
Identification of a Putative SARS-CoV-2 Main Protease Inhibitor through In Silico Screening of Self-Designed Molecular Library

 ,
,
Abstract
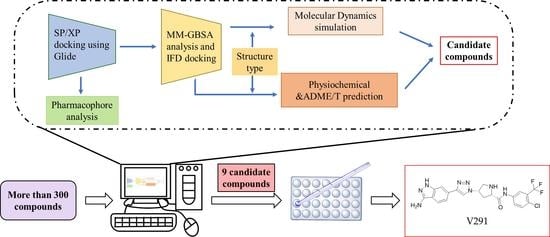
1. Introduction
2. Results and Discussion
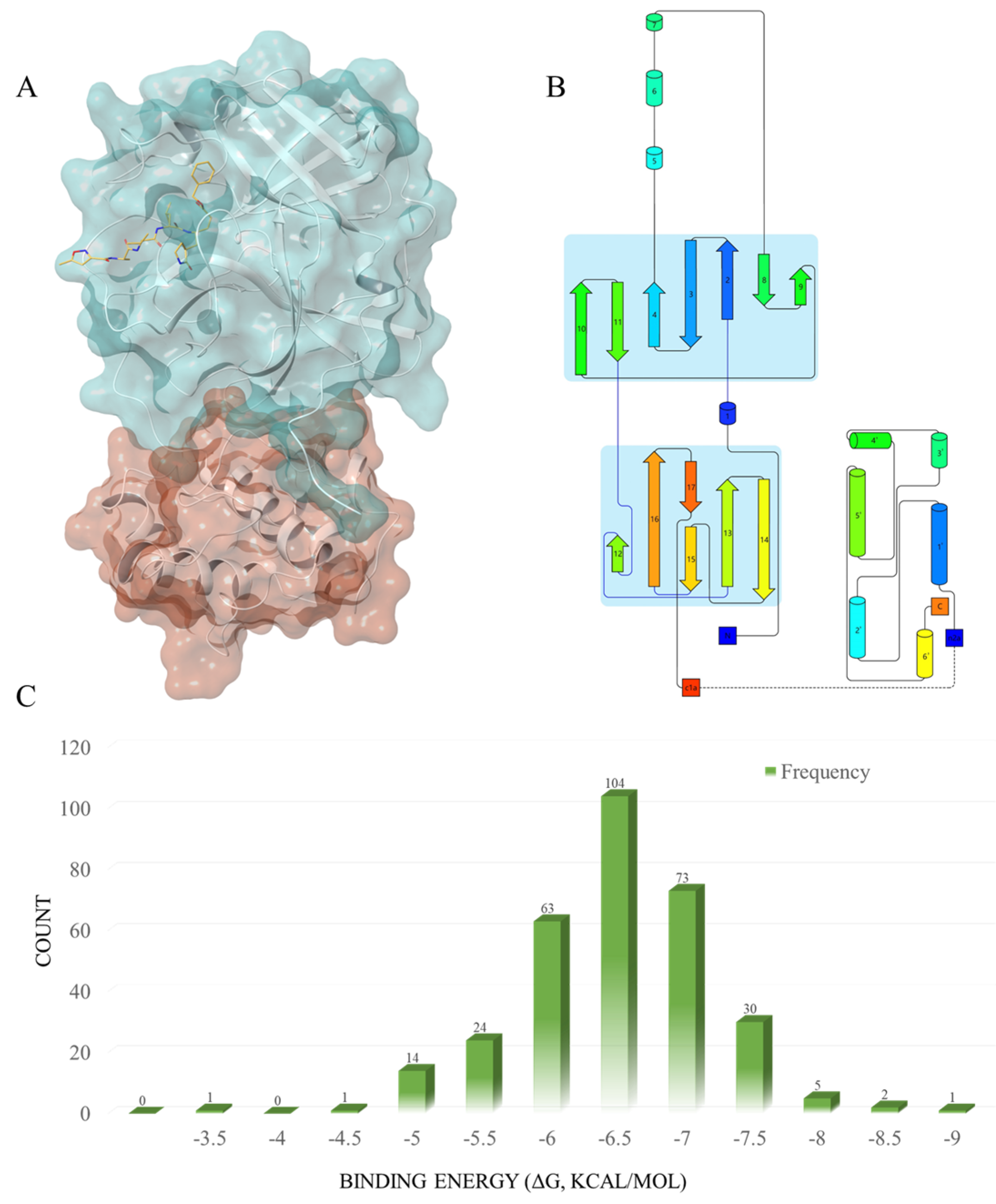
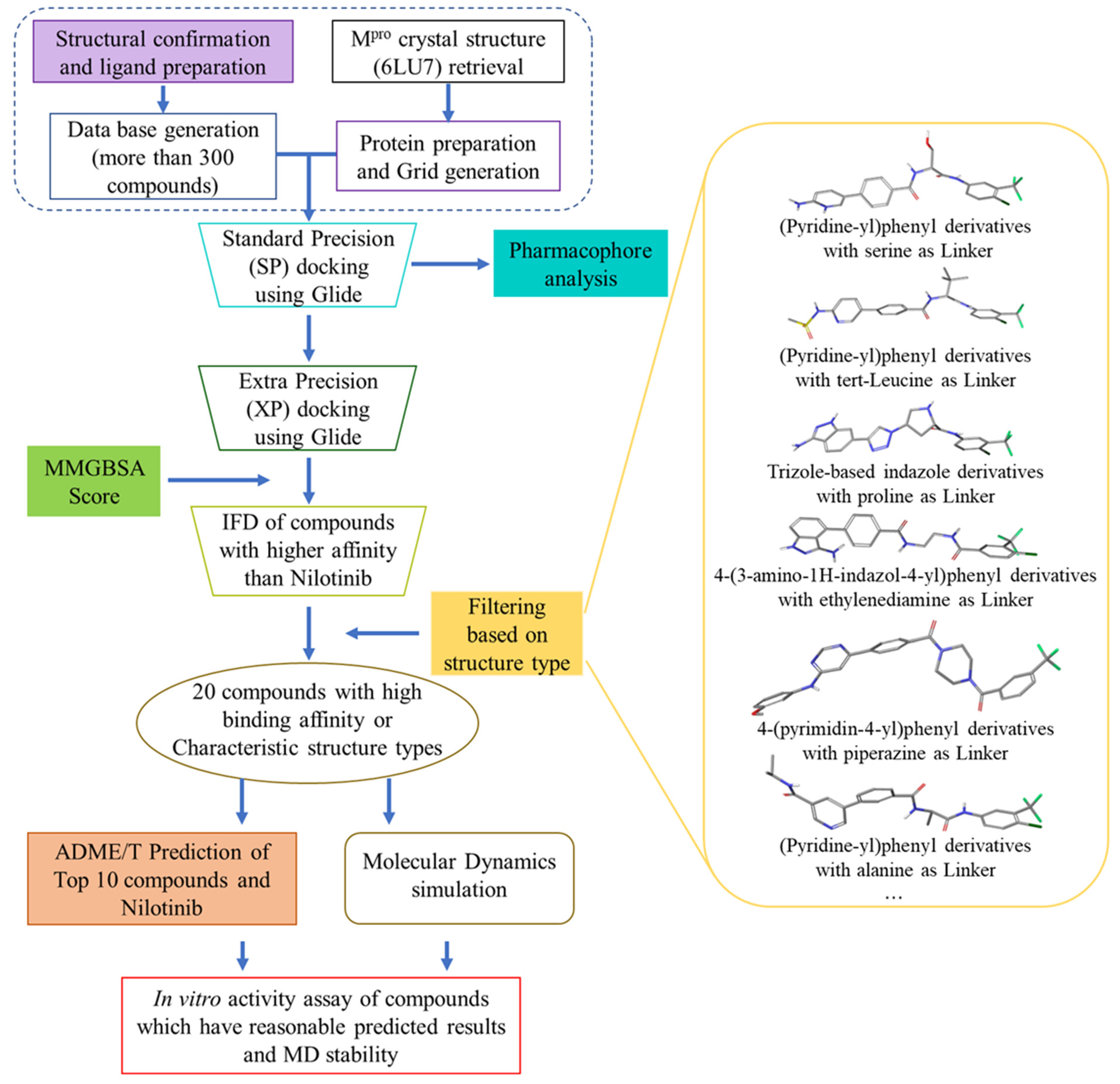
2.1. Virtual Screening
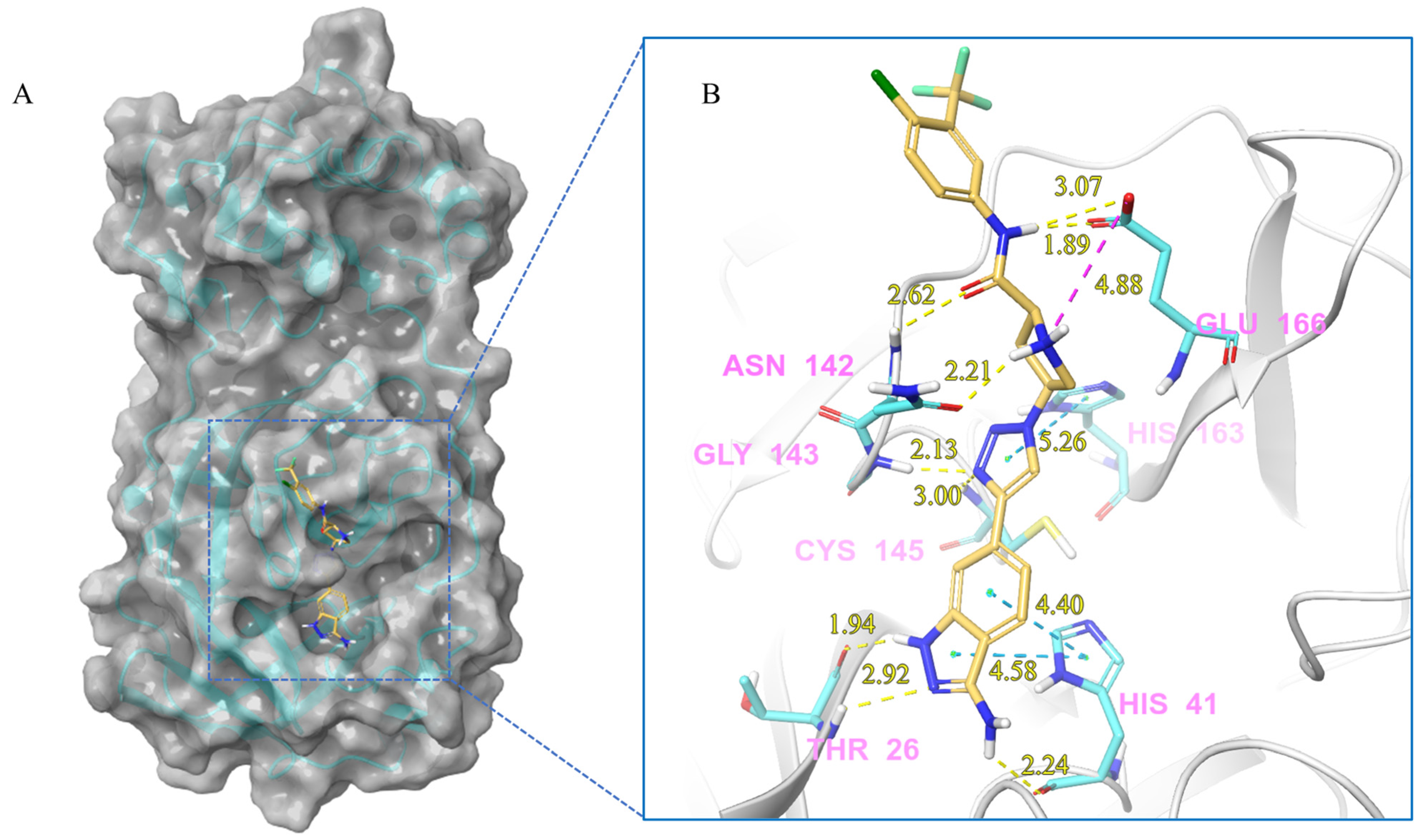
2.2. Further Screening through Induced-Fit Docking
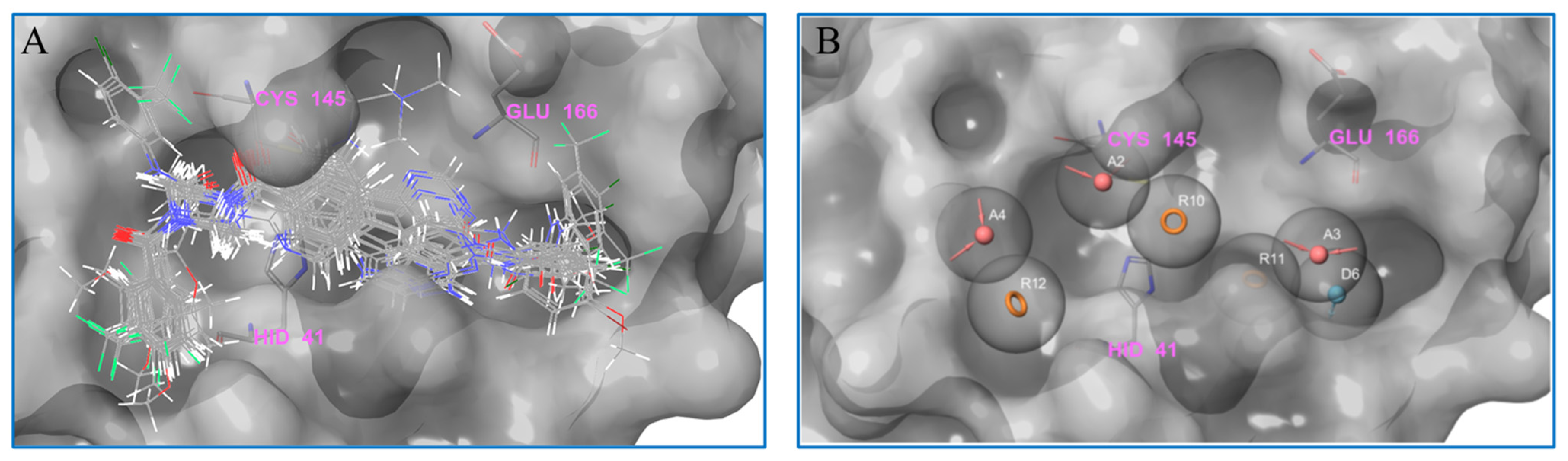
2.3. Pharmacophore Analysis
2.4. Molecular Dynamics Analysis
2.5. ADME/T Prediction
2.6. Compound Enzymatic Activity Assay
3. Materials and Methods
3.1. Experimental Procedures
3.2. Ligand Preparation
3.3. Protein Preparation and Grid Generation
3.4. Molecular Docking
3.5. MM-GBSA
3.6. Induced-Fit Docking (IFD)
3.7. Pharmacophore Analysis
3.8. ADME/T Prediction
3.9. Molecular Dynamics Analysis
3.10. In Vitro Enzymatic and Inhibition Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weekly Epidemiological Update on COVID-19—15 February 2023. Available online: https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19---15-february-2023 (accessed on 15 February 2023).
- Naming the Coronavirus Disease (COVID-19) and the Virus That Causes It [EB/OL]. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/technical-guidance/naming-the-coronavirus-disease-(covid-2019)-and-the-virus-that-causes-it (accessed on 13 January 2021).
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Ikbel, H.H. COVID-19 vaccines and variants of concern: A review. Rev. Med. Virol. 2022, 32, e2313. [Google Scholar]
- Thi, L.D.; Van, T.H.; Philippe, G. Recurrence of SARS-CoV-2 viral RNA in recovered COVID-19 patients: A narrative review. Eur. J. Clin. Microbiol. Infect. Dis. 2021, 40, 13–25. [Google Scholar]
- Mohamadian, M.; Chiti, H.; Shoghli, A.; Biglari, S.; Parsamanesh, N.; Esmaeilzadeh, A. COVID-19: Virology, biology and novel laboratory diagnosis. J. Gene Med. 2021, 23, e3303. [Google Scholar] [CrossRef] [PubMed]
- Abduljalil, J.M.; Abduljalil, B.M. Epidemiology, genome, and clinical features of the pandemic SARS-CoV-2: A recent view. New Microbes New Infect. 2020, 35, 100672. [Google Scholar] [CrossRef] [PubMed]
- Araf, Y.; Akter, F.; Tang, Y.D.; Fatemi, R.; Parvez, M.S.A.; Zheng, C.; Hossain, M.G. Omicron variant of SARS-CoV-2: Genomics, transmissibility, and responses to current COVID-19 vaccines. J. Med. Virol. 2022, 94, 1825–1832. [Google Scholar] [CrossRef]
- Chia, S.K.; Dinesh, S.R.; Syed, S.H. The effectiveness of mRNA-1273 vaccine against COVID-19 caused by Delta variant: A systematic review and meta-analysis. J. Med. Virol. 2022, 94, 2269–2274. [Google Scholar]
- Yu, C.; Qianyun, L.; Deyin, G. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 2249. [Google Scholar]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef]
- Agost-Beltrán, L.; de la Hoz-Rodríguez, S.; Bou-Iserte, L.; Rodríguez, S.; Fernández-de-la-Pradilla, A.; González, F.V. Advances in the development of SARS-CoV-2 Mpro inhibitor. Molecules 2022, 27, 2523. [Google Scholar] [CrossRef]
- Lv, Z.; Cano, K.E.; Jia, L.; Drag, M.; Huang, T.T.; Olsen, S.K. Targeting SARS-CoV-2 proteases for COVID-19 antiviral development. Front. Chem. 2022, 9, 819165. [Google Scholar] [CrossRef]
- Issa, S.S.; Sokornova, S.V.; Zhidkin, R.R.; Matveeva, T.V. The main protease of SARS-CoV-2 as a target for phytochemicals against coronavirus. Plants 2022, 11, 1862. [Google Scholar] [CrossRef]
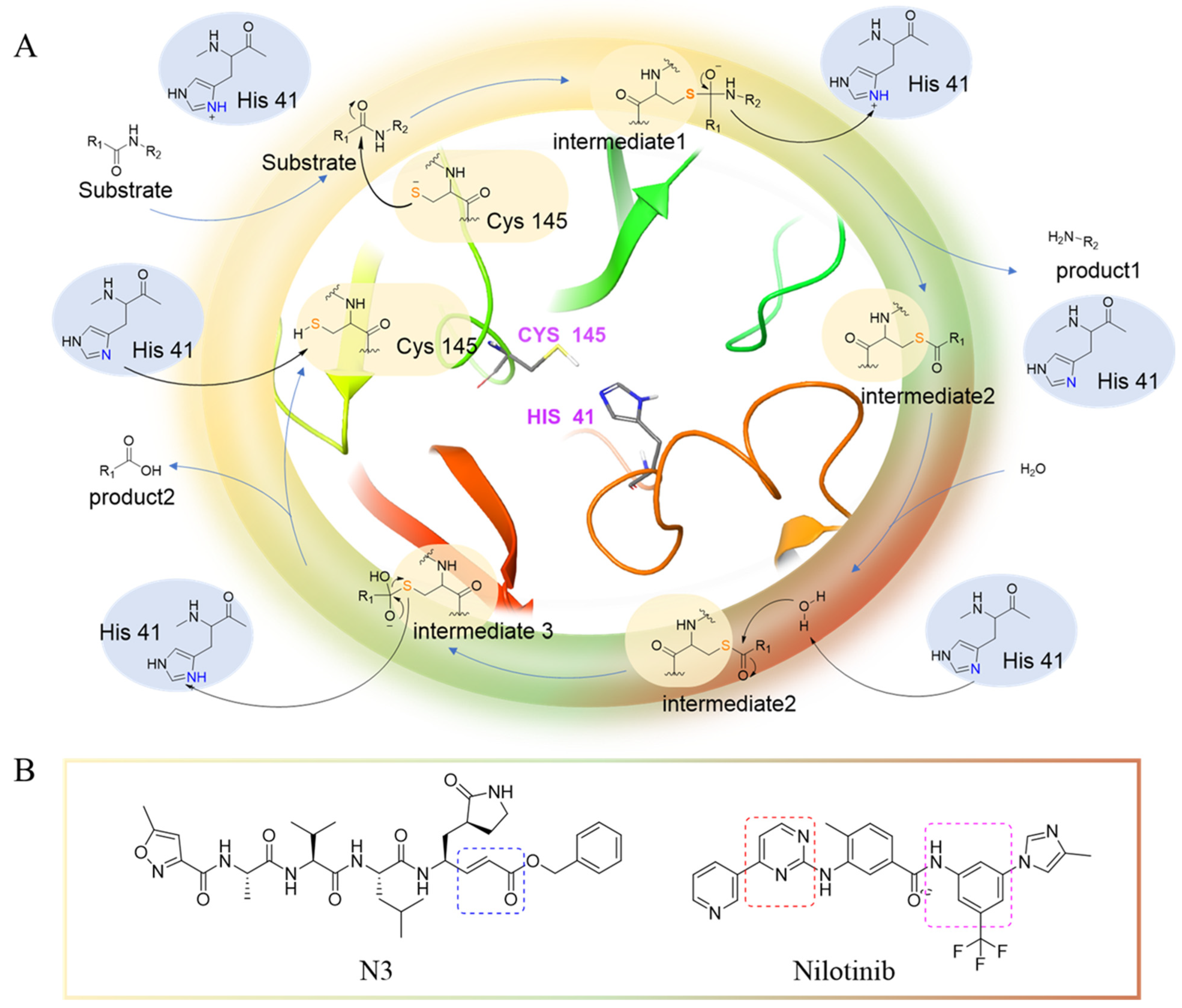
- Wang, H.; He, S.; Deng, W.; Zhang, Y.; Li, G.; Sun, J.; Zhao, W.; Guo, Y.; Yin, Z.; Li, D.; et al. Comprehensive insights into the catalytic mechanism of middle east respiratory syndrome 3C-Like protease and severe acute respiratory syndrome 3C-Like protease. ACS Catal. 2020, 10, 5871–5890. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.F.; Kuo, C.J.; Chang, K.T.; Chang, H.C.; Chou, C.C.; Ko, T.P.; Shr, H.L.; Chang, G.G.; Wang, A.H.J.; Liang, P.H. Mechanism of the maturation process of SARS-CoV 3CL protease. J. Biol. Chem. 2005, 280, 31257–31266. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wei, P.; Fan, K.; Liu, Y.; Lai, L. 3C-like proteinase from SARS coronavirus catalyzes substrate hydrolysis by a general base mechanism. Biochemistry 2004, 43, 4568–4574. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Kreutzer, A.G.; Krumberger, M.; Diessner, E.M.; Parrocha, C.M.T.; Morris, M.A.; Guaglianone, G.; Butts, C.T.; Nowick, J.S. A cyclic peptide inhibitor of the SARS-CoV-2 main protease. Eur. J. Med. Chem. 2021, 221, 113530. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Yadav, S.; Banerjee, S.; Fakayode, S.O.; Parvathareddy, J.; Reichard, W.; Surendranathan, S.; Mahmud, F.; Whatcott, R.; Thammathong, J.; et al. Drug repurposing to identify Nilotinib as a potential SARS-CoV-2 main protease inhibitor: Insights from a computational and in vitro study. J. Chem. Inf. Model. 2021, 61, 5469–5483. [Google Scholar] [CrossRef]
- Pan, X.; Liu, N.; Zhang, Q.; Wang, K.; Li, Y.; Shan, Y.; Li, Z.; Zhang, J. Design, synthesis, and biological evaluation of novel Bcr-Abl T315I inhibitors incorporating amino acids as flexible linker. Bioorg. Med. Chem. 2021, 48, 116398. [Google Scholar] [CrossRef]
- Pan, X.; Liu, N.; Liu, Y.; Zhang, Q.; Wang, K.; Liu, X.; Zhang, J. Design, synthesis, and biological evaluation of trizole-based heteroaromatic derivatives as Bcr-Abl kinase inhibitors. Eur. J. Med. Chem. 2022, 238, 114425. [Google Scholar] [CrossRef]
- Li, J.; Si, R.; Zhang, Q.; Li, Y.; Zhang, J.; Shan, Y. Novel indole-guanidine hybrids as potential anticancer agents: Design, synthesis and biological evaluation. Chem. Biol. Interact. 2022, 368, 110242. [Google Scholar] [CrossRef]
- Rehman, M.T.; AlAjmi, M.F.; Hussain, A.; Rather, G.M.; Khan, M.A. High-throughput virtual screening, molecular dynamics simulation, and enzyme kinetics identified ZINC84525623 as a potential inhibitor of NDM-1. Int. J. Mol. Sci. 2019, 20, 819. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Xie, W.; Xue, X.; Yang, K.; Ma, J.; Liang, W.; Zhao, Q.; Zhou, Z.; Pei, D.; Ziebuhr, J.; et al. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005, 3, e324. [Google Scholar]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Frishman, D.; Argos, P. Knowledge-based protein secondary structure assignment. Proteins 1995, 23, 566–579. [Google Scholar] [CrossRef]
- Hongyi, Z.; Bin, X.; Yaoqi, Z. DDOMAIN: Dividing structures into domains using a normalized domain-domain interaction profile. Protein Sci. 2007, 16, 947–955. [Google Scholar]
- Pearl, F.; Todd, A.; Sillitoe, I.; Dibley, M.; Redfern, O.; Lewis, T.; Bennett, C.; Marsden, R.; Grant, A.; Lee, D.; et al. The CATH domain structure database and related resources Gene3D and DHS provide comprehensive domain family information for genome analysis. Nucleic Acids Res. 2005, 33, D247–D251. [Google Scholar] [CrossRef] [PubMed]
- Mili, A.; Das, S.; Nandakumar., K.; Lobo, R. Molecular docking and dynamics guided approach to identify potential anti-inflammatory molecules as NRF2 activator to protect against drug-induced liver injury (DILI): A computational study. J. Biomol. Struct. Dyn. 2022, 3, 1–18. [Google Scholar] [CrossRef]
- Pearl, F.; Todd, A.; Sillitoe, I. The Key-Lock theory and the induced fit theory. Angew. Chem. 1995, 33, 2375–2378. [Google Scholar]
- Ylilauri, M.; Pentikäinen, O. MMGBSA as a tool to understand the binding affinities of filamin-peptide interactions. J. Chem. Inf. Model. 2013, 53, 2626–2633. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, B.; Ma, S.; Wang, H.; Shang, L.; Zhu, C.; Ye, S. Discovery of SARS-CoV-2 3CLPro Peptidomimetic inhibitors through the catalytic dyad histidine-specific protein-ligand interactions. Int. J. Mol. Sci. 2022, 23, 2392. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Li, J.; Song, S.; Xiao, Z.; Chen, X.; Huang, B.; Sun, M.; Su, G.; Zhou, D.; Wang, G.; et al. Effective inhibition of coronavirus replication by Polygonum cuspidatum. Front. Biosci. Landmark Ed. 2021, 26, 789–798. [Google Scholar] [PubMed]
- Qi, X.; Li, B.; Omarini, A.B.; Gand, M.; Zhang, X.; Wang, J. Discovery of TCMs and derivatives against the main protease of SARS-CoV-2 via high throughput screening, ADMET analysis, and inhibition assay in vitro. J. Mol. Struct. 2022, 1268, 133709. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.; Zhang, M.; Xu, Q.; Song, F.; Wang, L.; Gai, S.; Tang, H.; Wang, S.; Zhou, L.; Li, H. Exploration of molecular targets and mechanisms of Chinese medicinal formula Acacia Catechu -Scutellariae Radix in the treatment of COVID-19 by a systems pharmacology strategy. Phytother. Res. 2022, 36, 4210–4229. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.M.; Zhu, Y.; Zhang, L.; Zhong, G.; Tai, L.; Liu, S.; Yin, G.; Lu, J.; He, Q.; Li, M.J.; et al. Novel cleavage sites identified in SARS-CoV-2 spike protein reveal mechanism for cathepsin L-facilitated viral infection and treatment strategies. Cell. Discov. 2022, 8, 53. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rank | Compound | SP Docking Binding Energy (ΔG, kcal/mol) | XP Docking Binding Energy (ΔG, kcal/mol) | MM-GBSA dG Bind (kcal/mol) | IFD Binding Energy (kcal/mol) |
|---|---|---|---|---|---|
| 1 | V247 | −8.538 | −7.976 | −63.43 | −11.34 |
| 2 | V253 | −8.832 | −7.97 | −62.15 | −12.787 |
| 3 | V133 | −8.379 | −7.969 | −58.29 | −10.76 |
| 4 | V109 | −7.567 | −7.108 | −68.64 | −12.342 |
| 5 | V212 | −7.688 | −7.038 | −47.32 | -- a |
| 6 | V131 | −7.876 | −6.759 | −67.51 | −9.919 |
| 7 | V254 | −7.286 | −6.665 | −57.63 | −10.896 |
| 8 | V248 | −7.565 | −6.645 | −46.39 | -- a |
| 9 | V282 | −7.481 | −6.564 | −49.38 | −9.959 |
| 10 | V139 | −7.422 | −6.49 | −54.66 | −9.584 |
| 11 | V231 | −7.372 | −6.453 | −57.18 | −10.961 |
| 12 | V128 | −7.264 | −6.422 | −59.99 | −9.557 |
| 13 | V160 | −7.241 | −6.409 | −58.19 | −10.191 |
| 14 | V163 | −7.052 | −6.337 | −52.59 | −9.817 |
| 15 | V174 | −7.369 | −6.293 | −56.94 | −10.993 |
| 16 | V243 | −7.68 | −6.267 | −60.64 | −11.459 |
| 17 | V229 | −7.509 | −6.237 | −57.48 | −9.365 |
| 18 | V204 | −7.89 | −6.229 | −53.3 | −9.652 |
| 19 | V291 | −8.071 | −6.225 | −59.08 | −9.628 |
| 20 | V228 | −8.122 | −6.12 | −56.37 | −10.792 |
| 21 | V170 | −7.085 | −6.118 | −50.4 | −10.11 |
| 22 | V215 | −7.032 | −6.08 | −45.32 | -- a |
| 23 | V165 | −7.051 | −6.078 | −48.72 | -- a |
| 24 | V75 | −7.331 | −6.072 | −56.99 | −10.714 |
| 25 | V205 | −7.501 | −6.029 | −47.89 | -- a |
| 26 | V120 | −7.081 | −6.025 | −51.7 | −9.171 |
| 27 | V222 | −7.305 | −6.018 | −48.47 | -- a |
| 28 | V245 | −7.401 | −6.008 | −56.32 | −10.098 |
| 29 | V226 | −7.141 | −6 | −61.64 | −11.774 |
| 30 | V159 | −7.091 | −5.995 | −46.69 | -- a |
| 31 | V12 | −7.192 | −5.987 | −48.94 | -- a |
| 32 | V225 | −7.068 | −5.983 | −48.31 | -- a |
| 33 | V252 | −7.416 | −5.981 | −57.86 | −11.603 |
| 34 | V219 | −7.212 | −5.972 | −43.13 | -- a |
| 35 | V173 | −7.138 | −5.965 | −47.29 | -- a |
| 36 | V241 | −7.108 | −5.963 | −39.62 | -- a |
| 37 | V154 | −7.26 | −5.93 | −49.83 | −10.761 |
| 38 | V304 | −7.878 | −5.911 | −54.47 | −9.512 |
| 39 | V230 | −7.314 | −5.891 | −43.13 | -- a |
| 40 | V172 | −7.403 | −5.85 | −50.46 | −11.391 |
| 41 | V97 | −7.138 | −5.825 | −60.03 | −11.778 |
| 42 | V111 | −7.195 | −5.823 | −49.62 | −10.706 |
| 43 | V238 | −7.134 | −5.811 | −55.55 | −10.605 |
| 44 | V155 | −7.113 | −5.77 | −48.22 | -- a |
| 45 | V112 | −7.29 | −5.744 | −44.36 | -- a |
| 46 | V60 | −7.261 | −5.637 | −49.07 | −10.191 |
| 47 | V257 | −7.37 | −5.637 | −48.64 | -- a |
| 48 | V103 | −7.535 | −5.55 | −60.98 | −9.795 |
| 49 | Nilotinib | −7.026 | −5.476 | −48.96 | −9.179 |
| 50 | V74 | −7.225 | −5.262 | -- a | -- a |
| 51 | V175 | −7.538 | −5.258 | -- a | -- a |
| 52 | V283 | −7.532 | −5.159 | -- a | -- a |
| 53 | V306 | −7.335 | −5.136 | -- a | -- a |
| 54 | V293 | −7.205 | −5.042 | -- a | -- a |
| 55 | V286 | −7.432 | −5.041 | -- a | -- a |
| 56 | V168 | −7.182 | −5.023 | -- a | -- a |
| 57 | V144 | −7.195 | −4.988 | -- a | -- a |
| 58 | V122 | −7.095 | −4.935 | -- a | -- a |
| 59 | V150 | −7.145 | −4.932 | -- a | -- a |
| 60 | V240 | −7.211 | −4.738 | -- a | -- a |
| 61 | V70 | −7.299 | −4.731 | -- a | -- a |
| 62 | V86 | −7.085 | −4.152 | -- a | -- a |
| 63 | V147 | −7.364 | −3.493 | -- a | -- a |
| 64 | V303 | −7.458 | −3.164 | -- a | -- a |
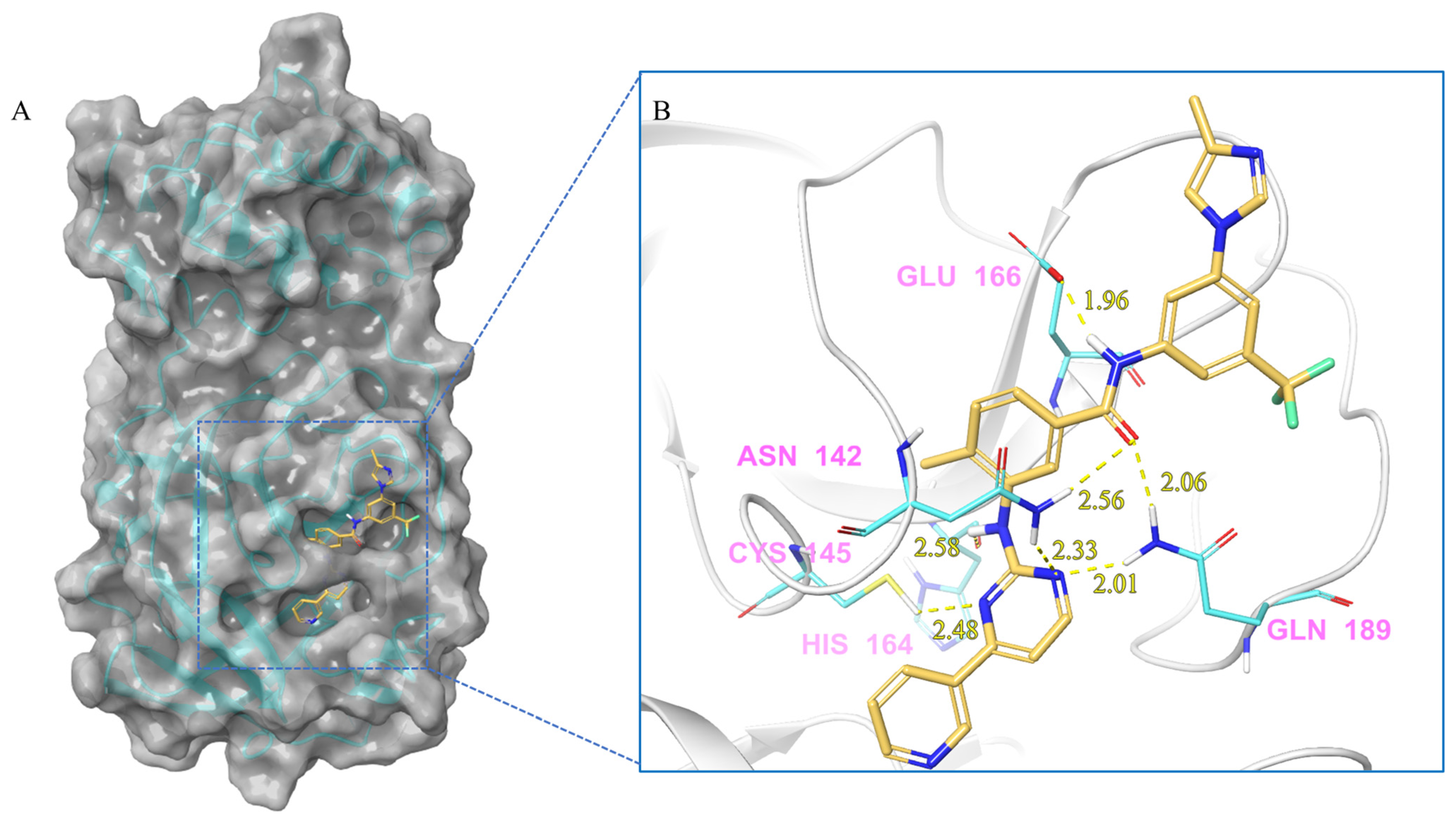
| Compound | Molecular Interactions | Nature of Interactions | Distance (Å) |
|---|---|---|---|
| Nilotinib | Asn142:HD22-Lig:O1 | Hydrogen Bond | 2.56 |
| Asn142:HD21-Lig:N6 | Hydrogen Bond | 2.33 | |
| Cys145:HG-Lig:N5 | Hydrogen Bond | 2.48 | |
| Lig:H16-His164:O | Hydrogen Bond | 2.58 | |
| Lig:H9-Glu166:OE2 | Hydrogen Bond | 1.96 | |
| Gln189:HE21-Lig:N6 | Hydrogen Bond | 2.01 | |
| Gln189:HE22-Lig:O1 | Hydrogen Bond | 2.06 | |
| V253 | Lig:N1-Glu166:OE2 | Salt bridge | 3.17 |
| Lig:H19-Leu141:O | Hydrogen Bond | 1.97 | |
| Gly143:H-Lig:O4 | Hydrogen Bond | 2.15 | |
| Ser144:H-Lig:O4 | Hydrogen Bond | 2.68 | |
| Cys145:H-Lig:O4 | Hydrogen Bond | 2.17 | |
| Lig:H15-His164:O | Hydrogen Bond | 1.78 | |
| Lig:H28-Glu166:OE2 | Hydrogen Bond | 2.43 | |
| Lig:H11-Glu166:O | Hydrogen Bond | 2.62 | |
| Gln189:HE21-Lig:O2 | Hydrogen Bond | 2.91 | |
| Gln192:H-Lig:N3 | Hydrogen Bond | 3.17 | |
| Gln192:HE21-Lig:N3 | Hydrogen Bond | 2.73 | |
| His41-Lig | Hydrophobic (pi-pi Stacking) | 5.47 | |
| His41-Lig | Hydrophobic (pi-pi Stacking) | 5.43 | |
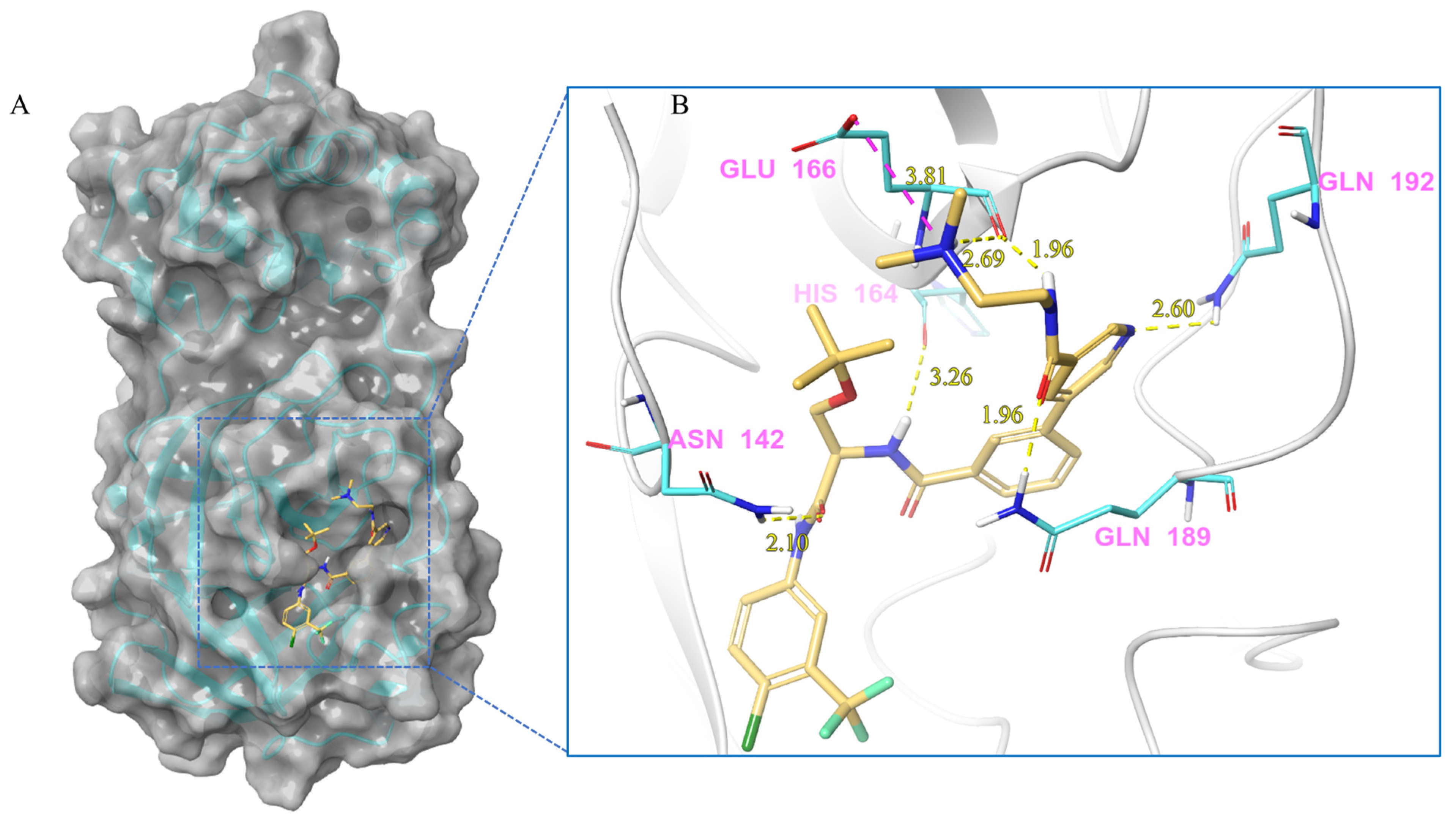
| V247 | Lig:N1-Glu166:OE2 | Salt bridge | 3.81 |
| Asn142:HD21-Lig:O4 | Hydrogen Bond | 2.10 | |
| Lig:H15-His164:O | Hydrogen Bond | 3.26 | |
| Lig:H11-Glu166:O | Hydrogen Bond | 1.96 | |
| Lig:H36-Glu166:O | Hydrogen Bond | 2.69 | |
| Gln189:HE21-Lig:O1 | Hydrogen Bond | 1.96 | |
| Gln192:HE21-Lig:N3 | Hydrogen Bond | 2.60 | |
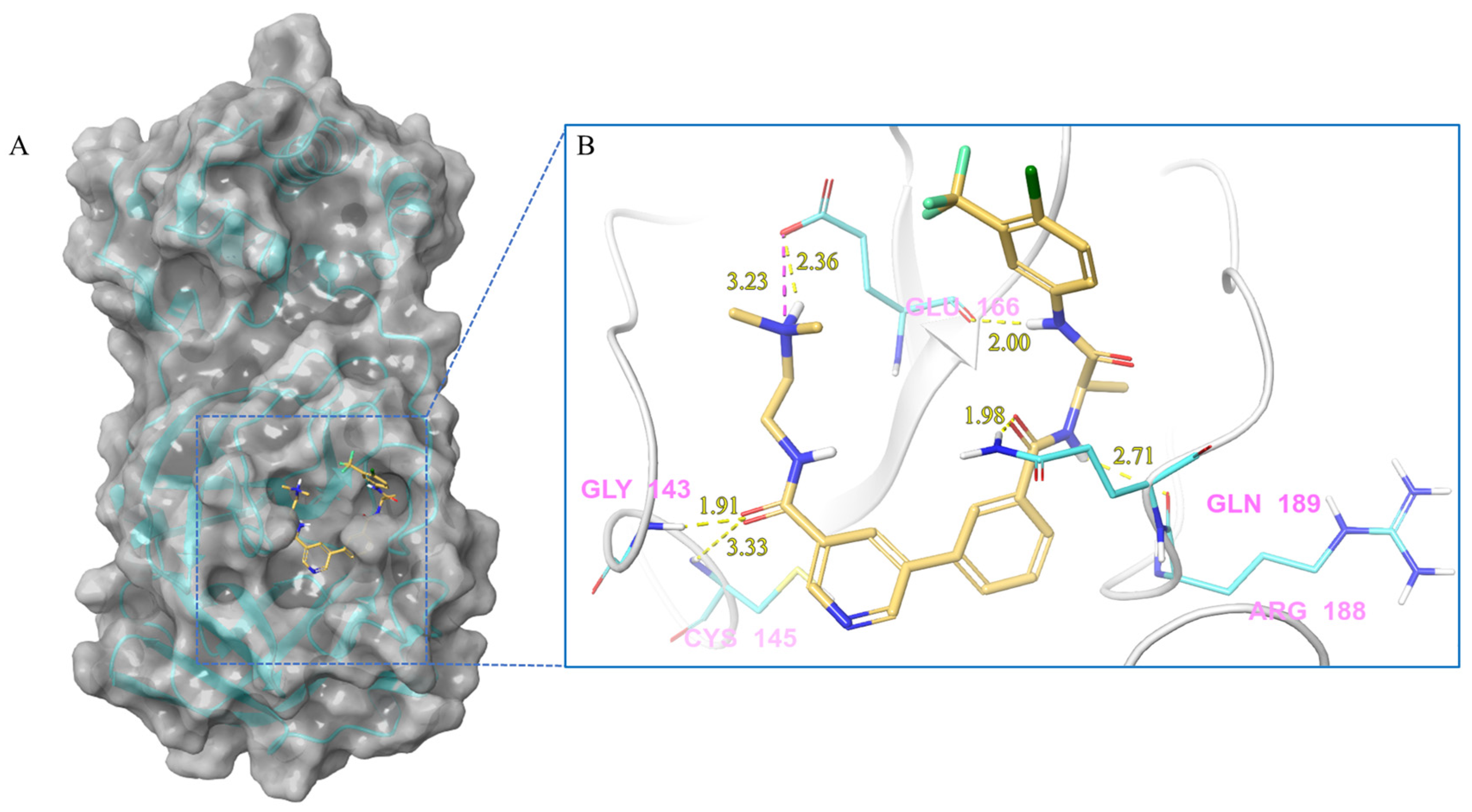
| V133 | Lig:N3-Glu166:OE2 | Salt bridge | 3.23 |
| Gly143:H-Lig:O2 | Hydrogen Bond | 1.91 | |
| Cys145:H-Lig:O2 | Hydrogen Bond | 3.33 | |
| Lig:H28-Glu166:OE2 | Hydrogen Bond | 2.36 | |
| Lig:H24-Glu166:O | Hydrogen Bond | 2.00 | |
| Lig:H5-Arg188:O | Hydrogen Bond | 2.71 | |
| Gln189:HE21-Lig:O1 | Hydrogen Bond | 1.98 | |
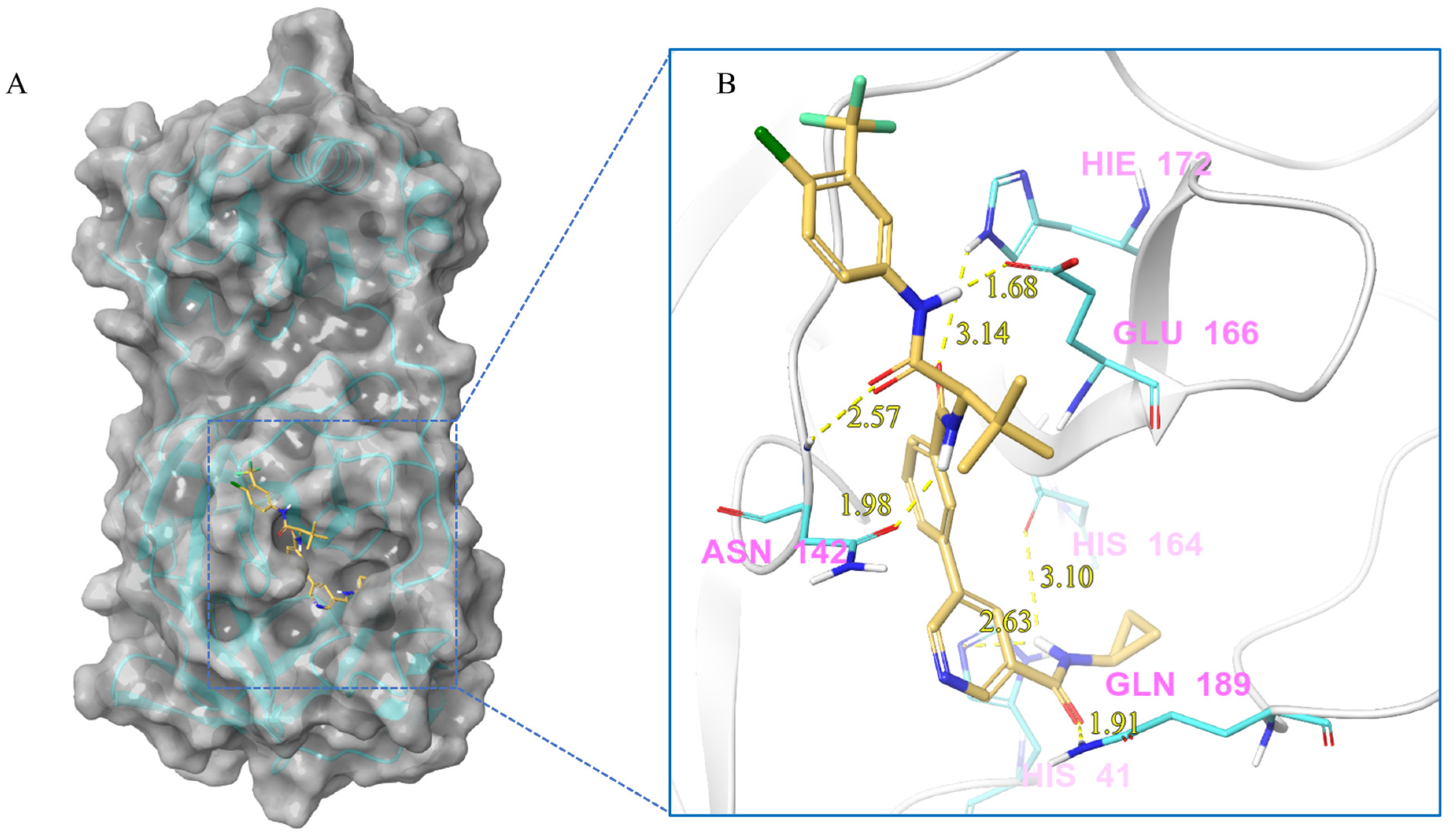
| V228 | Lig:H1-His41:NE2 | Hydrogen Bond | 2.63 |
| Asn142:H-Lig:O2 | Hydrogen Bond | 2.57 | |
| Lig:H10-Asn142:OD1 | Hydrogen Bond | 1.98 | |
| Lig:H1-His164:O | Hydrogen Bond | 3.10 | |
| Lig:H21-Glu166:OE1 | Hydrogen Bond | 1.68 | |
| His172:HE2-Lig:O3 | Hydrogen Bond | 3.14 | |
| Gln189:HE21-Lig:O1 | Hydrogen Bond | 1.91 | |
| Lig:H1-His41:NE2 | Hydrogen Bond | 2.63 | |
| V291 | Lig:N2-Glu166:OE2 | Salt bridge | 4.88 |
| Lig:H14-Thr26:O | Hydrogen Bond | 1.94 | |
| Thr26:H-Lig:N7 | Hydrogen Bond | 2.92 | |
| Lig:H12-His41:O | Hydrogen Bond | 2.24 | |
| Lig:H19-Asn142:OD1 | Hydrogen Bond | 2.21 | |
| Asn142:H-Lig:O1 | Hydrogen Bond | 2.62 | |
| Gly143:H-Lig:N5 | Hydrogen Bond | 2.13 | |
| Cys145:H-Lig:N5 | Hydrogen Bond | 3.00 | |
| Lig:H2-Glu166:OE1 | Hydrogen Bond | 1.89 | |
| Lig:H2-Glu166:OE2 | Hydrogen Bond | 3.07 | |
| His41-Lig | Hydrophobic (pi-pi Stacking) | 4.40 | |
| His41-Lig | Hydrophobic (pi-pi Stacking) | 4.58 | |
| His163-Lig | Hydrophobic (pi-pi Stacking) | 5.26 |
| Compounds | PSA | QPlogS | QPlogPo/w | donorHB | accptHB | CNS | #metab | Human Oral Absorption | QPlogBB | QPPMDCK | QPPCaco | QPlogHERG |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| V75 | 109.835 | −7.648 | 4.646 | 1 | 10.25 | −2 | 2 | 1 | −1.295 | 318.027 | 664.465 | −7.395 |
| V97 | 108.109 | −7.351 | 4.096 | 1 | 10 | −2 | 2 | 1 | −1.35 | 469.306 | 338.538 | −7.096 |
| V111 | 122.61 | −5.7 | 3.621 | 5 | 6.5 | −2 | 1 | 3 | −1.388 | 324.488 | 274.769 | −6.644 |
| V131 | 118.105 | −9.052 | 5.627 | 2.25 | 8.25 | −2 | 4 | 1 | −1.183 | 1829.769 | 487.844 | −7.478 |
| V133 | 121.912 | −7.656 | 4.886 | 2.25 | 10.25 | −1 | 5 | 1 | −0.944 | 458.546 | 123.765 | −8.379 |
| V139 | 131.795 | −7.531 | 4.288 | 4 | 10 | −1 | 4 | 1 | −0.999 | 332.432 | 91.695 | −8.504 |
| V159 | 89.316 | −8.698 | 6.02 | 1 | 8.5 | −1 | 1 | 1 | −0.568 | 3877.557 | 1007.174 | −7.822 |
| V172 | 89.548 | −10.162 | 6.837 | 1 | 8.5 | −1 | 3 | 1 | −0.58 | 7550.982 | 849.164 | −8.139 |
| V205 | 111.144 | −6.49 | 3.559 | 3 | 7.5 | −2 | 1 | 1 | −1.563 | 133.383 | 172.141 | −6.845 |
| V222 | 127.504 | −5.182 | 3.093 | 5 | 7.25 | −1 | 2 | 3 | −1.731 | 101.888 | 231.806 | −6.597 |
| V226 | 103.042 | −9.687 | 6.75 | 1.25 | 8.75 | −2 | 4 | 1 | −1.075 | 2761.001 | 715.442 | −7.297 |
| V231 | 113.094 | −9.105 | 5.992 | 2.25 | 7.75 | −2 | 3 | 1 | −1.085 | 1992.813 | 528.81 | −7.326 |
| V243 | 125.557 | −9.876 | 6.523 | 2.25 | 9 | −2 | 5 | 1 | −1.568 | 1356.918 | 372.469 | −7.538 |
| V245 | 116.326 | −9.815 | 6.884 | 1.25 | 9.5 | −2 | 5 | 1 | −1.416 | 1933.915 | 514.511 | −7.371 |
| V247 | 132.565 | −6.51 | 5.629 | 2.25 | 11 | −1 | 6 | 1 | −0.775 | 522.9 | 152.091 | −7.308 |
| V253 | 143.489 | −7.077 | 4.417 | 2.25 | 10.95 | −2 | 6 | 1 | −1.538 | 184.545 | 53.315 | −8.299 |
| V254 | 129.75 | −8.419 | 5.29 | 1.25 | 9.45 | −2 | 5 | 1 | −1.861 | 687.446 | 197.201 | −7.324 |
| V282 | 123.041 | −6.679 | 3.535 | 3 | 10 | −1 | 2 | 1 | −0.581 | 408.384 | 110.962 | −7.606 |
| V291 | 134.153 | −5.819 | 2.54 | 5 | 8 | −2 | 2 | 2 | −1.351 | 53.764 | 17.108 | −7.099 |
| V304 | 129.939 | −5.218 | 2.33 | 4 | 9.5 | −1 | 4 | 2 | −0.865 | 146.26 | 23.945 | −6.2 |
| Standard range | 7–200 | −6.5–0.5 | −2.0–6.5 | 0.0–6.0 | 2.0–20.0 | −2–+2 | 1–8 | 1, 2, or 3 for low, medium, or high | −3.0–1.2 | <25 poor, >500 great | <25 poor, >500 great | <−5 |
| Compounds | Structure | IC50 Value (μM) |
|---|---|---|
| V111 |  | >20 |
| V139 |  | >20 |
| V159 |  | >20 |
| V205 |  | >20 |
| V226 |  | >20 |
| V231 |  | >20 |
| V243 |  | >20 |
| V291 |  | 2.77 ± 0.56 |
| V304 |  | >20 |
| Nilotinib |  | 19.92 ± 1.27 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, N.; Yang, Z.; Liu, Y.; Dang, X.; Zhang, Q.; Wang, J.; Liu, X.; Zhang, J.; Pan, X. Identification of a Putative SARS-CoV-2 Main Protease Inhibitor through In Silico Screening of Self-Designed Molecular Library. Int. J. Mol. Sci. 2023, 24, 11390. https://doi.org/10.3390/ijms241411390
Liu N, Yang Z, Liu Y, Dang X, Zhang Q, Wang J, Liu X, Zhang J, Pan X. Identification of a Putative SARS-CoV-2 Main Protease Inhibitor through In Silico Screening of Self-Designed Molecular Library. International Journal of Molecular Sciences. 2023; 24(14):11390. https://doi.org/10.3390/ijms241411390
Chicago/Turabian StyleLiu, Nanxin, Zeyu Yang, Yuying Liu, Xintao Dang, Qingqing Zhang, Jin Wang, Xueying Liu, Jie Zhang, and Xiaoyan Pan. 2023. "Identification of a Putative SARS-CoV-2 Main Protease Inhibitor through In Silico Screening of Self-Designed Molecular Library" International Journal of Molecular Sciences 24, no. 14: 11390. https://doi.org/10.3390/ijms241411390
APA StyleLiu, N., Yang, Z., Liu, Y., Dang, X., Zhang, Q., Wang, J., Liu, X., Zhang, J., & Pan, X. (2023). Identification of a Putative SARS-CoV-2 Main Protease Inhibitor through In Silico Screening of Self-Designed Molecular Library. International Journal of Molecular Sciences, 24(14), 11390. https://doi.org/10.3390/ijms241411390