

Physiological Approaches Targeting Cellular and Mitochondrial Pathways Underlying Adipose Organ Senescence

 ,
,  ,
,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
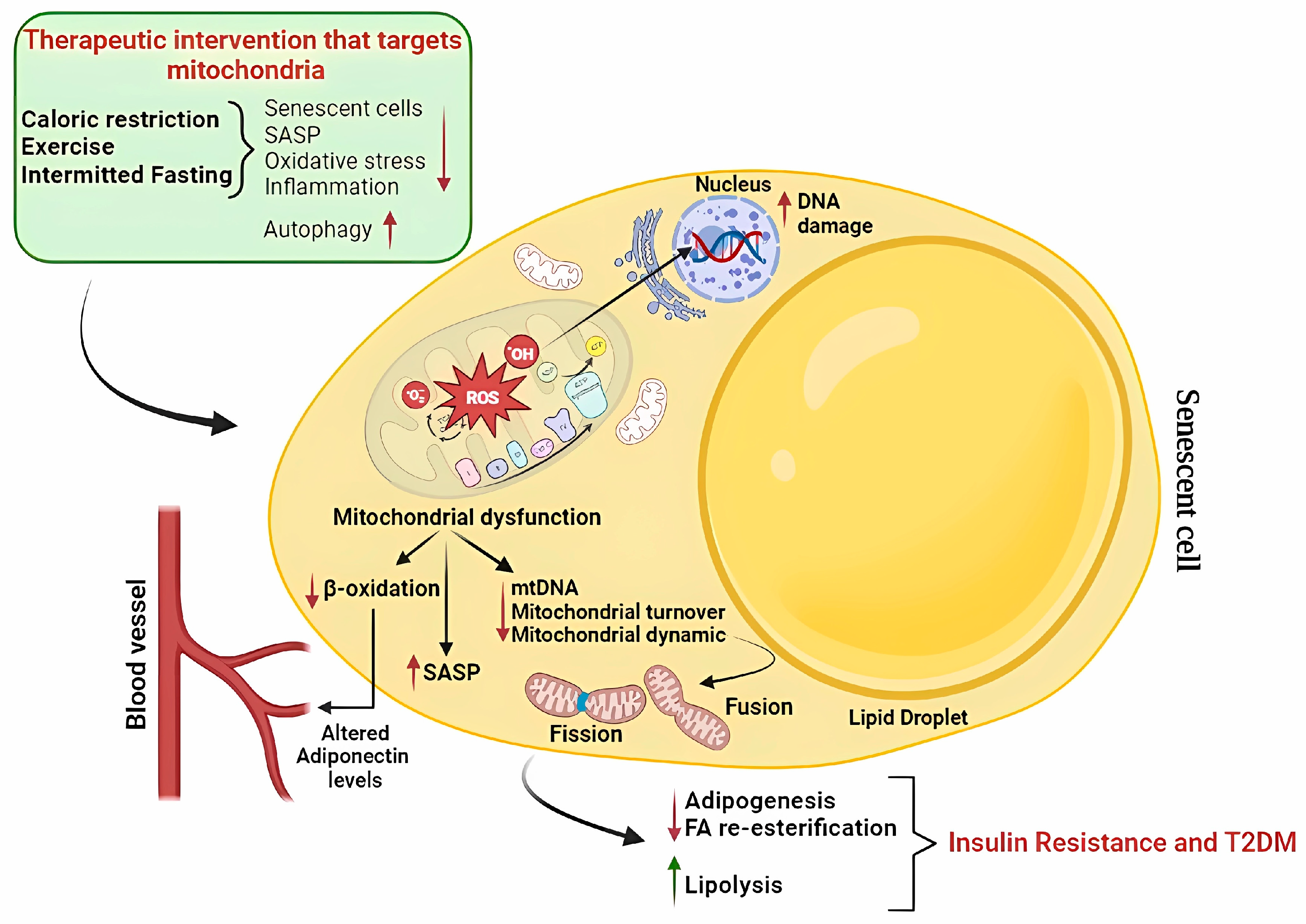{kind=link}
Abstract
1. Introduction
2. Age-Related Adipose Tissue Dysfunction
3. Adipose Tissue Inflammaging and Cell Senescence
4. Mitochondrial Dysfunction in Adipocyte Senescence
5. Adipose–Skeletal-Muscle Crosstalk in Aging
6. Targeting Adipose Senescence to Improve Metabolic Function
- Caloric restriction
- Physical Exercise
- Intermittent fasting
7. From Evidence That Adipose Senescence Drives Age-Related Metabolic Diseases to Approaches to Identify Senolytics
8. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Schaum, N.; Lehallier, B.; Hahn, O.; Pálovics, R.; Hosseinzadeh, S.; Lee, S.E.; Sit, R.; Lee, D.P.; Losada, P.M.; Zardeneta, M.E.; et al. Ageing hallmarks exhibit organ-specific temporal signatures. Nature 2020, 583, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Kim, D.H.; Lee, E.K.; Kim, N.D.; Im, D.S.; Lee, J.; Yu, B.P.; Chung, H.Y. Age-related inflammation and insulin resistance: A review of their intricate interdependency. Arch. Pharm. Res. 2014, 37, 1507–1514. [Google Scholar] [CrossRef] [PubMed]
- Camell, C.D.; Sander, J.; Spadaro, O.; Lee, A.; Nguyen, K.Y.; Wing, A.; Goldberg, E.L.; Youm, Y.H.; Brown, C.W.; Elsworth, J.; et al. Inflammasome-driven catecholamine catabolism in macrophages blunts lipolysis during ageing. Nature 2017, 550, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Zwick, R.K.; Guerrero-Juarez, C.F.; Horsley, V.; Plikus, M.V. Anatomical, physiological, and functional diversity of adipose tissue. Cell Metab. 2018, 27, 68–83. [Google Scholar] [CrossRef]
- Chusyd, D.E.; Wang, D.; Huffman, D.M.; Nagy, T.R. Relationships between rodent white adipose fat pads and human white adipose fat depots. Front. Nutr. 2016, 16, 3–10. [Google Scholar] [CrossRef]
- Funcke, J.B.; Scherer, P.E. Beyond adiponectin and leptin: Adipose tissue-derived mediators of inter-organ communication. J. Lipid Res. 2019, 60, 1648–1684. [Google Scholar] [CrossRef]
- Carpentier, A.C. 100th Anniversary of the discovery of insulin perspective: Insulin and adipose tissue fatty acid metabolism. Am. J. Physiol. Endocrinol. Metab. 2021, 320, E653–E670. [Google Scholar] [CrossRef]
- Angueira, A.R.; Shapira, S.N.; Ishibashi, J.; Sampat, S.; Sostre-Colo’n, J.; Emmett, M.J.; Titchenell, P.M.; Lazar, M.A.; Lim, H.W.; Seale, P. Early B cell factor activity controls developmental and adaptive thermogenic gene programming in adipocytes. Cell Rep. 2020, 30, 2869–2878.e4. [Google Scholar] [CrossRef]
- Gavaldà-Navarro, A.; Villarroya, J.; Cereijo, R.; Giralt, M.; Villarroya, F. The endocrine role of brown adipose tissue: An update on actors and actions. Rev. Endocr. Metab. Disord. 2022, 23, 31–41. [Google Scholar] [CrossRef]
- Sakers, A.; De Siqueira, M.K.; Seale, P.; Villanueva, C.J. Adipose-tissue plasticity in health and disease. Cell 2022, 185, 419–446. [Google Scholar] [CrossRef]
- Rui, L. Brown and Beige Adipose Tissues in Health and Disease. Compr. Physiol. 2017, 7, 1281–1306. [Google Scholar]
- Cheng, L.; Wang, J.; Dai, H.; Duan, Y.; An, Y.; Shi, L.; Lv, Y.; Li, H.; Wang, C.; Ma, Q.; et al. Brown and beige adipose tissue: A novel therapeutic strategy for obesity and type 2 diabetes mellitus. Adipocyte 2021, 10, 48–65. [Google Scholar] [CrossRef]
- Vishvanath, L.; Gupta, R.K. Contribution of adipogenesis to healthy adipose tissue expansion in obesity. J. Clin. Investig. 2019, 129, 4022–4031. [Google Scholar] [CrossRef]
- Boudina, S.; Graham, T.E. Mitochondrial function/dysfunction in white adipose tissue. Exp. Physiol. 2014, 99, 1168–1178. [Google Scholar] [CrossRef]
- Liu, Z.; Wu, K.K.L.; Jiang, X.; Xu, A.; Cheng, K.K.Y. The role of adipose tissue senescence in obesity- and ageing-related metabolic disorders. Clin. Sci. 2020, 134, 315–330. [Google Scholar] [CrossRef]
- Santos, A.L.; Sinha, S. Obesity, and aging: Molecular mechanisms and therapeutic approaches. Ageing Res. Rev. 2021, 67, 101268. [Google Scholar] [CrossRef]
- Trim, W.V.; Walhin, J.P.; Koumanov, F.; Bouloumie, A.; Lindsay, M.A.; Chen, Y.C.; Travers, R.L.; Turner, J.E.; Thompson, D. Divergent immunometabolic changes in adipose tissue and skeletal muscle with ageing in healthy humans. J. Physiol. 2022, 600, 921–947. [Google Scholar] [CrossRef]
- Liu, X.M.; Chan, H.C.; Ding, G.L.; Cai, J.; Song, Y.; Wang, T.T.; Zhang, D.; Chen, H.; Yu, M.K.; Wu, Y.T.; et al. FSH regulates fat accumulation and redistribution in aging through the Galphai/Ca(2+)/CREB pathway. Aging Cell 2015, 14, 409–420. [Google Scholar] [CrossRef]
- Huffman, D.M.; Barzilai, N. Role of visceral adipose tissue in aging. Biochim. Biophys. Acta 2009, 1790, 1117–1123. [Google Scholar] [CrossRef]
- Raguso, C.A.; Kyle, U.; Kossovsky, M.P.; Roynette, C.; Paoloni-Giacobino, A.; Hans, D.; Genton, L.; Pichard, C. A 3-year longitudinal study on body composition changes in the elderly: Role of physical exercise. Clin. Nutr. 2006, 25, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Caso, G.; McNurlan, M.A.; Mileva, I.; Zemlyak, A.; Mynarcik, D.C.; Gelato, M.C. Peripheral fat loss and decline in adipogenesis in older humans. Metab. Clin. Exp. 2013, 62, 337–340. [Google Scholar] [CrossRef] [PubMed]
- Zoico, E.; Rubele, S.; De Caro, A.; Nori, N.; Mazzali, G.; Fantin, F.; Rossi, A.; Zamboni, M. Brown and Beige Adipose Tissue and Aging. Front. Endocrinol. 2019, 10, 368. [Google Scholar] [CrossRef] [PubMed]
- Valle, A.; Guevara, R.; Garcia-Palmer, F.J.; Roca, P.; Oliver, J. Caloric restriction retards the age-related decline in mitochondrial function of brown adipose tissue. Rejuvenation Res. 2008, 11, 597–604. [Google Scholar] [CrossRef]
- McDonald, R.B.; Horwitz, B.A. Brown adipose tissue thermogenesis during aging and senescence. J. Bioenerg. Biomembr. 1999, 31, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Yoneshiro, T.; Ogawa, T.; Okamoto, N.; Matsushita, M.; Aita, S.; Kameya, T.; Iwanaga, T.; Saito, M. Impact of UCP1 and β3AR gene polymorphisms on age-related changes in brown adipose tissue and adiposity in humans. Int. J. Obes. 2013, 37, 993–998. [Google Scholar] [CrossRef]
- Ma, X.; Xu, L.; Gavrilova, O.; Mueller, E. Role of forkhead box protein A3 in age-associated metabolic decline. Proc. Natl. Acad. Sci. USA 2014, 111, 14289–14294. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Wang, L.; Kon, N.; Zhao, W.; Lee, S.; Zhang, Y.; Rosenbaum, M.; Zhao, Y.; Gu, W.; Farmer, S.R.; et al. Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of Pparγ. Cell 2012, 150, 620–632. [Google Scholar] [CrossRef]
- Zhu, M.; Kohan, E.; Bradley, J.; Hedrick, M.; Benhaim, P.; Zuk, P. The effect of age on osteogenic, adipogenic and proliferative potential of female adipose-derived stem cells. J. Tissue Eng. Regen. Med. 2009, 3, 290–301. [Google Scholar] [CrossRef]
- Kim, S.M.; Lun, M.; Wang, M.; Senyo, S.E.; Guillermier, C.; Patwari, P.; Steinhauser, M.L. Loss of white adipose hyperplastic potential is associated with enhanced susceptibility to insulin resistance. Cell Metab. 2014, 20, 1049–1058. [Google Scholar] [CrossRef]
- Mota de Sá, P.; Richard, A.J.; Hang, H.; Stephens, J.M. Transcriptional regulation of adipogenesis. Compr. Physiol. 2017, 7, 635–674. [Google Scholar] [PubMed]
- Tchkonia, T.; Pirtskhalava, T.; Thomou, T.; Cartwright, M.J.; Wise, B.; Karagiannides, I.; Shpilman, A.; Lash, T.L.; Becherer, J.D.; Kirkland, J.L. Increased TNFalpha and CCAAT/enhancer-binding protein homologous protein with aging predispose preadipocytes to resist adipogenesis. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E1810–E1819. [Google Scholar] [CrossRef]
- Fei, J.; Tamski, H.; Cook, C.; Santanam, N. MicroRNA regulation of adipose derived stem cells in aging rats. PLoS ONE 2013, 8, e59238. [Google Scholar] [CrossRef]
- Silva, G.D.N.; Amato, A.A. Thermogenic adipose tissue aging: Mechanisms and implications. Front. Cell. Dev. Biol. 2022, 10, 955612. [Google Scholar] [CrossRef]
- Bruder, J.; Fromme, T. Global Adipose Tissue Remodeling During the First Month of Postnatal Life in Mice. Front. Endocrinol. 2022, 13, 849877. [Google Scholar] [CrossRef]
- Cohen, P.; Kajimura, S. The cellular and functional complexity of thermogenic fat. Nat. Rev. Mol. Cell Biol. 2021, 22, 393–409. [Google Scholar] [CrossRef]
- Rogers, N.H. Brown adipose tissue during puberty and with aging. Ann. Med. 2015, 47, 142–149. [Google Scholar] [CrossRef]
- Gonçalves, L.F.; Machado, T.Q.; Castro-Pinheiro, C.; de Souza, N.G.; Oliveira, K.J.; Fernandes-Santos, C. Ageing is associated with Brown adipose tissue remodelling and loss of white fat browning in female C57BL/6 mice. Int. J. Exp. Pathol. 2017, 98, 100–108. [Google Scholar] [CrossRef]
- Berry, D.C.; Jiang, Y.; Arpke, R.W.; Close, E.L.; Uchida, A.; Reading, D.; Berglund, E.D.; Kyba, M.; Graff, J.M. Cellular aging contributes to failure of cold-induced beige adipocyte formation in old mice and humans. Cell. Metab. 2017, 25, 166–181. [Google Scholar] [CrossRef]
- de Jong, J.M.A.; Sun, W.; Pires, N.D.; Frontini, A.; Balaz, M.; Jespersen, N.Z.; Feizi, A.; Petrovic, K.; Fischer, A.W.; Bokhari, M.H.; et al. Human brown adipose tissue is phenocopied by classical brown adipose tissue in physiologically humanized mice. Nat. Metab. 2019, 1, 830–843. [Google Scholar] [CrossRef]
- Cannon, B.; de Jong, J.M.A.; Fischer, A.W.; Nedergaard, J.; Petrovic, N. Human Brown adipose tissue: Classical Brown rather than brite/beige? Exp. Physiol. 2020, 105, 1191–1200. [Google Scholar] [CrossRef] [PubMed]
- Miles, E.A.; Rees, D.; Banerjee, T.; Cazzola, R.; Lewis, S.; Wood, R.; Oates, R.; Tallant, A.; Cestaro, B.; Yaqoob, P.; et al. Age-related increases in circulating inflammatory markers in men are independent of BMI, blood pressure and blood lipid concentrations. Atherosclerosis 2008, 196, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Soro-Arnaiz, I.; Li, Q.O.Y.; Torres-Capelli, M.; Melendez-Rodriguez, F.; Veiga, S.; Veys, K.; Sebastian, D.; Elorza, A.; Tello, D.; Hernansanz-Agustin, P.; et al. Role of mitochondrial complex IV in age-dependent obesity. Cell. Rep. 2016, 16, 2991–3002. [Google Scholar] [CrossRef]
- Smith, U.; Li, Q.; Rydén, M.; Spalding, K.L. Cellular senescence, and its role in white adipose tissue. Int. J. Obes. 2021, 45, 934–943. [Google Scholar] [CrossRef]
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A.; et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010, 6, 347. [Google Scholar] [CrossRef]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- Teissier, T.; Boulanger, E.; Cox, L.S. Interconnections between Inflammageing and Immunosenescence during Ageing. Cells 2022, 11, 359. [Google Scholar] [CrossRef]
- Cai, Z.; He, B. Adipose tissue aging: An update on mechanisms and therapeutic strategies. Metabolism 2023, 138, 155328. [Google Scholar] [CrossRef]
- De Carvalho, F.G.; Justice, J.N.; Freitas, E.C.; Kershaw, E.E.; Sparks, L.M. Adipose Tissue Quality in Aging: How Structural and Functional Aspects of Adipose Tissue Impact Skeletal Muscle Quality. Nutrients 2019, 11, 2553. [Google Scholar] [CrossRef]
- Nerstedt, A.; Smith, U. The impact of cellular senescence in human adipose tissue. J. Cell Commun. Signal. 2023. [Google Scholar] [CrossRef]
- Xu, M.; Palmer, A.K.; Ding, H.; Weivoda, M.M.; Pirtskhalava, T.; White, T.A.; Sepe, A.; Johnson, K.O.; Stout, M.B.; Giorgadze, N.; et al. Targeting senescent cells enhances adipogenesis and metabolic function in old age. Elife 2015, 4, e12997. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Liu, Y.; Sun, C.; Yin, H. Transient p53 inhibition sensitizes aged white adipose tissue for beige adipocyte recruitment by blocking mitophagy. FASEB J. 2019, 33, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Le Pelletier, L.; Mantecon, M.; Gorwood, J.; Auclair, M.; Foresti, R.; Motterlini, R.; Laforge, M.; Atlan, M.; Feve, B.; Capeau, J.; et al. Metformin alleviates stress-induced cellular senescence of aging human adipose stromal cells and the ensuing adipocyte dysfunction. Elife 2021, 10, e62635. [Google Scholar] [CrossRef]
- Ziqubu, K.; Dludla, P.V.; Mthembu, S.X.H.; Nkambule, B.B.; Mabhida, S.E.; Jack, B.U.; Nyambuya, T.M.; Mazibuko-Mbeje, S.E. An insight into brown/beige adipose tissue whitening, a metabolic complication of obesity with the multifactorial origin. Front. Endocrinol. 2023, 14, 1114767. [Google Scholar] [CrossRef]
- Sarantopoulos, C.N.; Banyard, D.A.; Ziegler, M.E.; Sun, B.; Shaterian, A.; Widgerow, A.D. Elucidating the Preadipocyte and Its Role in Adipocyte Formation: A Comprehensive Review. Stem Cell. Rev. Rep. 2018, 14, 27–42. [Google Scholar] [CrossRef]
- Xu, M.; Tchkonia, T.; Ding, H.; Ogrodnik, M.; Lubbers, E.R.; Pirtskhalava, T.; White, T.A.; Johnson, K.O.; Stout, M.B.; Mezera, V.; et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc. Natl. Acad. Sci. USA 2015, 112, E6301–E6310. [Google Scholar] [CrossRef]
- Zhao, M.; Chen, X. Effect of lipopolysaccharides on adipogenic potential and premature senescence of adipocyte progenitors. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E334–E344. [Google Scholar] [CrossRef]
- Krstic, J.; Reinisch, I.; Schupp, M.; Schulz, T.J.; Prokesch, A. p53 functions in adipose tissue metabolism and homeostasis. Int. J. Mol. Sci. 2018, 19, 2622. [Google Scholar] [CrossRef]
- Vergoni, B.; Cornejo, P.J.; Gilleron, J.; Djedaini, M.; Ceppo, F.; Jacquel, A.; Bouget, G.; Ginet, C.; Gonzalez, T.; Maillet, J.; et al. DNA Damage and the Activation of the p53 Pathway Mediate Alterations in Metabolic and Secretory Functions of Adipocytes. Diabetes 2016, 65, 3062–3074. [Google Scholar] [CrossRef]
- Minamino, T.; Orimo, M.; Shimizu, I.; Kunieda, T.; Yokoyama, M.; Ito, T.; Nojima, A.; Nabetani, A.; Oike, Y.; Matsubara, H.; et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat. Med. 2009, 15, 1082–1087. [Google Scholar] [CrossRef]
- Ejaz, A.; Mattesich, M.; Zwerschke, W. Silencing of the small GTPase DIRAS3 induces cellular senescence in human white adipose stromal/progenitor cells. Aging 2017, 9, 860–879. [Google Scholar] [CrossRef] [PubMed]
- Tchkonia, T.; Morbeck, D.E.; Von Zglinicki, T.; Van Deursen, J.; Lustgarten, J.; Scrable, H.; Khosla, S.; Jensen, M.D.; Kirkland, J.L. Fat tissue, aging, and cellular senescence. Aging Cell 2010, 9, 667–684. [Google Scholar] [CrossRef]
- Suganami, T.; Nishida, J.; Ogawa, Y. A paracrine loop between adipocytes and macrophages aggravates inflammatory changes: Role of free fatty acids and tumor necrosis factor alpha. Arterioscler. Thromb. Vasc. Biol. 2015, 25, 2062–2068. [Google Scholar] [CrossRef]
- Gustafson, B.; Nerstedt, A.; Spinelli, R.; Beguinot, F.; Smith, U. Type 2 diabetes, independent of obesity and age, is characterized by senescent and dysfunctional mature human adipose cells. Diabetes 2022, 71, 2372–2383. [Google Scholar] [CrossRef]
- Zaragosi, L.E.; Wdziekonski, B.; Villageois, P.; Keophiphath, M.; Maumus, M.; Tchkonia, T.; Bourlier, V.; Mohsen-Kanson, T.; Ladoux, A.; Elabd, C.; et al. Activin a plays a critical role in proliferation and differentiation of human adipose progenitors. Diabetes 2010, 59, 2513–2521. [Google Scholar] [CrossRef]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef]
- Ikeda, K.; Maretich, P.; Kajimura, S. The Common and Distinct Features of Brown and Beige Adipocytes. Trends Endocrinol. Metab. 2018, 29, 191–200. [Google Scholar] [CrossRef]
- Lee, J.H.; Park, A.; Oh, K.J.; Lee, S.C.; Kim, W.K.; Bae, K.H. The Role of Adipose Tissue Mitochondria: Regulation of Mitochondrial Function for the Treatment of Metabolic Diseases. Int. J. Mol. Sci. 2019, 20, 4924. [Google Scholar] [CrossRef]
- Heinonen, S.; Jokinen, R.; Rissanen, A.; Pietiläinen, K.H. White adipose tissue mitochondrial metabolism in health and in obesity. Obes. Rev. 2020, 21, e12958. [Google Scholar] [CrossRef]
- Kladnická, I.; Čedíková, M.; Kripnerová, M.; Dvořáková, J.; Kohoutová, M.; Tůma, Z.; Müllerová, D.; Kuncová, J. Mitochondrial respiration of adipocytes differentiating from human mesenchymal stem cells derived from adipose tissue. Physiol. Res. 2019, 68 (Suppl. 3), S287–S296. [Google Scholar] [CrossRef]
- Joffin, N.; Paschoal, V.A.; Gliniak, C.M.; Crewe, C.; Elnwasany, A.; Szweda, L.I.; Zhang, Q.; Hepler, C.; Kusminski, C.M.; Gordillo, R.; et al. Mitochondrial metabolism is a key regulator of the fibro-inflammatory and adipogenic stromal subpopulations in white adipose tissue. Cell Stem Cell 2021, 28, 702–717.e8. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.; Wang, O.A.; Song, A.; Vishvanath, L.; Busbuso, N.C.; Scherer, P.E.; Gupta, R.K. Cellular origins of beige fat cells revisited. Diabetes 2019, 68, 1874–1885. [Google Scholar] [CrossRef] [PubMed]
- Forner, F.; Kumar, C.; Luber, C.A.; Fromme, T.; Klingenspor, M.; Mann, M. Proteome differences between brown and white fat mitochondria reveal specialized metabolic functions. Cell Metab. 2009, 10, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, A.; Krause, F.N.; Moran, A.; MacCannell, A.D.V.; Scragg, J.L.; McNally, B.D.; Boateng, E.; Murfitt, S.A.; Virtue, S.; Wright, J.; et al. Brown and beige adipose tissue regulate systemic metabolism through a metabolite interorgan signaling axis. Nat. Commun. 2021, 12, 1905. [Google Scholar] [CrossRef]
- MacCannell, A.D.V.; Roberts, L.D. Metabokines in the regulation of systemic energy metabolism. Curr. Opin. Pharmacol. 2022, 67, 102286–102360. [Google Scholar] [CrossRef]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Xin, Y.; Zhao, L.; Peng, R. HIF-1 signaling: An emerging mechanism for mitochondrial dynamics. J. Physiol. Biochem. 2023, 79, 489–500. [Google Scholar] [CrossRef]
- Skeie, J.M.; Nishimura, D.Y.; Wang, C.L.; Schmidt, G.A.; Aldrich, B.T.; Greiner, M.A. Mitophagy: An Emerging Target in Ocular Pathology. Invest. Ophthalmol. Vis. Sci. 2021, 62, 22. [Google Scholar] [CrossRef]
- Fielder, E.; Wan, T.; Alimohammadiha, G.; Ishaq, A.; Low, E.; Weigand, B.M.; Kelly, G.; Parker, C.; Griffin, B.; Jurk, D.; et al. Short senolytic or senostatic interventions rescue progression of radiation-induced frailty and premature ageing in mice. Elife 2022, 4, e75492. [Google Scholar] [CrossRef]
- Kang, H.T.; Lee, K.B.; Kim, S.Y.; Choi, H.R.; Park, S.C. Autophagy impairment induces premature senescence in primary human fibroblasts. PLoS ONE 2011, 6, e23367. [Google Scholar] [CrossRef]
- Yoon, Y.S.; Yoon, D.S.; Lim, I.K.; Yoon, S.H.; Chung, H.Y.; Rojo, M.; Malka, F.; Jou, M.J.; Martinou, J.C.; Yoon, G. Formation of elongated giant mitochondria in DFO-induced cellular senescence: Involvement of enhanced fusion process through modulation of Fis1. J. Cell Physiol. 2006, 209, 468–480. [Google Scholar] [CrossRef]
- Lee, S.; Jeong, S.Y.; Lim, W.C.; Kim, S.; Park, Y.Y.; Sun, X.; Youle, R.J.; Ch, H. Mitochondrial fission and fusion mediators, hFis1 and OPA1, modulate cellular senescence. J. Biol. Chem. 2007, 282, 22977–22983. [Google Scholar] [CrossRef]
- Martini, H.; Passos, J.F. Cellular senescence: All roads lead to mitochondria. FEBS J. 2022, 290, 1186–1202. [Google Scholar] [CrossRef]
- Protasoni, M.; Serrano, M. Targeting Mitochondria to Control Ageing and Senescence. Pharmaceutics 2023, 15, 352. [Google Scholar] [CrossRef]
- Macip, S.; Igarashi, M.; Fang, L.; Chen, A.; Pan, Z.Q.; Lee, S.W.; Aaronson, S.A. Inhibition of p21-mediated ROS accumulation can rescue p21-induced senescence. EMBO J. 2002, 21, 2180–2188. [Google Scholar] [CrossRef]
- Miwa, S.; Kashyap, S.; Chini, E.; von Zglinicki, T. Mitochondrial dysfunction in cell senescence and aging. J. Clin. Investig. 2022, 132, e158447. [Google Scholar] [CrossRef]
- Chapman, J.; Fielder, E.; Passos, J.F. Mitochondrial dysfunction, and cell senescence: Deciphering a complex relationship. FEBS Lett. 2019, 59, 1566–1579. [Google Scholar] [CrossRef]
- Chen, C.; Zhou, M.; Ge, Y.; Wang, X. SIRT1 and aging related signaling pathways. Mech. Ageing Dev. 2020, 187, 111215. [Google Scholar] [CrossRef]
- Wang, G.; Meyer, J.G.; Cai, W.; Softic, S.; Li, M.E.; Verdin, E.; Newgard, C.; Schilling, B.; Kahn, C.R. Regulation of UCP1 and Mitochondrial Metabolism in Brown Adipose Tissue by Reversible Succinylation. Mol. Cell 2019, 74, 844–857.e7. [Google Scholar] [CrossRef]
- Sidler, C.; Kovalchuk, O.; Kovalchuk, I. Epigenetic Regulation of Cellular Senescence and aging. Front. Genet. 2017, 8, 138. [Google Scholar]
- Parka, Y.J.; Leed, S.; Limd, S.; Nahmgoonga, H.; Jia, Y.; Huha, J.Y.; Alfaddae, A.A.; Kimd, S.; Kim, J.B. DNMT1 maintains metabolic fitness of adipocytes through acting as an epigenetic safeguard of mitochondrial dynamics. Proc. Natl. Acad. Sci. USA 2021, 118, e2021073118. [Google Scholar] [CrossRef] [PubMed]
- Lemecha, M.; Morino, K.; Imamura, T.; Iwasaki, H.; Ohashi, N.; Ida, S.; Sato, D.; Sekine, O.; Ugi, S.; Maegawa, H. MiR-494-3p regulates mitochondrial biogenesis and thermogenesis through PGC1-α signalling in beige adipocytes. Sci. Rep. 2018, 8, 15096. [Google Scholar] [CrossRef] [PubMed]
- Koh, E.H.; Chen, Y.; Bader, D.A.; Hamilton, M.P.; He, B.; York, B.; Kajimura, S.; McGuire, S.E.; Hartig, S.M. Mitochondrial Activity in Human White Adipocytes Is Regulated by the Ubiquitin Carrier Protein 9/microRNA-30a Axis. J. Biol. Chem. 2016, 291, 24747–24755. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Zhu, W.; Zheng, F.; Gui, W.; Zhang, W.; Lin, X.; Li, H. The Long Noncoding RNA Blnc1 Protects Against Diet-Induced Obesity by Promoting Mitochondrial Function in White Fat. Diabetes Metab. Syndr. Obes. 2020, 13, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Yue, F.; Jia, Z.; Gao, Y.; Jin, W.; Hu, K.; Zhang, Y.; Zhu, D.; Yang, G.; Kuang, S. A novel brown adipocyte-enriched long non-coding RNA that is required for brown adipocyte differentiation and sufficient to drive thermogenic gene program in white adipocytes. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 2018, 1863, 409–419. [Google Scholar] [CrossRef]
- Colitti, M.; Montanari, T. Brain-derived neurotrophic factor modulates mitochondrial dynamics and thermogenic phenotype on 3T3-L1 adipocytes. Tissue Cell 2020, 66, 101388. [Google Scholar] [CrossRef]
- Zhou, Z.; Moore, T.M.; Drew, B.G.; Ribas, V.; Wanagat, J.; Civelek, M.; Segawa, M.; Wolf, D.M.; Norheim, F.; Seldin, M.M.; et al. Estrogen receptor α controls metabolism in white and brown adipocytes by regulating Polg1 and mitochondrial remodeling. Sci. Transl. Med. 2020, 12, eaax8096. [Google Scholar] [CrossRef]
- Bauzá-Thorbrügge, M.; Rodríguez-Cuenca, S.; Vidal-Puig, A.; Galmés-Pascual, B.M.; Sbert-Roig, M.; Gianotti, M.; Lladó, I.; Proenza, A.M. GPER and ERα mediate estradiol enhancement of mitochondrial function in inflamed adipocytes through a PKA dependent mechanism. J. Steroid Biochem. Mol. Biol. 2019, 185, 256–267. [Google Scholar] [CrossRef]
- Norman-Burgdolf, H.; Li, D.; Sullivan, P.; Wang, S. CD47 differentially regulates white and brown fat function. Biol. Open 2020, 9, bio056747. [Google Scholar] [CrossRef]
- Qian, S.; Pan, J.; Su, Y.; Tang, Y.; Wang, Y.; Zou, Y.; Zhao, Y.; Ma, H.; Zhang, Y.; Liu, Y.; et al. BMPR2 promotes fatty acid oxidation and protects white adipocytes from cell death in mice. Commun. Biol. 2020, 3, 200. [Google Scholar] [CrossRef]
- Brestoff, J.R.; Wilen, C.B.; Moley, J.R.; Li, Y.; Zou, W.; Malvin, N.P.; Rowen, M.N.; Saunders, B.T.; Ma, H.; Mack, M.R.; et al. Intercellular Mitochondria Transfer to Macrophages Regulates White Adipose Tissue Homeostasis and Is Impaired in Obesity. Cell. Metab. 2021, 33, 270–282.e8. [Google Scholar] [CrossRef]
- Crewe, C.; Scherer, P.E. Intercellular and interorgan crosstalk through adipocyte extracellular vesicles. Rev. Endocr. Metab. Disord. 2022, 23, 61–69. [Google Scholar] [CrossRef]
- Puhm, F.; Afonyushkin, T.; Resch, U.; Obermayer, G.; Rohde, M.; Penz, T.; Schuster, M.; Wagner, G.; Rendeiro, A.F.; Melki, I.; et al. Mitochondria Are a Subset of Extracellular Vesicles Released by Activated Monocytes and Induce Type I IFN and TNF Responses in Endothelial Cells. Circ. Res. 2019, 125, 43–52. [Google Scholar] [CrossRef]
- Clement, E.; Lazar, I.; Attané, C.; Carrié, L.; Dauvillier, S.; Ducoux-Petit, M.; Esteve, D.; Menneteau, T.; Moutahir, M.; Le Gonidec, S.; et al. Adipocyte extracellular vesicles carry enzymes and fatty acids that stimulate mitochondrial metabolism and remodeling in tumor cells. EMBO J. 2020, 39, e102525. [Google Scholar] [CrossRef]
- Fu, A.; Shi, X.; Zhang, H.; Fu, B. Mitotherapy for Fatty Liver by Intravenous Administration of Exogenous Mitochondria in Male Mice. Front. Pharmacol. 2017, 8, 241. [Google Scholar] [CrossRef]
- Batsis, J.A. Obesity in the Older Adult: Special Issue. J. Nutr. Geront. Geriatr. 2019, 38, 1–5. [Google Scholar] [CrossRef]
- Zhang, Y.X.; Ou, M.Y.; Yang, Z.H.; Sun, Y.; Li, Q.F.; Zhou, S.B. Adipose tissue aging is regulated by an altered immune system. Front. Immunol. 2023, 14, 1125395. [Google Scholar] [CrossRef]
- Sachs, S.; Zarini, S.; Kahn, D.E.; Harrison, K.A.; Perreault, L.; Phang, T.; Newsom, S.A.; Strauss, A.; Kerege, A.; Schoen, J.A.; et al. Intermuscular adipose tissue directly modulates skeletal muscle insulin sensitivity in humans. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E866–E879. [Google Scholar] [CrossRef]
- Foryst-Ludwig, A.; Kreissl, M.C.; Benz, V.; Brix, S.; Smeir, E.; Ban, Z.; Januszewicz, E.; Salatzki, J.; Grune, J.; Schwanstecher, A.K.; et al. Adipose Tissue Lipolysis Promotes Exercise-induced Cardiac Hypertrophy Involving the Lipokine C16:1n7-Palmitoleate. J. Biol. Chem. 2015, 290, 23603–23615. [Google Scholar] [CrossRef]
- Goodpaster, B.H.; Bergman, B.C.; Brennan, A.M.; Sparks, L.M. Intermuscular adipose tissue in metabolic disease. Nat. Rev. Endocrinol. 2023, 19, 285–298. [Google Scholar] [CrossRef]
- Pellegrinelli, V.; Rouault, C.; Rodriguez-Cuenca, S.; Albert, V.; Edom-Vovard, F.; Vidal-Puig, A.; Clement, K.; Butler-Browne, G.S.; Lacasa, D. Human Adipocytes Induce Inflammation and Atrophy in Muscle Cells During Obesity. Diabetes 2015, 64, 3121–3134. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.S.; Justice, J.N.; Thompson, L. Lipotoxicity, aging, and muscle contractility: Does fiber type matter? Geroscience 2019, 41, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Stanford, K.I.; Goodyear, L.J. Muscle-Adipose Tissue Cross Talk. Cold Spring Harb. Perspect. Med. 2018, 8, a029801. [Google Scholar] [CrossRef]
- Li, F.; Li, Y.; Duan, Y.; Hu, C.A.; Tang, Y.; Yin, Y. Myokines and adipokines: Involvement in the crosstalk between skeletal muscle and adipose tissue. Cytokine Growth Factor Rev. 2017, 33, 73–82. [Google Scholar] [CrossRef]
- Mau, T.; Yung, R. Adipose tissue inflammation in aging. Exp. Gerontol. 2018, 105, 27–31. [Google Scholar] [CrossRef]
- Trim, W.; Turner, J.E.; Thompson, D. Parallels in Immunometabolic Adipose Tissue Dysfunction with Ageing and Obesity. Front. Immunol. 2018, 9, 169. [Google Scholar] [CrossRef]
- Zhu, S.; Tian, Z.; Torigoe, D.; Zhao, J.; Xie, P.; Sugizaki, T.; Sato, M.; Horiguchi, H.; Terada, K.; Kadomatsu, T.; et al. Aging- and obesity-related peri-muscular adipose tissue accelerates muscle atrophy. PLoS ONE 2019, 14, e0221366. [Google Scholar] [CrossRef]
- Parise, G.; Brose, A.N.; Tarnopolsky, M.A. Resistance exercise training decreases oxidative damage to DNA and increases cytochrome oxidase activity in older adults. Exp. Gerontol. 2005, 40, 173–180. [Google Scholar] [CrossRef]
- Parise, G.; Phillips, S.M.; Kaczor, J.J.; Tarnopolsky, M.A. Antioxidant enzyme activity is up-regulated after unilateral resistance exercise training in older adults. Free Radic. Biol. Med. 2005, 39, 289–295. [Google Scholar] [CrossRef]
- Ljubicic, V.; Joseph, A.M.; Adhihetty, P.J.; Huang, J.H.; Saleem, A.; Uguccioni, G.; Hood, D.A. Molecular basis for an attenuated mitochondrial adaptive plasticity in aged skeletal muscle. Aging 2009, 1, 818–830. [Google Scholar] [CrossRef]
- Fealy, C.E.; Grevendonk, L.; Hoeks, J.; Hesselink, M.K.C. Skeletal muscle mitochondrial network dynamics in metabolic disorders and aging. Trends Mol. Med. 2021, 27, 1033–1044. [Google Scholar] [CrossRef]
- Ferri, E.; Marzetti, E.; Calvani, R.; Picca, A.; Cesari, M.; Arosio, B. Role of Age-Related Mitochondrial Dysfunction in Sarcopenia. Int. J. Mol. Sci. 2020, 21, 5236. [Google Scholar] [CrossRef]
- Palmer, A.K.; Tchkonia, T.; Kirkland, J.L. Targeting cellular senescence in metabolic disease. Mol. Metab. 2022, 66, 101601. [Google Scholar] [CrossRef]
- Conley, S.M.; Hickson, L.J.; Kellogg, T.A.; McKenzie, T.; Heimbach, J.K.; Taner, T.; Lerman, L.O. Human Obesity Induces Dysfunction and Early Senescence in Adipose Tissue-Derived Mesenchymal Stromal/Stem Cells. Front. Cell. Dev. Biol. 2020, 8, 197. [Google Scholar] [CrossRef]
- Gustafson, B.; Nerstedt, A.; Smith, U. Reduced subcutaneous adipogenesis in human hypertrophic obesity is linked to senescent precursor cells. Nat. Commun. 2019, 10, 2757. [Google Scholar] [CrossRef]
- Li, Q.; Hagberg, C.E.; Silva Cascales, H.; Lang, S.; Hyvönen, M.T.; Salehzadeh, F.; Chen, P.; Alexandersson, I.; Terezaki, E.; Harms, M.J.; et al. Obesity and hyperinsulinemia drive adipocytes to activate a cell cycle program and senesce. Nat. Med. 2021, 27, 1941–1953. [Google Scholar] [CrossRef]
- Xie, H.; Liu, X.; Zhou, Q.; Huang, T.; Zhang, L.; Gao, J.; Wang, Y.; Liu, Y.; Yan, T.; Zhang, S.; et al. DNA Methylation Modulates Aging Process in Adipocytes. Aging Dis. 2022, 13, 433–446. [Google Scholar] [CrossRef]
- Castellano-Castillo, D.; Denechaud, P.D.; Fajas, L.; Moreno-Indias, I.; Oliva-Olivera, W.; Tinahones, F.; Queipo-Ortuño, M.I.; Cardona, F. Human adipose tissue H3K4me3 histone mark in adipogenic, lipid metabolism and inflammatory genes is positively associated with BMI and HOMA-IR. PLoS ONE 2019, 14, e0215083. [Google Scholar] [CrossRef]
- Zhang, Z.; Luo, K.; Zou, Z.; Qiu, M.; Tian, J.; Sieh, L.; Shi, H.; Zou, Y.; Wang, G.; Morrison, J.; et al. Genetic analyses support the contribution of mRNA N6-methyladenosine (m6A) modification to human disease heritability. Nat. Genet. 2020, 52, 939–949. [Google Scholar] [CrossRef]
- Wang, L.; Wang, B.; Gasek, N.S.; Zhou, Y.; Cohn, R.L.; Martin, D.E.; Zuo, W.; Flynn, W.F.; Guo, C.; Jellison, E.R.; et al. Targeting p21Cip1 highly expressing cells in adipose tissue alleviates insulin resistance in obesity. Cell. Metab. 2022, 34, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.T.; Tuday, E.; Allen, S.; Kim, J.; Trott, D.W.; Holland, W.L.; Donato, A.J.; Lesniewski, L.A. Senolytic drugs, dasatinib and quercetin, attenuate adipose tissue inflammation, and ameliorate metabolic function in old age. Aging Cell 2023, 22, e13767. [Google Scholar] [CrossRef] [PubMed]
- Bartelt, A.; Bruns, O.T.; Reimer, R.; Hohenberg, H.; Ittrich, H.; Peldschus, K.; Kaul, M.G.; Tromsdorf, U.I.; Weller, H.; Waurisch, C.; et al. Brown adipose tissue activity controls triglyceride clearance. Nat. Med. 2011, 17, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Gentile, A.; Magnacca, N.; De Matteis, R.; Moreno, M.; Cioffi, F.; Giacco, A.; Lanni, A.; de Lange, P.; Senese, R.; Goglia, F.; et al. Ablation of uncoupling protein 3 affects interrelated factors leading to lipolysis and insulin resistance in visceral white adipose tissue. FASEB J. 2022, 36, e22325. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, E.; Senese, R.; De Matteis, R.; Cioffi, F.; Moreno, M.; Lanni, A.; Gentile, A.; Busiello, R.A.; Salzano, A.M.; Scaloni, A.; et al. Absence of uncoupling protein 3 at thermoneutrality influences brown adipose tissue mitochondrial functionality in mice. FASEB J. 2020, 34, 15146–15163. [Google Scholar] [CrossRef] [PubMed]
- Vegiopoulos, A.; Müller-Decker, K.; Strzoda, D.; Schmitt, I.; Chichelnitskiy, E.; Ostertag, A.; Berriel Diaz, M.; Rozman, J.; Hrabe de Angelis, M.; Nüsing, R.M.; et al. Cyclooxygenase-2 controls energy homeostasis in mice by de novo recruitment of brown adipocytes. Science 2010, 328, 1158–1161. [Google Scholar] [CrossRef]
- Seale, P.; Conroe, H.M.; Estall, J.; Kajimura, S.; Frontini, A.; Ishibashi, J.; Cohen, P.; Cinti, S.; Spiegelman, B.M. Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. J. Clin. Investig. 2011, 121, 96–105. [Google Scholar] [CrossRef]
- Wu, J.; Boström, P.; Sparks, L.M.; Ye, L.; Choi, J.H.; Giang, A.H.; Khandekar, M.; Virtanen, K.A.; Nuutila, P.; Schaart, G.; et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 2012, 150, 366–376. [Google Scholar] [CrossRef]
- Sidossis, L.; Kajimura, S. Brown and beige fat in humans: Thermogenic adipocytes that control energy and glucose homeostasis. J. Clin. Investig. 2015, 125, 478–486. [Google Scholar] [CrossRef]
- Gasek, N.S.; Kuchel, G.A.; Kirkland, J.L.; Xu, M. Strategies for Targeting Senescent Cells in Human Disease. Nat. Aging 2021, 1, 870–879. [Google Scholar] [CrossRef]
- Chaib, S.; Tchkonia, T.; Kirkland, J.L. Cellular senescence and senolytics: The path to the clinic. Nat. Med. 2022, 28, 1556–1568. [Google Scholar] [CrossRef]
- Chimienti, G.; Picca, A.; Fracasso, F.; Russo, F.; Orlando, A.; Riezzo, G.; Leeuwenburgh, C.; Pesce, V.; Lezza, A.M.S. The age-sensitive efficacy of calorie restriction on mitochondrial biogenesis and mtdna damage in rat liver. Int. J. Mol. Sci. 2021, 22, 1665. [Google Scholar] [CrossRef]
- Fontana, L.; Nehme, J.; Demaria, M. Caloric restriction and cellular senescence. Mech. Ageing Dev. 2018, 176, 19–23. [Google Scholar] [CrossRef]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef]
- Herranz, D.; Serrano, M. SIRT1: Recent lessons from mouse models. Nat. Rev. Cancer 2010, 10, 819–823. [Google Scholar] [CrossRef]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-Dependent Regulation of Foxo Transcription Factors by the SIRT1 Deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef]
- Zhang, S.; Sun, S.; Wei, X.; Zhang, M.; Chen, Y.; Mao, X.; Chen, G.; Liu, C. Short-term moderate caloric restriction in a high-fat diet alleviates obesity via AMPK/SIRT1 signaling in white adipocytes and liver. Food Nutr. Res. 2022, 3, 66. [Google Scholar] [CrossRef]
- Muzumdar, R.; Allison, D.B.; Huffman, D.M.; Ma, X.; Atzmon, G.; Einstein, F.H.; Fishman, S.; Poduval, A.D.; McVei, T.; Keith, S.W.; et al. Visceral adipose tissue modulates mammalian longevity. Aging Cell 2008, 7, 438–440. [Google Scholar] [CrossRef]
- Corrales, P.; Vivas, Y.; Izquierdo-Lahuerta, A.; Horrillo, D.; Seoane-Collazo, P.; Velasco, I.; Torres, L.; Lopez, Y.; Martínez, C.; López, M.; et al. Long-term caloric restriction ameliorates deleterious effects of aging on white and brown adipose tissue plasticity. Aging Cell 2019, 18, e12948. [Google Scholar] [CrossRef]
- Spadaro, O.; Youm, Y.; Shchukina, I.; Ryu, S.; Sidorov, S.; Ravussin, A.; Nguyen, K.; Aladyeva, E.; Predeus, A.N.; Smith, S.R.; et al. Caloric restriction in humans reveals immunometabolic regulators of health span. Science 2022, 375, 671–677. [Google Scholar] [CrossRef]
- Veech, R.L.; Bradshaw, P.C.; Clarke, K.; Curtis, W.; Pawlosky, R.; King, M.T. Ketone bodies mimic the life span extending properties of caloric restriction. IUBMB Life 2017, 69, 305–314. [Google Scholar] [CrossRef]
- Wang, W.; Ishibashi, J.; Trefely, S.; Shao, M.; Cowan, A.J.; Sakers, A.; Lim, H.W.; O’Connor, S.; Doan, M.T.; Cohen, P.; et al. A PRDM16-Driven Metabolic Signal from Adipocytes Regulates Precursor Cell Fate. Cell Metab. 2019, 30, 174–189.e5. [Google Scholar] [CrossRef] [PubMed]
- Madeo, F.; Carmona-Gutierrez, D.; Hofer, S.J.; Guido Kroemer, G. Caloric Restriction Mimetics against Age-Associated Disease: Targets, Mechanisms, and Therapeutic Potential. Cell. Metab. 2019, 29, 592–610. [Google Scholar] [CrossRef] [PubMed]
- Green, C.L.; Lamming, D.W.; Fontana, L. Molecular mechanisms of dietary restriction promoting health and longevity. Nat. Rev. Mol. Cell. Biol. 2022, 23, 56–73. [Google Scholar] [CrossRef] [PubMed]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef]
- Andrade, J.M.; Frade, A.C.; Guimarães, J.B.; Freitas, K.M.; Lopes, M.T.; Guimarães, A.L.; de Paula, A.M.; Coimbra, C.C.; Santos, S.H. Resveratrol increases brown adipose tissue thermogenesis markers by increasing SIRT1 and energy expenditure and decreasing fat accumulation in adipose tissue of mice fed a standard diet. Eur. J. Nutr. 2014, 53, 1503–1510. [Google Scholar] [CrossRef]
- Flachs, P.; Horakova, O.; Brauner, P.; Rossmeisl, M.; Pecina, P.; Franssen-van Hal, N.; Ruzickova, J.; Sponarova, J.; Drahota, Z.; Vlcek, C.; et al. Polyunsaturated fatty acids of marine origin upregulate mitochondrial biogenesis and induce beta-oxidation in white fat. Diabetologia 2005, 48, 2365–2375. [Google Scholar] [CrossRef]
- Englund, D.A.; Sakamoto, A.E.; Fritsche, C.M.; Heeren, A.A.; Zhang, X.; Kotajarvi, B.R.; Lecy, D.R.; Yousefzadeh, M.J.; Schafer, M.J.; White, T.A.; et al. Exercise reduces circulating biomarkers of cellular senescence in humans. Aging Cell 2021, 20, e13415. [Google Scholar] [CrossRef]
- Justice, J.N.; Gregory, H.; Tchkonia, T.; LeBrasseur, N.K.; Kirkland, J.L.; Kritchevsky, S.B.; Nicklas, B.J. Cellular Senescence Biomarker p16INK4a+ Cell Burden in Thigh Adipose is Associated with Poor Physical Function in Older Women. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 939–945. [Google Scholar] [CrossRef]
- Chien, Y.; Scuoppo, C.; Wang, X.; Fang, X.; Balgley, B.; Bolden, J.E.; Premsrirut, P.; Luo, W.; Chicas, A.; Lee, C.S.; et al. Control of the senescence-associated secretory phenotype by NF-kappaB promotes senescence and enhances chemosensitivity. Genes Dev. 2011, 25, 2125–2136. [Google Scholar] [CrossRef]
- Herranz, N.; Gallage, S.; Mellone, M.; Wuestefeld, T.; Klotz, S.; Hanley, C.J.; Raguz, S.; Acosta, J.C.; Innes, A.J.; Banito, A.; et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell. Biol. 2015, 17, 1205–1217. [Google Scholar] [CrossRef]
- Laberge, R.M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell. Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef]
- Sorriento, D.; Di Vaia, E.; Iaccarino, G. Physical Exercise: A Novel Tool to Protect Mitochondrial Health. Front. Physiol. 2021, 12, 660068. [Google Scholar] [CrossRef]
- McGee, S.L.; Hargreaves, M. Exercise adaptations: Molecular mechanisms and potential targets for therapeutic benefit. Nat. Rev. Endocrinol. 2020, 16, 495–505. [Google Scholar] [CrossRef]
- Li, J.; Wang, Z.; Li, C.; Song, Y.; Wang, Y.; Bo, H.; Zhang, Y. Impact of Exercise and Aging on Mitochondrial Homeostasis in Skeletal Muscle: Roles of ROS and Epigenetics. Cells 2022, 11, 2086. [Google Scholar] [CrossRef]
- Mori, M.A.; Raghavan, P.; Thomou, T.; Boucher, J.; Robida-Stubbs, S.; Macotela, Y.; Russell, S.J.; Kirkland, J.L.; Blackwell, T.K.; Kahn, C.R. Role of microRNA processing in adipose tissue in stress defense and longevity. Cell Metab. 2012, 16, 336–347. [Google Scholar] [CrossRef]
- Oliverio, M.; Schmidt, E.; Mauer, J.; Baitzel, C.; Hansmeier, N.; Khani, S.; Konieczka, S.; Pradas-Juni, M.; Brodesser, S.; Van, T.M.; et al. Dicer1-miR-328-Bace1 signalling controls brown adipose tissue differentiation and function. Nat. Cell. Biol. 2016, 18, 328–336. [Google Scholar] [CrossRef]
- Brandão, B.B.; Madsen, S.; Rabiee, A.; Oliverio, M.; Ruiz, G.P.; Ferrucci, D.L.; Branquinho, J.L.; Razolli, D.; Pinto, S.; Nielsen, T.S.; et al. Dynamic changes in DICER levels in adipose tissue control metabolic adaptations to exercise. Proc. Natl. Acad. Sci. USA 2020, 117, 23932–23941. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, Z.; Zeng, T.; Lim, Y.C.; Wang, Y.; Xie, X.; Yang, S.; Huang, C.; Xu, M.; Tao, L.; et al. cAMP-MicroRNA-203-IFNγ network regulates subcutaneous white fat browning and glucose tolerance. Mol. Metab. 2019, 28, 36–47. [Google Scholar] [CrossRef]
- Liu, B.; Page, A.J.; Hatzinikolas, G.; Chen, M.; Wittert, G.A.; Heilbronn, L.K. Intermittent fasting improves glucose tolerance and promotes adipose tissue remodeling in male mice fed a high-fat diet. Endocrinology 2019, 160, 169–180. [Google Scholar] [CrossRef]
- Liu, B.; Page, A.J.; Hutchison, A.T.; Wittert, G.A.; Heilbronn, L.K. Intermittent fasting increases energy expenditure and promotes adipose tissue browning in mice. Nutrition 2019, 66, 38–43. [Google Scholar] [CrossRef] [PubMed]
- de Souza Marinho, T.; Ornellas, F.; Aguila, M.B.; Mandarim-De-Lacerda, C.A. Browning of the subcutaneous adipocytes in diet-induced obese mouse submitted to intermittent fasting. Mol. Cell. Endocrinol. 2020, 513, 110872. [Google Scholar] [CrossRef] [PubMed]
- de Cabo, R.; Mattson, M.P. Effects of intermittent fasting on health, aging, and disease. N. Engl. J. Med. 2019, 381, 2541–2551. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, X.; Luo, L.; Luo, Y.; Yang, X.; Ding, X.; Wang, L.; Le, H.; Feldman, L.E.R.; Men, X.; et al. Adipocyte-derived PGE2 is required for intermittent fasting-induced Treg proliferation and improvement of insulin sensitivity. JCI Insight 2022, 7, e153755. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liang, J.; Tian, X.; Chen, Q.; Zhu, L.; Wang, H.; Liu, Z.; Dai, X.; Bian, C.; Sun, C. Intermittent fasting promotes adipocyte mitochondrial fusion through Sirt3-mediated deacetylation of Mdh2. Br. J. Nutr. 2023, 23, 1–14. [Google Scholar] [CrossRef]
- Tinsley, G.M.; La Bounty, P.M. Effects of intermittent fasting on body composition and clinical health markers in humans. Nutr. Rev. 2015, 73, 661–674. [Google Scholar] [CrossRef]
- Wang, X.; Yang, Q.; Liao, Q.; Li, M.; Zhang, P.; Santos, H.O.; Kord-Varkaneh, H.; Abshirini, M. Effects of intermittent fasting diets on plasma concentrations of inflammatory biomarkers: A systematic review and meta-analysis of randomized controlled trials. Nutrition 2020, 79–80, 110974. [Google Scholar] [CrossRef]
- Lee, S.H.; Lee, J.H.; Lee, H.Y.; Min, K.J. Sirtuin signaling in cellular senescence and aging. BMB Rep. 2019, 52, 24–34. [Google Scholar] [CrossRef]
- Maissan, P.; Mooij, E.J.; Barberis, M. Sirtuins-mediated system-level regulation of mammalian tissues at the interface between metabolism and cell cycle: A systematic review. Biology 2021, 10, 194. [Google Scholar] [CrossRef]
- Hutchison, A.T.; Liu, B.; Wood, R.E.; Vincent, A.D.; Thompson, C.H.; O’Callaghan, N.J.; Wittert, G.A.; Heilbronn, L.K. Effects of Intermittent Versus Continuous Energy Intakes on Insulin Sensitivity and Metabolic Risk in Women with Overweight. Obesity 2019, 27, 50–58. [Google Scholar] [CrossRef]
- Nøhr, M.K.; Bobba, N.; Richelsen, B.; Lund, S.; Pedersen, S.B. Inflammation downregulates UCP1 expression in brown adipocytes potentially via SIRT1 and DBC1 interaction. Int. J. Mol. Sci. 2017, 18, 1006. [Google Scholar] [CrossRef]
- Bijland, S.; Mancini, S.J.; Salt, I.P. Role of AMP-activated protein kinase in adipose tissue metabolism and inflammation. Clin. Sci. 2013, 124, 491–507. [Google Scholar] [CrossRef]
- Weir, H.J.; Yao, P.; Huynh, F.K.; Escoubas, C.C.; Goncalves, R.L.; Burkewitz, K.; Laboy, R.; Hirschey, M.D.; Mair, W.B. Dietary Restriction and AMPK Increase Lifespan via Mitochondrial Network and Peroxisome Remodeling. Cell Metab. 2017, 26, 884–896.e5. [Google Scholar] [CrossRef]
- Jaspers, R.T.; Zillikens, M.C.; Friesema, E.C.M.; delli Paoli, G.; Bloch, W.; Uitterlinden, A.G.; Goglia, F.; Lanni, A.; de Lange, P. Exercise, fasting, and mimetics: Toward beneficial combinations? FASEB J. 2017, 31, 14–28. [Google Scholar] [CrossRef]
- Haganes, K.L.; Silva, C.P.; Eyjólfsdóttir, S.K.; Steen, S.; Grindberg, M.; Lydersen, S.; Hawley, J.A.; Moholdt, T. Time-restricted eating and exercise training improve HbA1c and body composition in women with overweight/obesity: A randomized controlled trial. Cell. Metab. 2022, 34, 1457–1471.e4. [Google Scholar] [CrossRef]
- Delli Paoli, G.; van de Laarschot, D.; Friesema, E.C.H.; Verkaik, R.; Giacco, A.; Senese, R.; Arp, P.P.; Jhamai, P.M.; Pagnotta, S.M.; Broer, L.; et al. Short-Term, Combined Fasting and Exercise Improves Body Composition in Healthy Males. Int. J. Sport. Nutr. Exerc. Metab. 2020, 30, 386–395. [Google Scholar] [CrossRef]
- Palmer, A.K.; Xu, M.; Zhu, Y.; Pirtskhalava, T.; Weivoda, M.M.; Hachfeld, C.M.; Prata, L.G.; van Dijk, T.H.; Verkade, E.; Casaclang-Verzosa, G.; et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 2019, 18, e12950. [Google Scholar] [CrossRef]
- Anderson, R.; Lagnado, A.; Maggiorani, D.; Walaszczyk, A.; Dookun, E.; Chapman, J.; Birch, J.; Salmonowicz, H.; Ogrodnik, M.; Jurk, D.; et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. 2019, 38, e100492. [Google Scholar] [CrossRef]
- Christopher, L.J.; Cui, D.; Li, W.; Barros, A., Jr.; Arora, V.K.; Zhang, H.; Wang, L.; Zhang, D.; Manning, J.A.; He, K.; et al. Biotransformation of [14C]dasatinib: In vitro studies in rat, monkey, and human and disposition after administration to rats and monkeys. Drug. Metab. Dispos. 2008, 36, 1341–1356. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Gregg, S.Q.; Robinson, A.R.; Niedernhofer, L.J. Physiological consequences of defects in ERCC1-XPF DNA repair endonuclease. DNA Repair 2011, 10, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Yousefzadeh, M.J.; Zhao, J.; Bukata, C.; Wade, E.A.; McGowan, S.J.; Angelini, L.A.; Bank, M.P.; Gurkar, A.U.; McGuckian, C.A.; Calubag, M.F.; et al. Tissue specificity of senescent cell accumulation during physiologic and accelerated aging of mice. Aging Cell 2020, 19, e13094. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.S.; Richardson, A.; Huffman, D.M.; Austad, S. Bring back the rat! J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Yousefzadeh, M.J.; Zhu, Y.; McGowan, S.J.; Angelini, L.; Fuhrmann-Stroissnigg, H.; Xu, M.; Ling, Y.Y.; Melos, K.I.; Pirtskhalava, T.; Inman, C.L.; et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 2018, 36, 18–28. [Google Scholar] [CrossRef]
- Gregoire, F.M.; Smas, C.M.; Sul, H.S. Understanding adipocyte differentiation. Physiol. Rev. 1998, 78, 783–809. [Google Scholar] [CrossRef]
- Ruiz-Ojeda, F.J.; Rupérez, A.I.; Gomez-Llorente, C.; Gil, A.; Aguilera, C.M. Cell Models and Their Application for Studying Adipogenic Differentiation in Relation to Obesity: A Review. Int. J. Mol. Sci. 2016, 17, 1040. [Google Scholar] [CrossRef]
- Zoico, E.; Nori, N.; Darra, E.; Tebon, M.; Rizzatti, V.; Policastro, G.; De Caro, A.; Rossi, A.P.; Fantin, F.; Zamboni, M. Senolytic effects of quercetin in an in vitro model of pre-adipocytes and adipocytes induced senescence. Sci. Rep. 2021, 11, 23237. [Google Scholar] [CrossRef]
- Kudlova, N.; De Sanctis, J.B.; Hajduch, M. Cellular Senescence: Molecular Targets, Biomarkers, and Senolytic Drugs. Int. J. Mol. Sci. 2022, 23, 4168. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Lange, P.; Lombardi, A.; Silvestri, E.; Cioffi, F.; Giacco, A.; Iervolino, S.; Petito, G.; Senese, R.; Lanni, A.; Moreno, M. Physiological Approaches Targeting Cellular and Mitochondrial Pathways Underlying Adipose Organ Senescence. Int. J. Mol. Sci. 2023, 24, 11676. https://doi.org/10.3390/ijms241411676
de Lange P, Lombardi A, Silvestri E, Cioffi F, Giacco A, Iervolino S, Petito G, Senese R, Lanni A, Moreno M. Physiological Approaches Targeting Cellular and Mitochondrial Pathways Underlying Adipose Organ Senescence. International Journal of Molecular Sciences. 2023; 24(14):11676. https://doi.org/10.3390/ijms241411676
Chicago/Turabian Stylede Lange, Pieter, Assunta Lombardi, Elena Silvestri, Federica Cioffi, Antonia Giacco, Stefania Iervolino, Giuseppe Petito, Rosalba Senese, Antonia Lanni, and Maria Moreno. 2023. "Physiological Approaches Targeting Cellular and Mitochondrial Pathways Underlying Adipose Organ Senescence" International Journal of Molecular Sciences 24, no. 14: 11676. https://doi.org/10.3390/ijms241411676
APA Stylede Lange, P., Lombardi, A., Silvestri, E., Cioffi, F., Giacco, A., Iervolino, S., Petito, G., Senese, R., Lanni, A., & Moreno, M. (2023). Physiological Approaches Targeting Cellular and Mitochondrial Pathways Underlying Adipose Organ Senescence. International Journal of Molecular Sciences, 24(14), 11676. https://doi.org/10.3390/ijms241411676