

Suppression of TGFβ-Induced Interleukin-6 Secretion by Sinulariolide from Soft Corals through Attenuation of the p38–NF-kB Pathway in Carcinoma Cells
 , ,
, ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Effects of Sinulariolide on Cell Viability
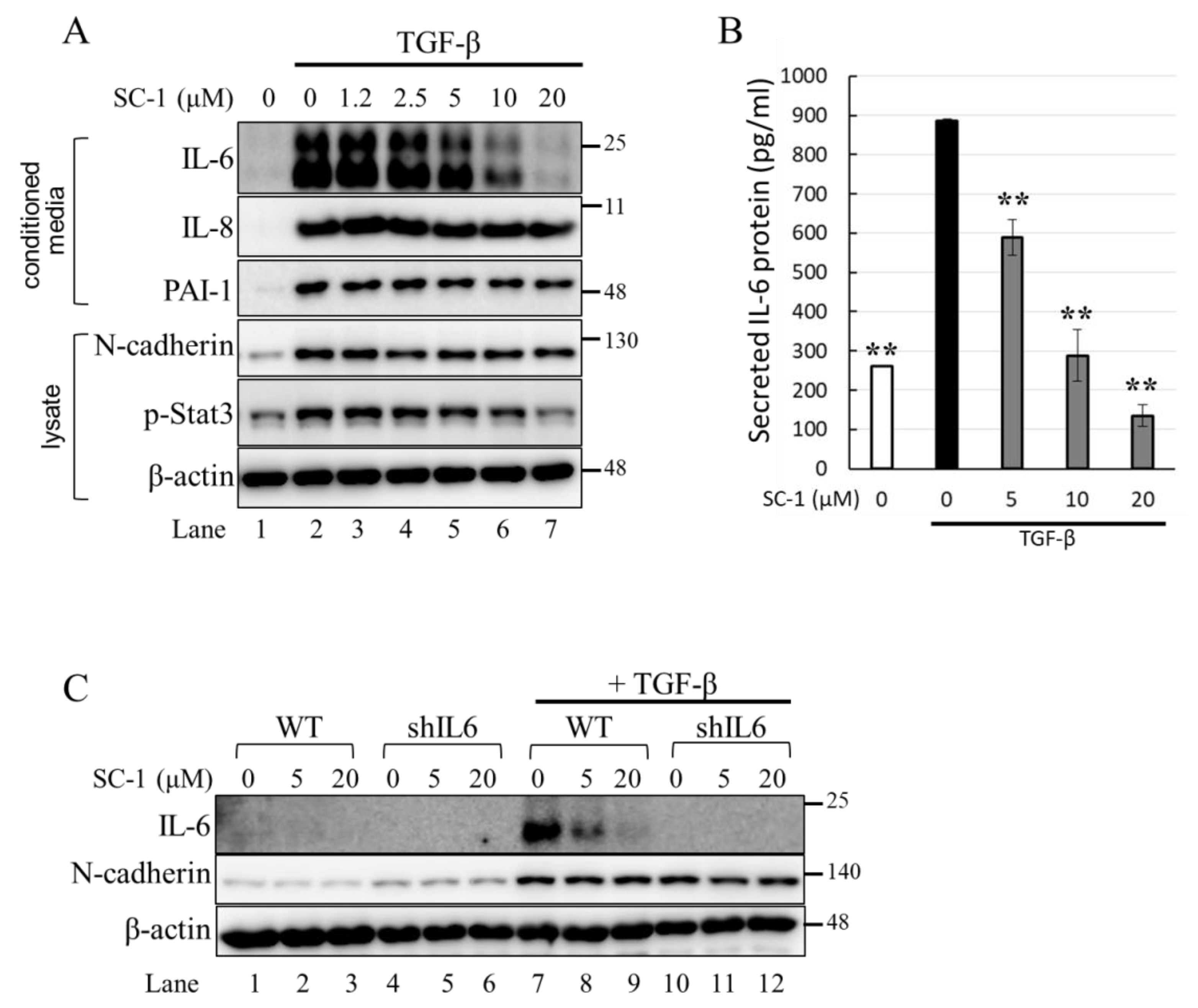
2.2. SC-1 Specifically Blocked TGFβ-Induced IL-6 Expression in A549 and HepG2 Cells
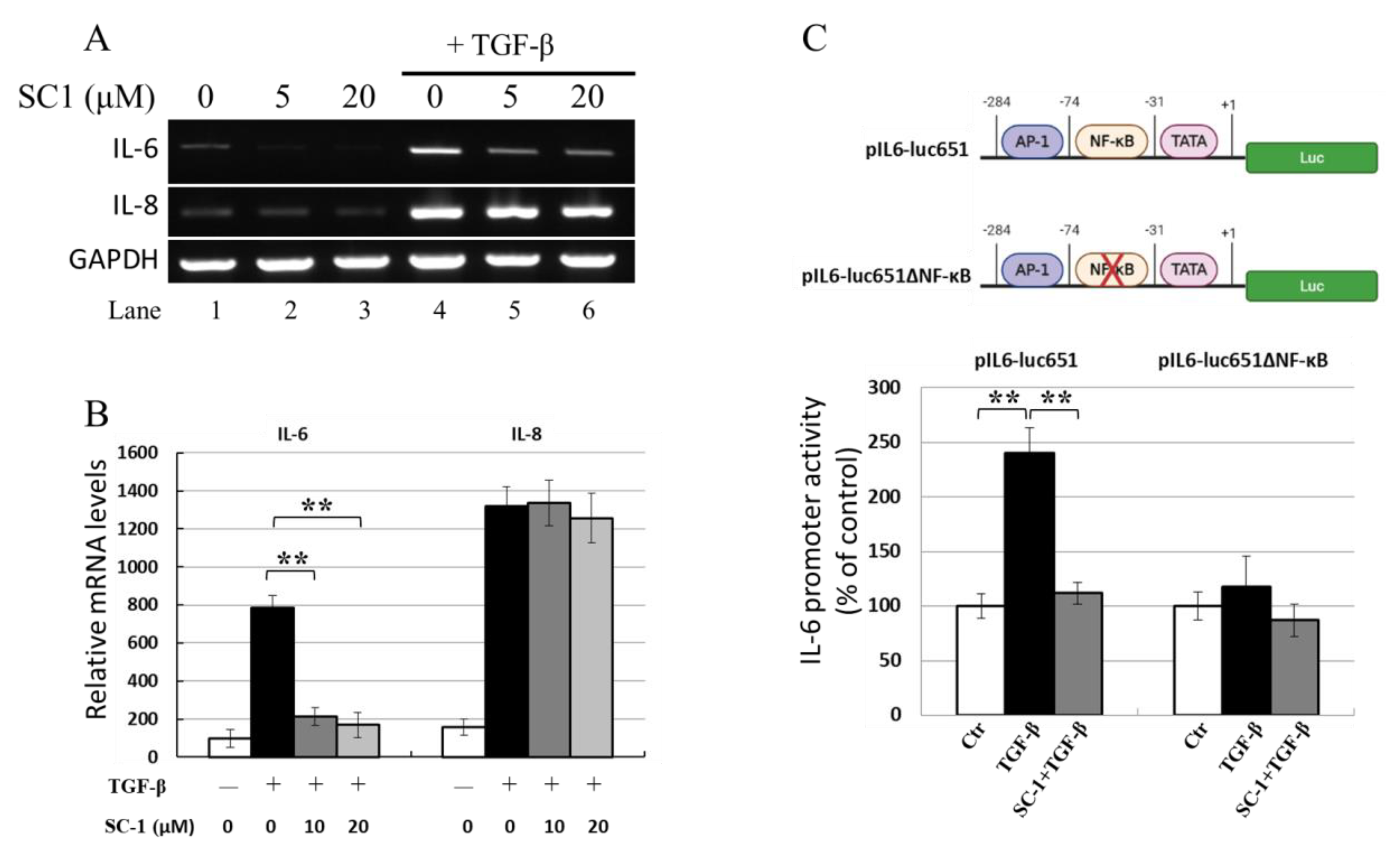
2.3. SC-1 Blocked TGFβ-Stimulated IL-6 mRNA Production
2.4. SC-1 Inhibited TGF-Mediated the IL-6 Transcription Potentially via an NF-κB-Responsive Element
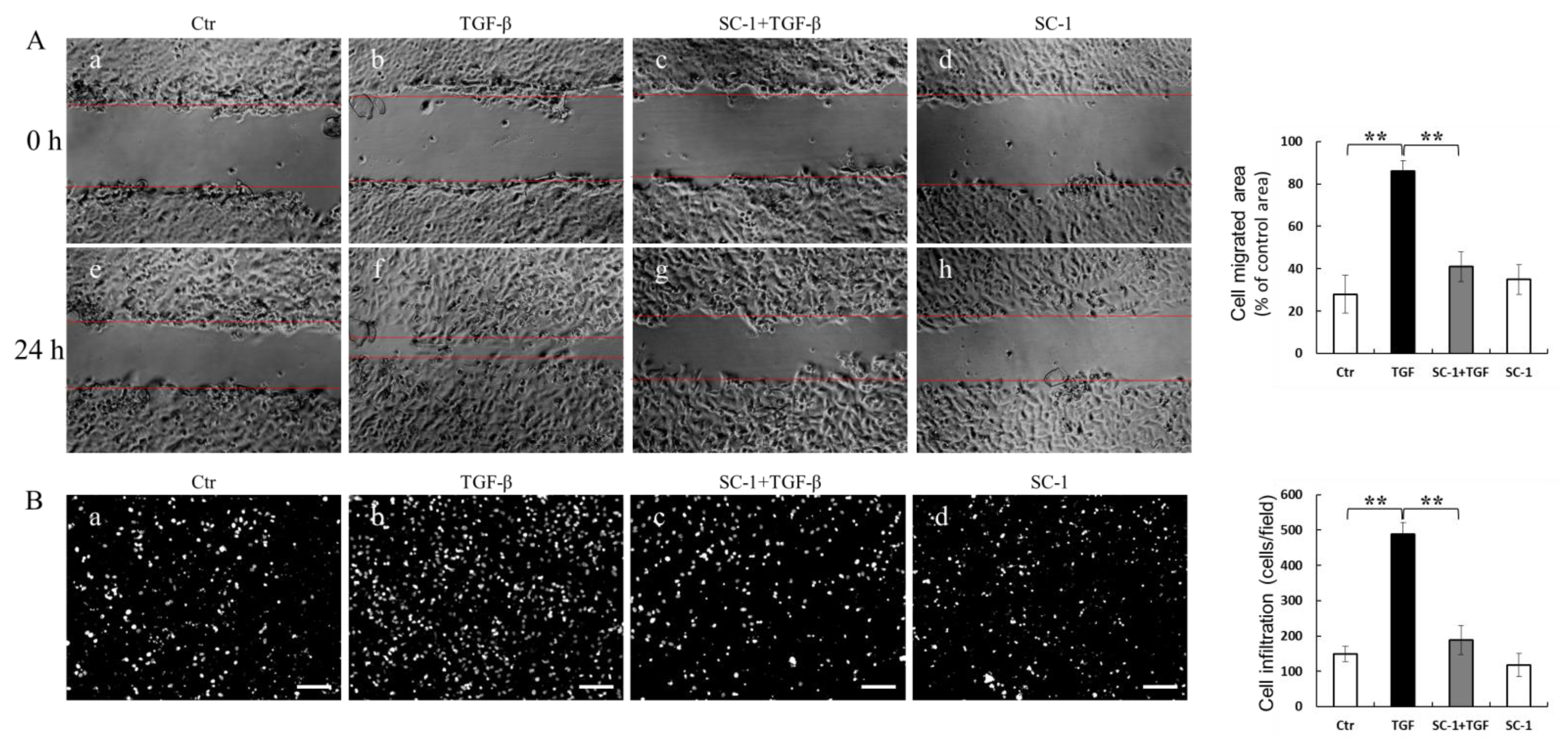
2.5. SC-1 Blocked TGFβ-Induced Cell Migration and Invasion
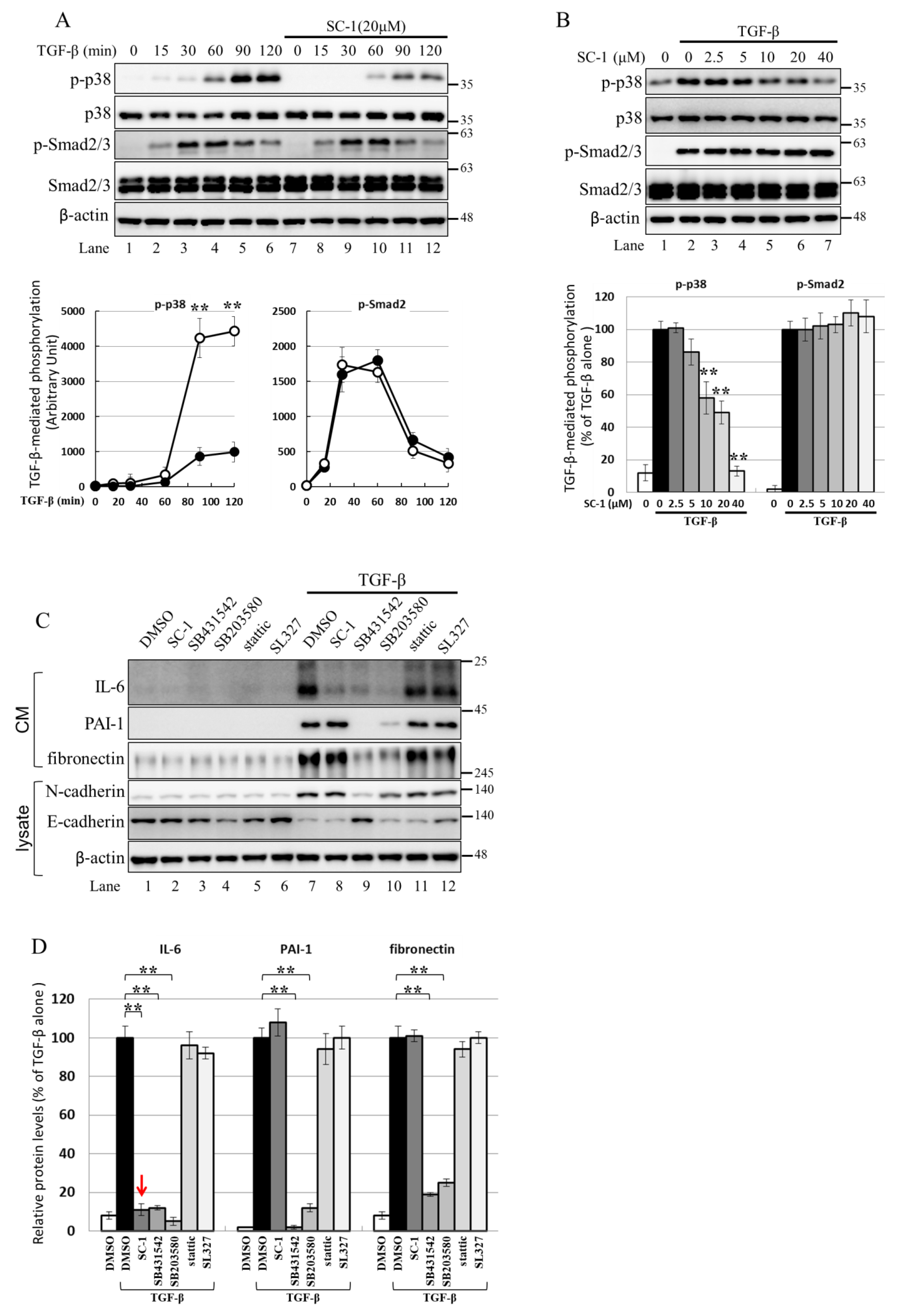
2.6. Using the Cell-Based Assay, p38 MAPK Is Activated by TGFβ and Involved in IL-6 Induction by TGFβ
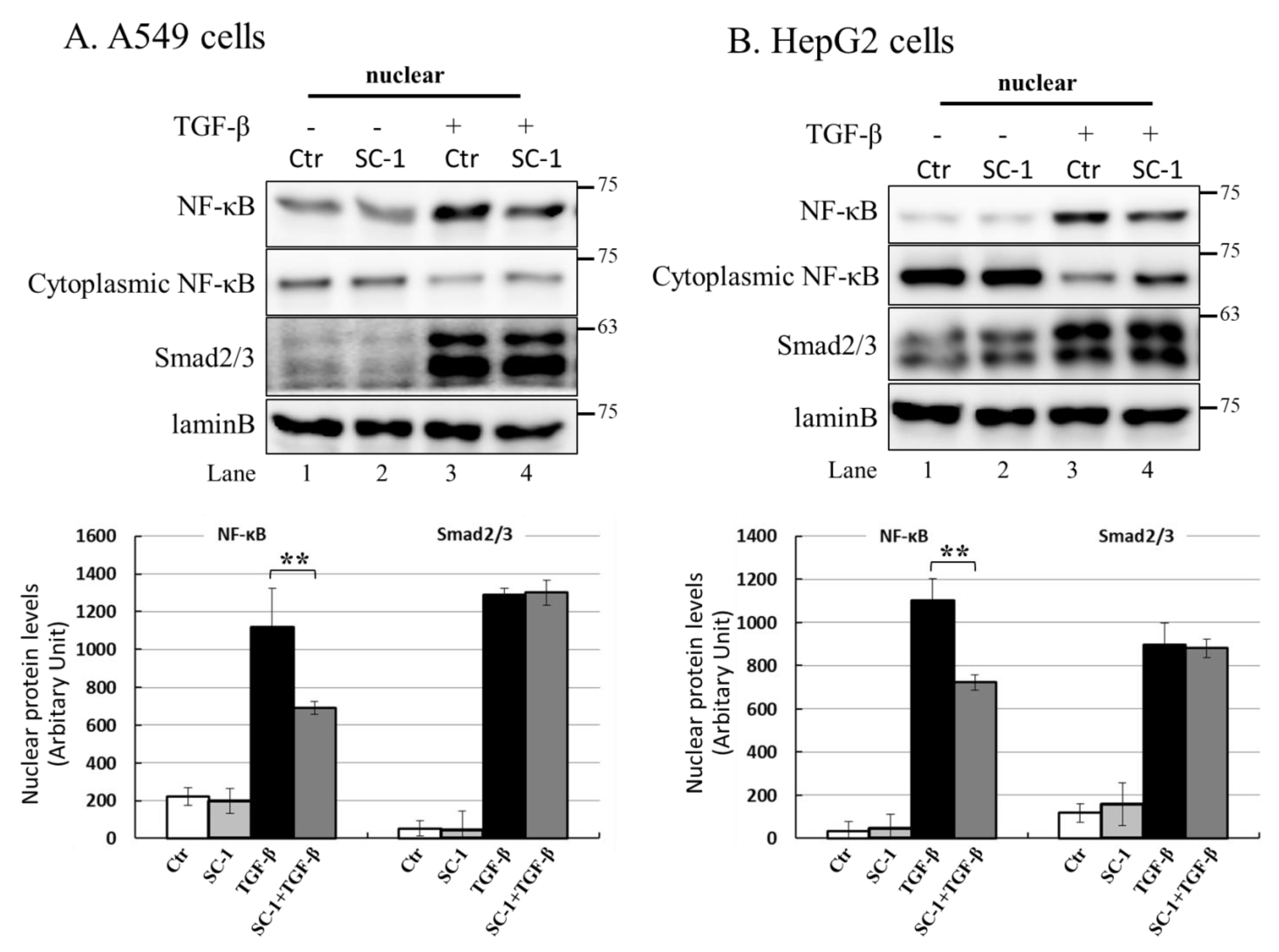
2.7. NF-kB Is Activated by p38 MAPK and Required for TGFβ-Mediated IL-6 Induction
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture
4.3. Preparation of Secretory Proteins and Nuclear Extracts
4.4. Western Blotting
4.5. Immunofluorescent Staining
4.6. Cell Viability
4.7. Enzyme-Linked Immunosorbent Assay (ELISA)
4.8. Cell Migration Assays
4.9. Transwell Migration Assay
4.10. Cell Transfection and Luciferase Assays
4.11. Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR) and Quantitative Polymerase Chain Reaction (qPCR)
4.12. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nguyen, N.B.A.; Chen, L.Y.; El-Shazly, M.; Peng, B.R.; Su, J.H.; Wu, H.C.; Lee, I.T.; Lai, K.H. Towards Sustainable Medicinal Resources through Marine Soft Coral Aquaculture: Insights into the Chemical Diversity and the Biological Potential. Mar. Drugs 2022, 20, 640. [Google Scholar] [CrossRef] [PubMed]
- Neoh, C.A.; Wang, R.Y.; Din, Z.H.; Su, J.H.; Chen, Y.K.; Tsai, F.J.; Weng, S.H.; Wu, Y.J. Induction of apoptosis by sinulariolide from soft coral through mitochondrial-related and p38MAPK pathways on human bladder carcinoma cells. Mar. Drugs 2012, 10, 2893–2911. [Google Scholar] [CrossRef]
- Hsiao, K.Y.; Wu, Y.J.; Liu, Z.N.; Chuang, C.W.; Huang, H.H.; Kuo, S.M. Anticancer Effects of Sinulariolide-Conjugated Hyaluronan Nanoparticles on Lung Adenocarcinoma Cells. Molecules 2016, 21, 297. [Google Scholar] [CrossRef] [PubMed]
- Li, H.H.; Su, J.H.; Chiu, C.C.; Lin, J.J.; Yang, Z.Y.; Hwang, W.I.; Chen, Y.K.; Lo, Y.H.; Wu, Y.J. Proteomic investigation of the sinulariolide-treated melanoma cells A375: Effects on the cell apoptosis through mitochondrial-related pathway and activation of caspase cascade. Mar. Drugs 2013, 11, 2625–2642. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Su, J.H.; Tsao, C.Y.; Hung, C.T.; Chao, H.H.; Lin, J.J.; Liao, M.H.; Yang, Z.Y.; Huang, H.H.; Tsai, F.J.; et al. Sinulariolide induced hepatocellular carcinoma apoptosis through activation of mitochondrial-related apoptotic and PERK/eIF2alpha/ATF4/CHOP pathway. Molecules 2013, 18, 10146–10161. [Google Scholar] [CrossRef]
- Wu, Y.J.; Neoh, C.A.; Tsao, C.Y.; Su, J.H.; Li, H.H. Sinulariolide Suppresses Human Hepatocellular Carcinoma Cell Migration and Invasion by Inhibiting Matrix Metalloproteinase-2/-9 through MAPKs and PI3K/Akt Signaling Pathways. Int. J. Mol. Sci. 2015, 16, 16469–16482. [Google Scholar] [CrossRef]
- Lu, Y.; Su, H.J.; Chen, Y.H.; Wen, Z.H.; Sheu, J.H.; Su, J.H. Anti-inflammatory cembranoids from the Formosan soft coral Sinularia discrepans. Arch. Pharm. Res. 2011, 34, 1263–1267. [Google Scholar] [CrossRef]
- Hsu, W.L.; Chiu, S.J.; Tsai, Y.T.; Chang, C.M.; Wang, J.Y.; Wang, E.T.; Hou, M.F.; Huang, C.Y.; Sheu, J.H.; Chang, W.C. A soft coral natural product, 11-episinulariolide acetate, inhibits gene expression of cyclooxygenase-2 and interleukin-8 through attenuation of calcium signaling. Molecules 2013, 18, 7023–7034. [Google Scholar] [CrossRef]
- Chung, T.W.; Li, Y.R.; Huang, W.Y.; Su, J.H.; Chan, H.L.; Lin, S.H.; Liu, C.S.; Lin, S.C.; Lin, C.C.; Lin, C.H. Sinulariolide suppresses LPS-induced phenotypic and functional maturation of dendritic cells. Mol. Med. Rep. 2017, 16, 6992–7000. [Google Scholar] [CrossRef]
- Davidson, S.; Coles, M.; Thomas, T.; Kollias, G.; Ludewig, B.; Turley, S.; Brenner, M.; Buckley, C.D. Fibroblasts as immune regulators in infection, inflammation and cancer. Nat. Rev. Immunol. 2021, 21, 704–717. [Google Scholar] [CrossRef]
- Piotrowski, I.; Kulcenty, K.; Suchorska, W. Interplay between inflammation and cancer. Rep. Pract. Oncol. Radiother. 2020, 25, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.; Kumar, B.; Datta, J.; Teknos, T.N.; Kumar, P. IL-6 promotes head and neck tumor metastasis by inducing epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling pathway. Mol. Cancer Res. 2011, 9, 1658–1667. [Google Scholar] [CrossRef] [PubMed]
- Tawara, K.; Oxford, J.T.; Jorcyk, C.L. Clinical significance of interleukin (IL)-6 in cancer metastasis to bone: Potential of anti-IL-6 therapies. Cancer Manag. Res. 2011, 3, 177–189. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Yamada, D.; Kobayashi, S.; Wada, H.; Kawamoto, K.; Marubashi, S.; Eguchi, H.; Ishii, H.; Nagano, H.; Doki, Y.; Mori, M. Role of crosstalk between interleukin-6 and transforming growth factor-beta 1 in epithelial-mesenchymal transition and chemoresistance in biliary tract cancer. Eur. J. Cancer 2013, 49, 1725–1740. [Google Scholar] [CrossRef] [PubMed]
- Mumm, J.B.; Oft, M. Cytokine-based transformation of immune surveillance into tumor-promoting inflammation. Oncogene 2008, 27, 5913–5919. [Google Scholar] [CrossRef]
- Park, J.I.; Lee, M.G.; Cho, K.; Park, B.J.; Chae, K.S.; Byun, D.S.; Ryu, B.K.; Park, Y.K.; Chi, S.G. Transforming growth factor-beta1 activates interleukin-6 expression in prostate cancer cells through the synergistic collaboration of the Smad2, p38-NF-kappaB, JNK, and Ras signaling pathways. Oncogene 2003, 22, 4314–4332. [Google Scholar] [CrossRef]
- Moller, A.; Schwarz, A.; Neuner, P.; Schwarz, T.; Luger, T.A. Regulation of monocyte and keratinocyte interleukin 6 production by transforming growth factor beta. Exp. Dermatol. 1994, 3, 314–320. [Google Scholar] [CrossRef]
- Mazzarelli, P.; Scuderi, F.; Mistretta, G.; Provenzano, C.; Bartoccioni, E. Effect of transforming growth factor-beta1 on interleukin-6 secretion in human myoblasts. J. Neuroimmunol. 1998, 87, 185–188. [Google Scholar] [CrossRef]
- Kuppner, M.C.; McKillop-Smith, S.; Forrester, J.V. TGF-beta and IL-1 beta act in synergy to enhance IL-6 and IL-8 mRNA levels and IL-6 production by human retinal pigment epithelial cells. Immunology 1995, 84, 265–271. [Google Scholar]
- Junn, E.; Lee, K.N.; Ju, H.R.; Han, S.H.; Im, J.Y.; Kang, H.S.; Lee, T.H.; Bae, Y.S.; Ha, K.S.; Lee, Z.W.; et al. Requirement of hydrogen peroxide generation in TGF-beta 1 signal transduction in human lung fibroblast cells: Involvement of hydrogen peroxide and Ca2+ in TGF-beta 1-induced IL-6 expression. J. Immunol. 2000, 165, 2190–2197. [Google Scholar] [CrossRef] [PubMed]
- Franchimont, N.; Rydziel, S.; Canalis, E. Transforming growth factor-beta increases interleukin-6 transcripts in osteoblasts. Bone 2000, 26, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Eickelberg, O.; Pansky, A.; Mussmann, R.; Bihl, M.; Tamm, M.; Hildebrand, P.; Perruchoud, A.P.; Roth, M. Transforming growth factor-beta1 induces interleukin-6 expression via activating protein-1 consisting of JunD homodimers in primary human lung fibroblasts. J. Biol. Chem. 1999, 274, 12933–12938. [Google Scholar] [CrossRef] [PubMed]
- Aoki, H.; Ohnishi, H.; Hama, K.; Shinozaki, S.; Kita, H.; Yamamoto, H.; Osawa, H.; Sato, K.; Tamada, K.; Sugano, K. Existence of autocrine loop between interleukin-6 and transforming growth factor-beta1 in activated rat pancreatic stellate cells. J. Cell. Biochem. 2006, 99, 221–228. [Google Scholar] [CrossRef]
- Teixeira, A.F.; Ten Dijke, P.; Zhu, H.J. On-Target Anti-TGF-beta Therapies Are Not Succeeding in Clinical Cancer Treatments: What Are Remaining Challenges? Front. Cell Dev. Biol. 2020, 8, 605. [Google Scholar] [CrossRef]
- Colak, S.; Ten Dijke, P. Targeting TGF-beta Signaling in Cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef]
- Chattopadhyay, I.; Ambati, R.; Gundamaraju, R. Exploring the Crosstalk between Inflammation and Epithelial-Mesenchymal Transition in Cancer. Mediat. Inflamm. 2021, 2021, 9918379. [Google Scholar] [CrossRef]
- Naugler, W.E.; Karin, M. The wolf in sheep’s clothing: The role of interleukin-6 in immunity, inflammation and cancer. Trends Mol. Med. 2008, 14, 109–119. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Karin, M. Inflammatory cytokines in cancer: Tumour necrosis factor and interleukin 6 take the stage. Ann. Rheum. Dis. 2011, 70 (Suppl. S1), i104–i108. [Google Scholar] [CrossRef]
- Dominguez, C.; David, J.M.; Palena, C. Epithelial-mesenchymal transition and inflammation at the site of the primary tumor. Semin. Cancer Biol. 2017, 47, 177–184. [Google Scholar] [CrossRef]
- Lin, Y.Y.; Jean, Y.H.; Lee, H.P.; Chen, W.F.; Sun, Y.M.; Su, J.H.; Lu, Y.; Huang, S.Y.; Hung, H.C.; Sung, P.J.; et al. A soft coral-derived compound, 11-epi-sinulariolide acetate suppresses inflammatory response and bone destruction in adjuvant-induced arthritis. PLoS ONE 2013, 8, e62926. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.J.; Lin, S.H.; Din, Z.H.; Su, J.H.; Liu, C.I. Sinulariolide Inhibits Gastric Cancer Cell Migration and Invasion through Downregulation of the EMT Process and Suppression of FAK/PI3K/AKT/mTOR and MAPKs Signaling Pathways. Mar. Drugs 2019, 17, 668. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Milstein, M.; Bliss, J.M.; Thai, M.; Malhotra, G.; Huynh, L.C.; Colicelli, J. Integration of transforming growth factor beta and RAS signaling silences a RAB5 guanine nucleotide exchange factor and enhances growth factor-directed cell migration. Mol. Cell. Biol. 2008, 28, 1573–1583. [Google Scholar] [CrossRef]
- Finger, E.C.; Turley, R.S.; Dong, M.; How, T.; Fields, T.A.; Blobe, G.C. TbetaRIII suppresses non-small cell lung cancer invasiveness and tumorigenicity. Carcinogenesis 2008, 29, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.L.; Huang, C.C.; Tzeng, T.T.; Liu, S.C.; Tsai, C.H.; Fong, Y.C.; Tang, C.H. S1P promotes IL-6 expression in osteoblasts through the PI3K, MEK/ERK and NF-kappaB signaling pathways. Int. J. Med. Sci. 2020, 17, 1207–1214. [Google Scholar] [CrossRef]
- Wery-Zennaro, S.; Zugaza, J.L.; Letourneur, M.; Bertoglio, J.; Pierre, J. IL-4 regulation of IL-6 production involves Rac/Cdc42- and p38 MAPK-dependent pathways in keratinocytes. Oncogene 2000, 19, 1596–1604. [Google Scholar] [CrossRef][Green Version]
- Craig, R.; Larkin, A.; Mingo, A.M.; Thuerauf, D.J.; Andrews, C.; McDonough, P.M.; Glembotski, C.C. p38 MAPK and NF-kappa B collaborate to induce interleukin-6 gene expression and release. Evidence for a cytoprotective autocrine signaling pathway in a cardiac myocyte model system. J. Biol. Chem. 2000, 275, 23814–23824. [Google Scholar] [CrossRef]
- Hanafusa, H.; Ninomiya-Tsuji, J.; Masuyama, N.; Nishita, M.; Fujisawa, J.; Shibuya, H.; Matsumoto, K.; Nishida, E. Involvement of the p38 mitogen-activated protein kinase pathway in transforming growth factor-beta-induced gene expression. J. Biol. Chem. 1999, 274, 27161–27167. [Google Scholar] [CrossRef]
- Asschert, J.G.; Vellenga, E.; Ruiters, M.H.; de Vries, E.G. Regulation of spontaneous and TNF/IFN-induced IL-6 expression in two human ovarian-carcinoma cell lines. Int. J. Cancer 1999, 82, 244–249. [Google Scholar] [CrossRef]
- Shintani, Y.; Fujiwara, A.; Kimura, T.; Kawamura, T.; Funaki, S.; Minami, M.; Okumura, M. IL-6 Secreted from Cancer-Associated Fibroblasts Mediates Chemoresistance in NSCLC by Increasing Epithelial-Mesenchymal Transition Signaling. J. Thorac. Oncol. 2016, 11, 1482–1492. [Google Scholar] [CrossRef]
- Yao, Z.; Fenoglio, S.; Gao, D.C.; Camiolo, M.; Stiles, B.; Lindsted, T.; Schlederer, M.; Johns, C.; Altorki, N.; Mittal, V.; et al. TGF-beta IL-6 axis mediates selective and adaptive mechanisms of resistance to molecular targeted therapy in lung cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 15535–15540. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.H.; Chang, M.C.; Su, Y.H.; Tsai, Y.T.; Kuo, M.L. Interleukin-6 inhibits transforming growth factor-beta-induced apoptosis through the phosphatidylinositol 3-kinase/Akt and signal transducers and activators of transcription 3 pathways. J. Biol. Chem. 1999, 274, 23013–23019. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Matsuda, T.; Muraguchi, A.; Miyazono, K.; Kawabata, M. Cross-talk between IL-6 and TGF-beta signaling in hepatoma cells. FEBS Lett. 2001, 492, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Dubois-Pot-Schneider, H.; Fekir, K.; Coulouarn, C.; Glaise, D.; Aninat, C.; Jarnouen, K.; Le Guevel, R.; Kubo, T.; Ishida, S.; Morel, F.; et al. Inflammatory cytokines promote the retrodifferentiation of tumor-derived hepatocyte-like cells to progenitor cells. Hepatology 2014, 60, 2077–2090. [Google Scholar] [CrossRef]
- Mitra, A.; Yan, J.; Xia, X.; Zhou, S.; Chen, J.; Mishra, L.; Li, S. IL6-mediated inflammatory loop reprograms normal to epithelial-mesenchymal transition(+) metastatic cancer stem cells in preneoplastic liver of transforming growth factor beta-deficient beta2-spectrin(+/−) mice. Hepatology 2017, 65, 1222–1236. [Google Scholar] [CrossRef]
- Kang, T.H.; Han, N.R.; Kim, H.M.; Jeong, H.J. Blockade of IL-6 secretion pathway by the sesquiterpenoid atractylenolide III. J. Nat. Prod. 2011, 74, 223–227. [Google Scholar] [CrossRef]
- Jain, S.K.; Singh, S.; Khajuria, A.; Guru, S.K.; Joshi, P.; Meena, S.; Nadkarni, J.R.; Singh, A.; Bharate, S.S.; Bhushan, S.; et al. Pyrano-isochromanones as IL-6 inhibitors: Synthesis, in vitro and in vivo antiarthritic activity. J. Med. Chem. 2014, 57, 7085–7097. [Google Scholar] [CrossRef]
- Kaur, S.; Bansal, Y.; Kumar, R.; Bansal, G. A panoramic review of IL-6: Structure, pathophysiological roles and inhibitors. Bioorg. Med. Chem. 2020, 28, 115327. [Google Scholar] [CrossRef]
- Wang, W.; Lee, T.G.; Patil, R.S.; Mun, B.; Yang, I.; Kim, H.; Hahn, D.; Won, D.H.; Lee, J.; Lee, Y.; et al. Monanchosterols A and B, bioactive bicyclo[4.3.1]steroids from a Korean sponge Monanchora sp. J. Nat. Prod. 2015, 78, 368–373. [Google Scholar] [CrossRef]
- Kahlon, D.K.; Lansdell, T.A.; Fisk, J.S.; Hupp, C.D.; Friebe, T.L.; Hovde, S.; Jones, A.D.; Dyer, R.D.; Henry, R.W.; Tepe, J.J. Nuclear factor-kappaB mediated inhibition of cytokine production by imidazoline scaffolds. J. Med. Chem. 2009, 52, 1302–1309. [Google Scholar] [CrossRef]
- Crusz, S.M.; Balkwill, F.R. Inflammation and cancer: Advances and new agents. Nat. Rev. Clin. Oncol. 2015, 12, 584–596. [Google Scholar] [CrossRef]
- Vanden Berghe, W.; Francesconi, E.; De Bosscher, K.; Resche-Rigon, M.; Haegeman, G. Dissociated glucocorticoids with anti-inflammatory potential repress interleukin-6 gene expression by a nuclear factor-kappaB-dependent mechanism. Mol. Pharmacol. 1999, 56, 797–806. [Google Scholar] [PubMed]
- Massague, J. TGF-beta signal transduction. Annu. Rev. Biochem. 1998, 67, 753–791. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Li, H.; Sun, L.; Brigstock, D.R.; Gao, R. Interleukin-6 participates in human pancreatic stellate cell activation and collagen I production via TGF-beta1/Smad pathway. Cytokine 2021, 143, 155536. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.L.; Topley, N.; Ito, T.; Phillips, A. Interleukin-6 regulation of transforming growth factor (TGF)-beta receptor compartmentalization and turnover enhances TGF-beta1 signaling. J. Biol. Chem. 2005, 280, 12239–12245. [Google Scholar] [CrossRef]
- Mitra, M.S.; Lancaster, K.; Adedeji, A.O.; Palanisamy, G.S.; Dave, R.A.; Zhong, F.; Holdren, M.S.; Turley, S.J.; Liang, W.C.; Wu, Y.; et al. A Potent Pan-TGFbeta Neutralizing Monoclonal Antibody Elicits Cardiovascular Toxicity in Mice and Cynomolgus Monkeys. Toxicol. Sci. 2020, 175, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Simoes, E.S.A.C.; Teixeira, M.M. ACE inhibition, ACE2 and angiotensin-(1-7) axis in kidney and cardiac inflammation and fibrosis. Pharmacol. Res. 2016, 107, 154–162. [Google Scholar] [CrossRef]
- Hsieh, P.W.; Chang, F.R.; McPhail, A.T.; Lee, K.H.; Wu, Y.C. New cembranolide analogues from the formosan soft coral Sinularia flexibilis and their cytotoxicity. Nat. Prod. Res. 2003, 17, 409–418. [Google Scholar] [CrossRef]
- Huang, S.S.; Chen, C.L.; Huang, F.W.; Hou, W.H.; Huang, J.S. DMSO Enhances TGF-beta Activity by Recruiting the Type II TGF-beta Receptor From Intracellular Vesicles to the Plasma Membrane. J. Cell. Biochem. 2016, 117, 1568–1579. [Google Scholar] [CrossRef]
- Pare, B.; Deschenes, L.T.; Pouliot, R.; Dupre, N.; Gros-Louis, F. An Optimized Approach to Recover Secreted Proteins from Fibroblast Conditioned-Media for Secretomic Analysis. Front. Cell. Neurosci. 2016, 10, 70. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.-L.; Lin, W.-L.; Tai, S.-B.; Ciou, Y.-S.; Chung, C.-L.; Chen, J.-J.; Liu, P.-F.; Lin, M.-W.; Chen, C.-L. Suppression of TGFβ-Induced Interleukin-6 Secretion by Sinulariolide from Soft Corals through Attenuation of the p38–NF-kB Pathway in Carcinoma Cells. Int. J. Mol. Sci. 2023, 24, 11656. https://doi.org/10.3390/ijms241411656
Yang J-L, Lin W-L, Tai S-B, Ciou Y-S, Chung C-L, Chen J-J, Liu P-F, Lin M-W, Chen C-L. Suppression of TGFβ-Induced Interleukin-6 Secretion by Sinulariolide from Soft Corals through Attenuation of the p38–NF-kB Pathway in Carcinoma Cells. International Journal of Molecular Sciences. 2023; 24(14):11656. https://doi.org/10.3390/ijms241411656
Chicago/Turabian StyleYang, Jenq-Lin, Weng-Ling Lin, Shun-Ban Tai, Yi-Siang Ciou, Chih-Ling Chung, Jih-Jung Chen, Pei-Feng Liu, Ming-Wei Lin, and Chun-Lin Chen. 2023. "Suppression of TGFβ-Induced Interleukin-6 Secretion by Sinulariolide from Soft Corals through Attenuation of the p38–NF-kB Pathway in Carcinoma Cells" International Journal of Molecular Sciences 24, no. 14: 11656. https://doi.org/10.3390/ijms241411656
APA StyleYang, J.-L., Lin, W.-L., Tai, S.-B., Ciou, Y.-S., Chung, C.-L., Chen, J.-J., Liu, P.-F., Lin, M.-W., & Chen, C.-L. (2023). Suppression of TGFβ-Induced Interleukin-6 Secretion by Sinulariolide from Soft Corals through Attenuation of the p38–NF-kB Pathway in Carcinoma Cells. International Journal of Molecular Sciences, 24(14), 11656. https://doi.org/10.3390/ijms241411656