A.; Almirón-Santacruz, J.; Barrios, I.; O’Higgins, M.; García, O.; Navarro, R.; Melgarejo, O.; Jafferany, M. Alopecia areata: A psychodermatological perspective. J. Cosmet. Dermatol. 2022, 21, 2318–2323. [Google Scholar] [CrossRef] [PubMed]
- Pezet, S.; Onténiente, B.; Grannec, G.; Calvino, B. Chronic pain is associated with increased TrkA immunoreactivity in spinoreticular neurons. J. Neurosci. 1999, 19, 5482–5492. [Google Scholar] [CrossRef] [PubMed]
- Bamji, S.X.; Majdan, M.; Pozniak, C.D.; Belliveau, D.J.; Aloyz, R.; Kohn, J.; Causing, C.G.; Miller, F.D. The p75 neurotrophin receptor mediates neuronal apoptosis and is essential for naturally occurring sympathetic neuron death. J. Cell. Biol. 1998, 140, 911–923. [Google Scholar] [CrossRef]
- Zhao, X.Y.; Li, Z.B.; Yuan, H.J.; Han, X.; Wu, J.S.; Feng, X.Y.; Zhang, M.; Tan, J.H. Restraint stress and elevation of corticotrophin-releasing hormone in female mice impair oocyte competence through activation of the tumour necrosis factor α (TNF-α) system. Reprod. Fertil. Dev. 2020, 32, 862–872. [Google Scholar] [CrossRef]
- Bono, F.; Lamarche, I.; Bornia, J.; Savi, P.; Della, V.G.; Herbert, J.M. Nerve growth factor (NGF) exerts its pro-apoptotic effect via the P75NTR receptor in a cell cycle-dependent manner. FEBS Lett. 1999, 457, 93–97. [Google Scholar] [CrossRef]
- Shen, J.; Maruyama, I.N. Nerve growth factor receptor TrkA exists as a preformed, yet inactive, dimer in living cells. FEBS Lett. 2011, 585, 295–299. [Google Scholar] [CrossRef]
- Fahnestock, M.; Shekari, A. ProNGF and Neurodegeneration in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 129. [Google Scholar] [CrossRef]
- Slominski, A.T.; Zmijewski, M.A.; Zbytek, B.; Tobin, D.J.; Theoharides, T.C.; Rivier, J. Key role of CRF in the skin stress response system. Endocr. Rev. 2013, 34, 827–884. [Google Scholar] [CrossRef]
- Gohary, Y.M.; Abdel, F.D.S. Detection of Tumor Necrosis Factor-alpha in Nonlesional Tissues of Alopecia Areata Patients: A Prove for a Systemic Disease. Int. J. Trichology 2017, 9, 154–159. [Google Scholar] [CrossRef]
- Ting, A.T.; Bertrand, M.J.M. More to Life than NF-κB in TNFR1 Signaling. Trends. Immunol. 2016, 37, 535–545. [Google Scholar] [CrossRef]
- Oceandy, D.; Amanda, B.; Ashari, F.Y.; Faizah, Z.; Azis, M.A.; Stafford, N. The Cross-Talk Between the TNF-α and RASSF-Hippo Signalling Pathways. Int. J. Mol. Sci. 2019, 20, 2346. [Google Scholar] [CrossRef]
- Kasumagic-Halilovic, E.; Prohic, A.; Cavaljuga, S. Tumor necrosis factor-alpha in patients with alopecia areata. Indian J. Dermatol. 2011, 56, 494–496. [Google Scholar] [CrossRef]
- Lis, A.; Pierzchała, E.; Brzezińska-Wcisło, L. The role of cell-mediated immune response in pathogenesis of alopecia areata. Wiad. Lek. 2001, 54, 159–163. [Google Scholar]
- Alysandratos, K.D.; Asadi, S.; Angelidou, A.; Zhang, B.; Sismanopoulos, N.; Yang, H.; Critchfield, A.; Theoharides, T.C. Neurotensin and CRH interactions augment human mast cell activation. PLoS ONE 2012, 7, e48934. [Google Scholar] [CrossRef]
- Choi, J.E.; Nardo, D.A. Skin neurogenic inflammation. Semin. Immunopathol. 2018, 40, 249–259. [Google Scholar] [CrossRef]
- Bertolini, M.; Zilio, F.; Rossi, A.; Kleditzsch, P.; Emelianov, V.E.; Gilhar, A.; Keren, A.; Meyer, K.C.; Wang, E.; Funk, W.; et al. Abnormal interactions between perifollicular mast cells and CD8+ T-cells may contribute to the pathogenesis of alopecia areata. PLoS ONE 2014, 9, e94260. [Google Scholar] [CrossRef]
- Suzuki, T.; Ito, T.; Gilhar, A.; Tokura, Y.; Reich, K.; Paus, R. The hair follicle-psoriasis axis: Shared regulatory mechanisms and therapeutic targets. Exp. Dermatol. 2022, 31, 266–279. [Google Scholar] [CrossRef]
- Rosenblum, M.D.; Yancey, K.B.; Olasz, E.B.; Truitt, R.L. CD200, a “no danger” signal for hair follicles. J. Dermatol. Sci. 2006, 41, 165–174. [Google Scholar] [CrossRef]
- Napolitano, M.; Fabbrocini, G.; Gencom, L.; Martora, F.; Potestio, L.; Patruno, C. Rapid improvement in pruritus in atopic dermatitis patients treated with upadacitinib: A real-life experience. J. Eur. Acad. Dermatol. Venereol. 2022, 36, 1497–1498. [Google Scholar] [CrossRef] [PubMed]
- Craiglow, B.G.; King, B.A. Killing two birds with one stone: Oral tofacitinib reverses alopecia universalis in a patient with plaque psoriasis. J. Investig. Dermatol. 2014, 134, 2988–2990. [Google Scholar] [CrossRef] [PubMed]
- Olsen, E.A.; Kornacki, D.; Sun, K.; Hordinsky, M.K. Ruxolitinib cream for the treatment of patients with alopecia areata: A 2-part, double-blind, randomized, vehicle-controlled phase 2 study. J. Am. Acad. Dermatol. 2020, 82, 412–419. [Google Scholar] [CrossRef]
- King, B.; Guttman-Yassky, E.; Peeva, E.; Banerjee, A.; Sinclair, R.; Pavel, A.B.; Zhu, L.; Cox, L.A.; Craiglow, B.; Chen, L.; et al. A phase 2a randomized, placebo-controlled study to evaluate the efficacy and safety of the oral Janus kinase inhibitors ritlecitinib and brepocitinib in alopecia areata: 24-week results. J. Am. Acad. Dermatol. 2021, 85, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Martora, F.; Villani, A.; Ocampo-Garza, S.; Fabbrocini, G.; Megna, M. Alopecia universalis improvement following risankizumab in a psoriasis patient. J. Eur. Acad. Dermatol. Venereol. 2022, 36, e543–e545. [Google Scholar] [CrossRef]
- Gupta, A.K.; Wang, T.; Polla, R.S.; Bamimore, M.A.; Piguet, V.; Tosti, A. Systematic review of newer agents for the management of alopecia areata in adults: Janus kinase inhibitors, biologics and phosphodiesterase-4 inhibitors. J. Eur. Acad. Dermatol. Venereol. 2023, 37, 666–679. [Google Scholar] [CrossRef]
- Żeberkiewicz, M.; Rudnicka, L.; Malejczyk, J. Immunology of alopecia areata. Cent. Eur. J. Immunol. 2020, 45, 325–333. [Google Scholar] [CrossRef]
- Steiner, J.; Bernstein, H.G.; Schiltz, K.; Müller, U.J.; Westphal, S.; Drexhage, H.A.; Bogerts, B. Immune system and glucose metabolism interaction in schizophrenia: A chicken-egg dilemma. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 48, 287–294. [Google Scholar] [CrossRef]
- Stelekati, E.; Bahri, R.; D’Orlando, O.; Orinska, Z.; Mittrücker, H.W.; Langenhaun, R.; Glatzel, M.; Bollinger, A.; Paus, R.; Bulfone-Paus, S. Mast cell-mediated antigen presentation regulates CD8+ T cell effector functions. Immunity 2009, 31, 665–676. [Google Scholar] [CrossRef]
- Ito, T.; Ito, N.; Saathoff, M.; Bettermann, A.; Takigawa, M.; Paus, R. Interferon-gamma is a potent inducer of catagen-like changes in cultured human anagen hair follicles. Br. J. Dermatol. 2005, 152, 623–631. [Google Scholar] [CrossRef]
- Peters, E.M.; Hendrix, S.; Gölz, G.; Klapp, B.F.; Arck, P.C.; Paus, R. Nerve growth factor and its precursor differentially regulate hair cycle progression in mice. J. Histochem. Cytochem. 2006, 54, 275–288. [Google Scholar] [CrossRef]
- Adly, M.A.; Assaf, H.A.; Hussein, M.R. Expression pattern of p75 neurotrophin receptor protein in human scalp skin and hair follicles: Hair cycle-dependent expression. J. Am. Acad. Dermatol. 2009, 60, 99–109. [Google Scholar] [CrossRef]
- Huelsken, J.; Vogel, R.; Erdmann, B.; Cotsarelis, G.; Birchmeier, W. beta-Catenin controls hair follicle morphogenesis and stem cell differentiation in the skin. Cell 2001, 105, 533–545. [Google Scholar] [CrossRef]
- Collins, C.A.; Kretzschmar, K.; Watt, F.M. Reprogramming adult dermis to a neonatal state through epidermal activation of beta-catenin. Development 2011, 138, 5189–5199. [Google Scholar] [CrossRef]
- Dong, L.; Hao, H.; Xia, L.; Liu, J.; Ti, D.; Tong, C.; Hou, Q.; Han, Q.; Zhao, Y.; Liu, H.; et al. Treatment of MSCs with Wnt1a-conditioned medium activates DP cells and promotes hair follicle regrowth. Sci. Rep. 2014, 4, 5432. [Google Scholar] [CrossRef]
- Ahmed, N.S.; Ghatak, S.; Masry, E.M.S.; Gnyawali, S.C.; Roy, S.; Amer, M.; Everts, H.; Sen, C.K.; Khanna, S. Epidermal E-Cadherin Dependent beta-Catenin Pathway Is Phytochemical Inducible and Accelerates Anagen Hair Cycling. Mol. Ther. 2017, 25, 2502–2512. [Google Scholar] [CrossRef]
- Castellana, D.; Paus, R.; Perez-Moreno, M. Macrophages Contribute to the Cyclic Activation of Adult Hair Follicle Stem Cells. PLoS. Biol. 2014, 12, e1002002. [Google Scholar] [CrossRef]
- Flores, A.; Schell, J.; Krall, A.S.; Jelinek, D.; Miranda, M.; Grigorian, M.; Braas, D.; White, A.C.; Zhou, J.L.; Graham, N.A.; et al. Lactate Dehydrogenase Activity Drives Hair Follicle Stem Cell Activation. Nat. Cell. Biol. 2017, 19, 1017–1026. [Google Scholar] [CrossRef]
- Gund, R.; Christiano, A.M. Impaired autophagy promotes hair loss in the C3H/HeJ mouse model of alopecia areata. Autophagy 2023, 19, 296–305. [Google Scholar] [CrossRef]
- Parodi, C.; Hardman, J.A.; Allavena, G.; Marotta, R.; Catelani, T.; Bertolini, M.; Paus, R.; Grimaldi, B. Autophagy is essential for maintaining the growth of a human (mini-)organ: Evidence from scalp hair follicle organ culture. PLoS Biol. 2018, 16, e2002864. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Current Status of Clinical Trial | Intervention: Drug Only | Study Title | Drug Target | Drug Application |
|---|---|---|---|---|
| Phase 4 | Tofacitinib (Xeljanz®) | Effectiveness and safety of Tofacitinib in patients with extensive and recalcitrant alopecia areata | JAK-1 and JAK-3 (JAK-STAT inhibition) | Oral medication, (tablet) |
| Phase 2 | Ruxolitinib (Jakavi®) | A study with ruxolitinib phosphate cream applied topically to subjects with alopecia areata (AA) | JAK-1 and JAK-2 (JAK-STAT inhibition) | Ointment |
| Phase 2 Phase 3 | Ritlecitinib (PF-06651600) | PF-06651600 for the treatment of alopecia areata | JAK-3 (JAK-STAT inhibition) | Oral medication, (tablet) |
| Phase 4 | Apremilast (Otezla®) | Apremilast in the treatment of central centrifugal cicatricial alopecia (CCCA) | PDE-4 inhibitor, TNF-α inhibitor | Oral medication, (tablet) |
| Phase 2 | Secukinumab | A study of secukinumab for the Treatment of alopecia Areata | PDE-4 inhibitor | Patches |
| Phase 2 | Dupilumab (Dupixent®) | Treatment of alopecia areata (AA) with dupilumab in patients with and without atopic dermatitis (AD) | IL-4 and IL-13 inhibitor | Subcutaneous injection |
| Phase 3 | Diphencyprone | DPCP for the Treatment of Alopecia Areata | IL-10 and TGF-β1 inhibitor | Ointment |
| Phase 4 | Imiquimod (Zyclara®) | Characteristics of T cells from alopecia areata scalp skin before and after treatment with aldara 5% | TLR-7 activation | Ointment |
| Phase 2 | Tralokinumab (Adtralza®, Adbry®) | A pilot study of tralokinumab in subjects with moderate to severe alopecia areata | IL-13 inhibitor | Subcutaneous injection |
| Phase 2 | Triamcinolone (Kenalog®) | Adrenal function and use of intralesional triamcinolone acetonide 10 mg/mL (Kenalog-10) in patients with alopecia areata | Glucocorticoid receptor agonist | Oral medication, subcutaneous/muscle injection, and inhalation |
| Phase 2 | Naltrexone (Revia®) | Oral low-dose naltrexone for lichen planopilaris and frontal fibrosing alopecia | μ-opioid receptor antagonist | Oral medication (tablet), and intramuscular injection |
| Phase 3 | Ingenol | Efficacy and safety of ingenol mebutate gel for actinic keratosis applied on large area on face, scalp or chest | Not fully understood | Ointment |
| Phase 3 | Mebutate | Efficacy and safety of ingenol mebutate gel for actinic keratosis applied on large area on face, scalp or chest | Not fully understood | Ointment |
| Phase 2 | LEO43204 | LEO 124249 ointment in the treatment of alopecia areata | Not fully understood | Ointment |
| Not applicable | Alefacept (Amevive®) | Alefacept in patients with severe scalp alopecia areata | CD2 inhibition | Intramuscular injection |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahn, D.; Kim, H.; Lee, B.; Hahm, D.-H. Psychological Stress-Induced Pathogenesis of Alopecia Areata: Autoimmune and Apoptotic Pathways. Int. J. Mol. Sci. 2023, 24, 11711. https://doi.org/10.3390/ijms241411711
Ahn D, Kim H, Lee B, Hahm D-H. Psychological Stress-Induced Pathogenesis of Alopecia Areata: Autoimmune and Apoptotic Pathways. International Journal of Molecular Sciences. 2023; 24(14):11711. https://doi.org/10.3390/ijms241411711
Chicago/Turabian StyleAhn, Dongkyun, Hyungjun Kim, Bombi Lee, and Dae-Hyun Hahm. 2023. "Psychological Stress-Induced Pathogenesis of Alopecia Areata: Autoimmune and Apoptotic Pathways" International Journal of Molecular Sciences 24, no. 14: 11711. https://doi.org/10.3390/ijms241411711
APA StyleAhn, D., Kim, H., Lee, B., & Hahm, D.-H. (2023). Psychological Stress-Induced Pathogenesis of Alopecia Areata: Autoimmune and Apoptotic Pathways. International Journal of Molecular Sciences, 24(14), 11711. https://doi.org/10.3390/ijms241411711