
Synergistic Inhibitory Effect of Quercetin and Cyanidin-3O-Sophoroside on ABCB1
 , ,
, ,  , and
, and
Abstract
1. Introduction
2. Results
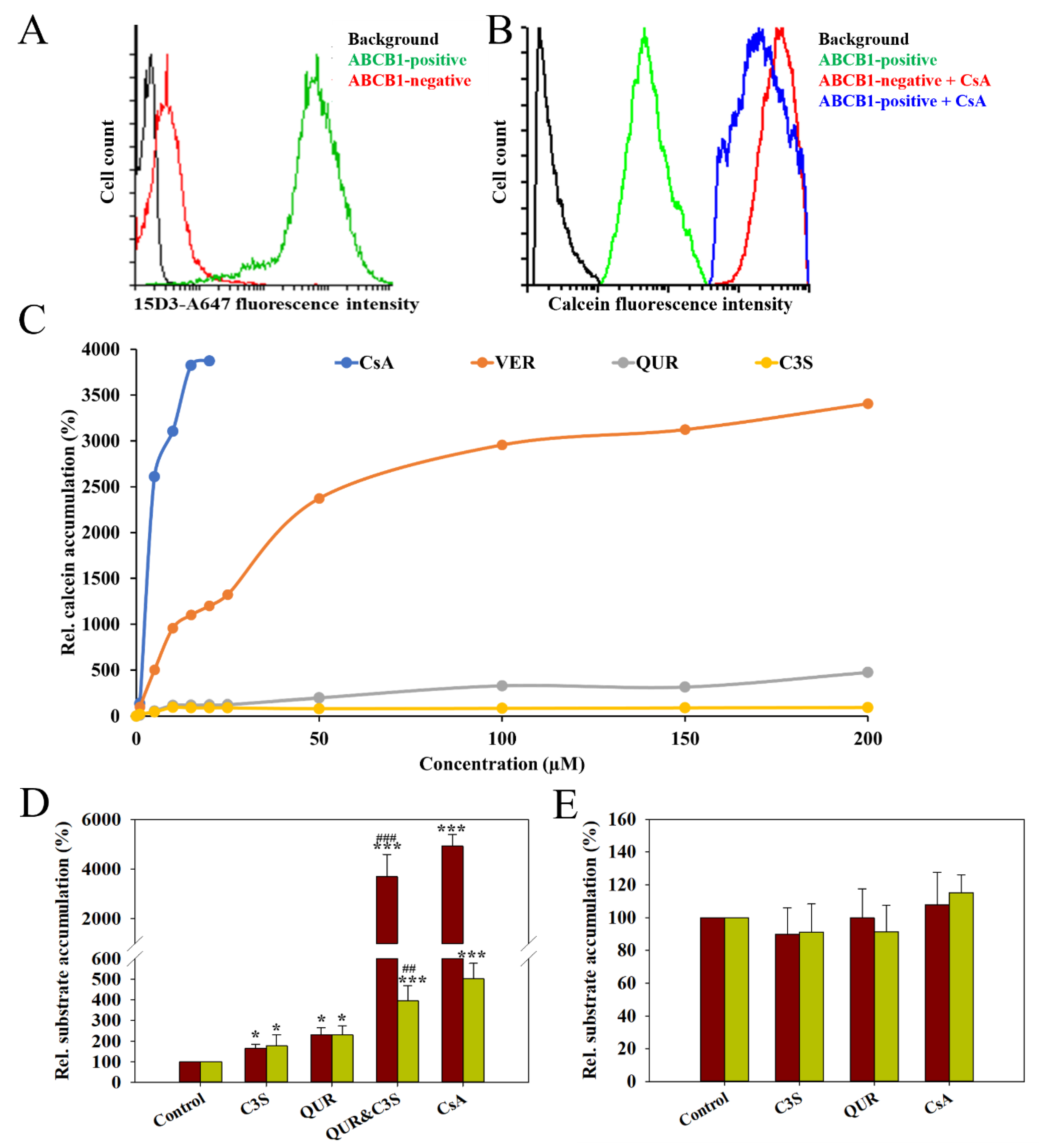
2.1. Co-Treatment with Quercetin and Cyanidin-3O-Sophoroside Inhibits Human ABCB1-Mediated Transport
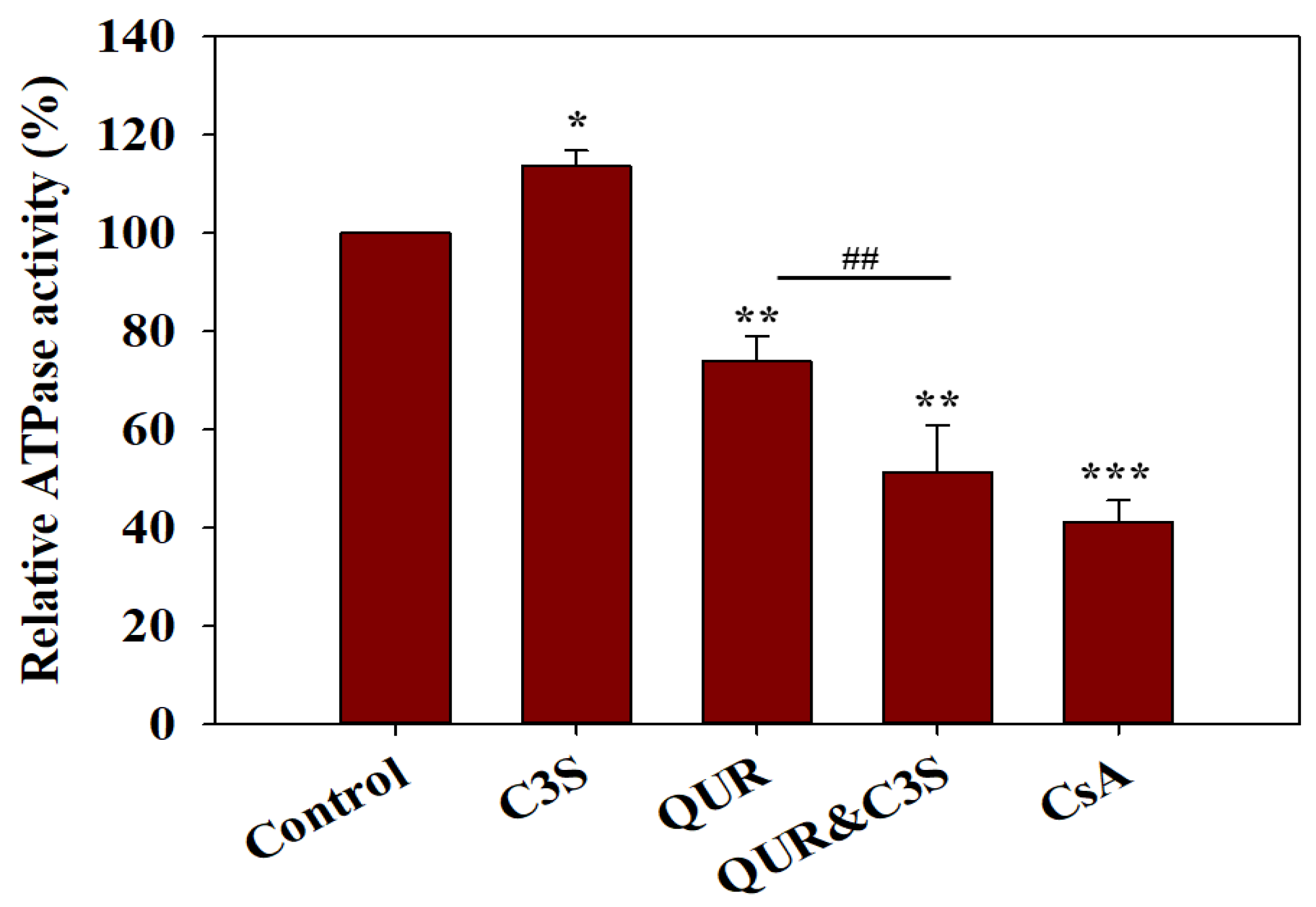
2.2. Quercetin and Cyanidin-3O-Sophoroside Synergistically Lower the ATPase Activity of ABCB1
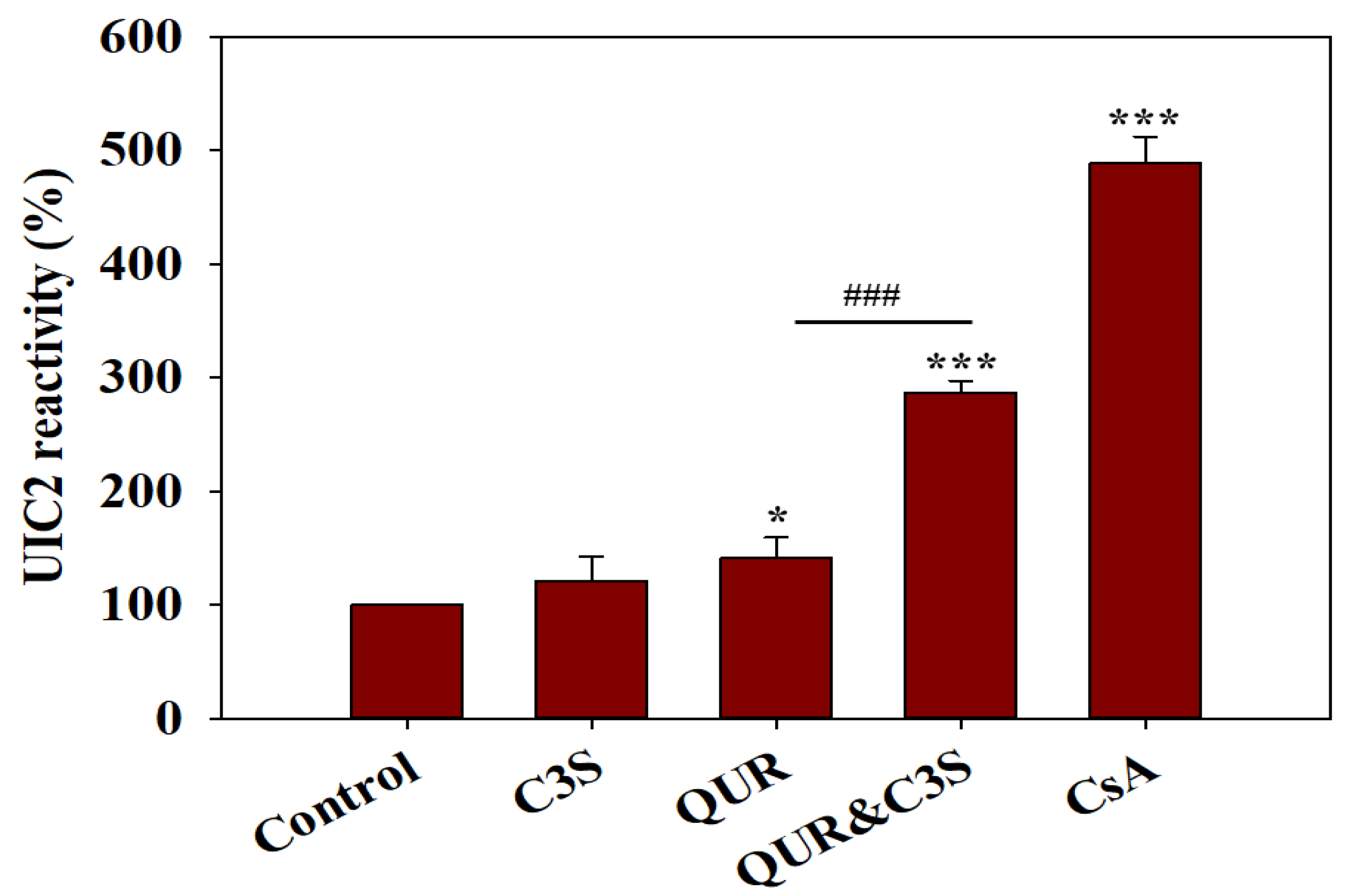
2.3. Synergistic Effects of Quercetin and Cyanidin-3O-Sophoroside on the Conformational Transitions Detected by UIC2
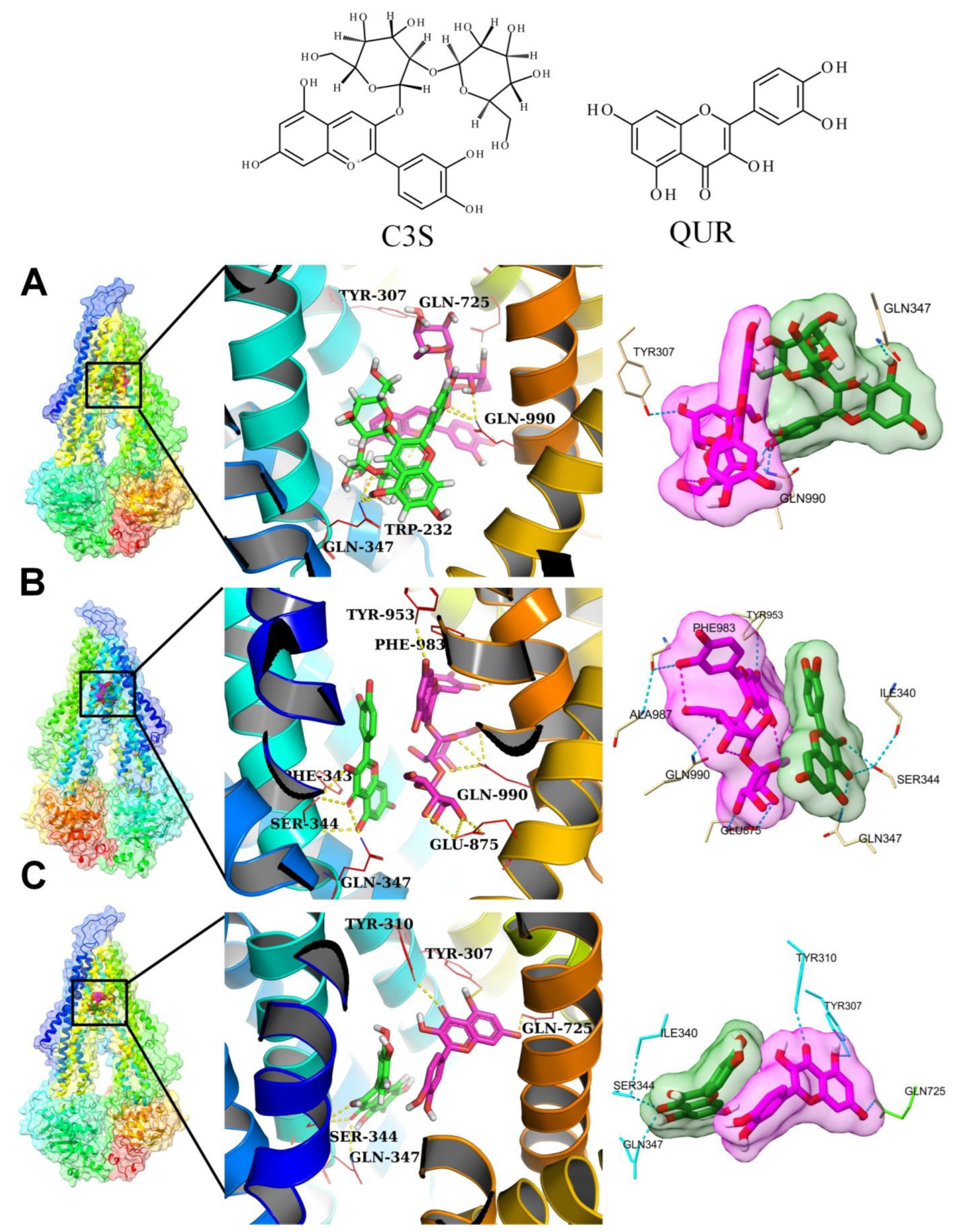
2.4. Molecular Docking Experiments
2.5. Molecular Dynamics Simulations
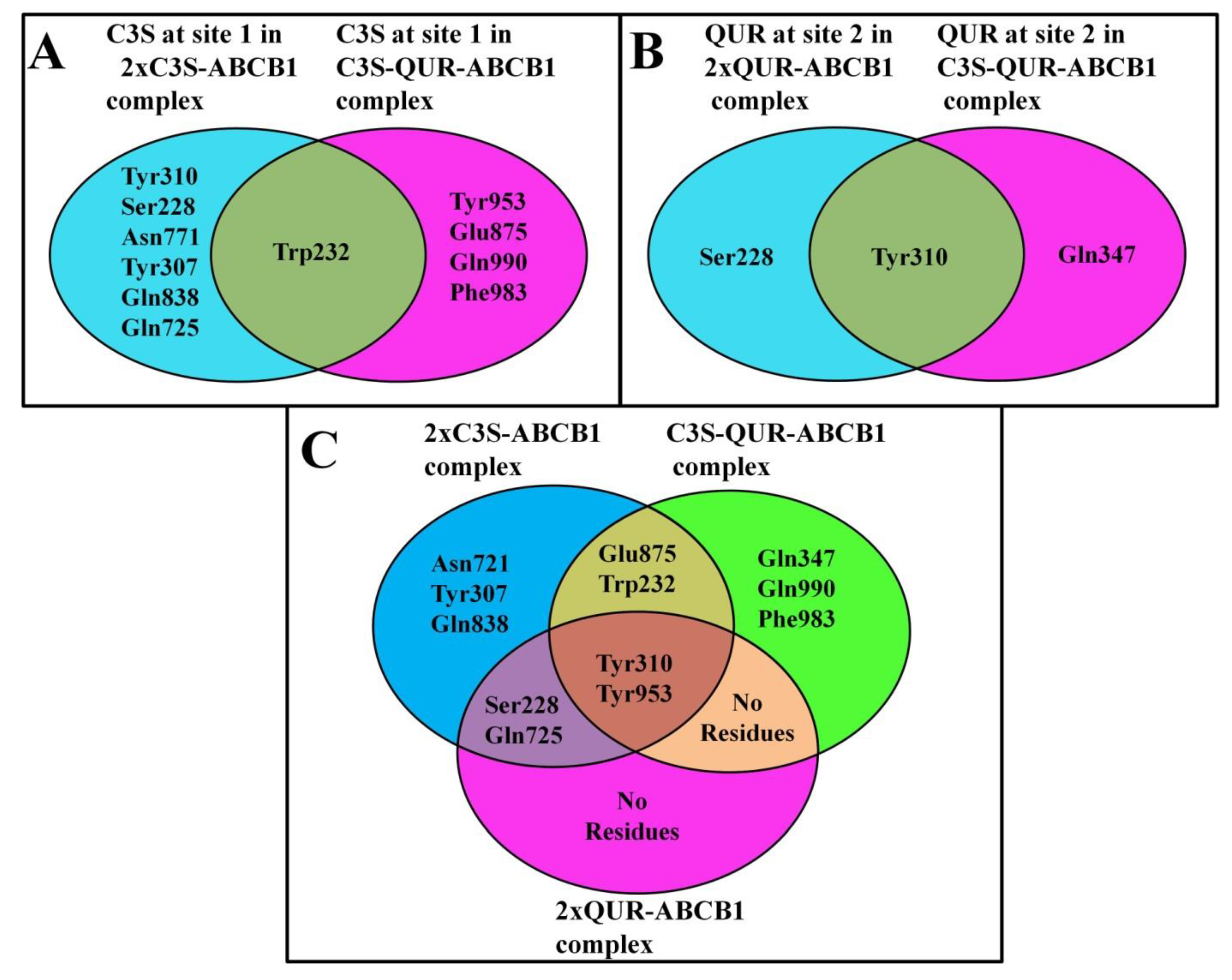
2.5.1. Analysis of Hydrogen Bonding between Ligands and Amino Acid Residues of the Substrate-Binding Pocket
2×C3S–ABCB1 Complex
C3S-QUR–ABCB1 Complex
2×QUR–ABCB1 Complex
The Same Ligand Exhibits Different Hydrogen Bonding Patterns in Hetero-Liganded and Homo-Liganded Complexes
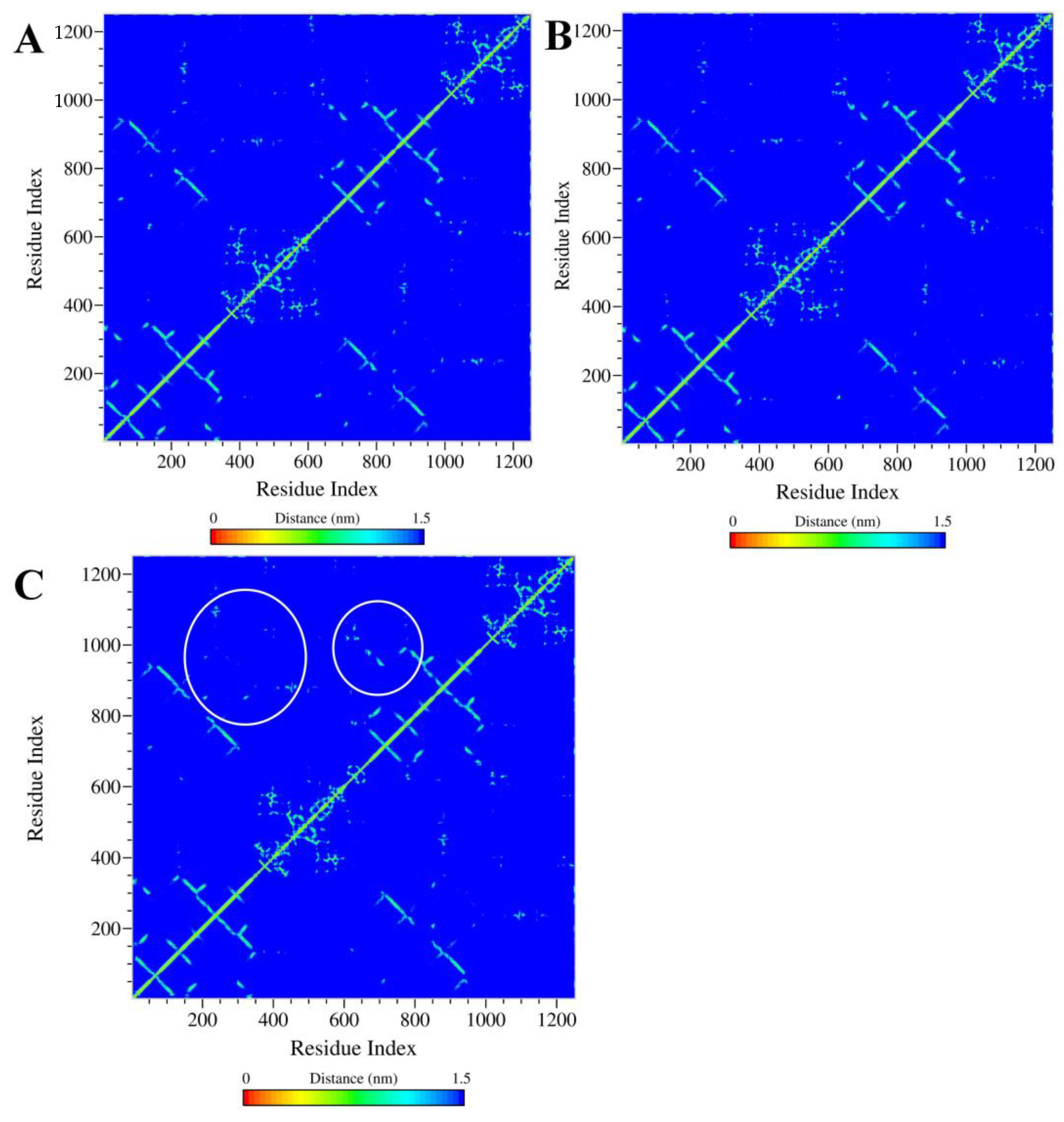
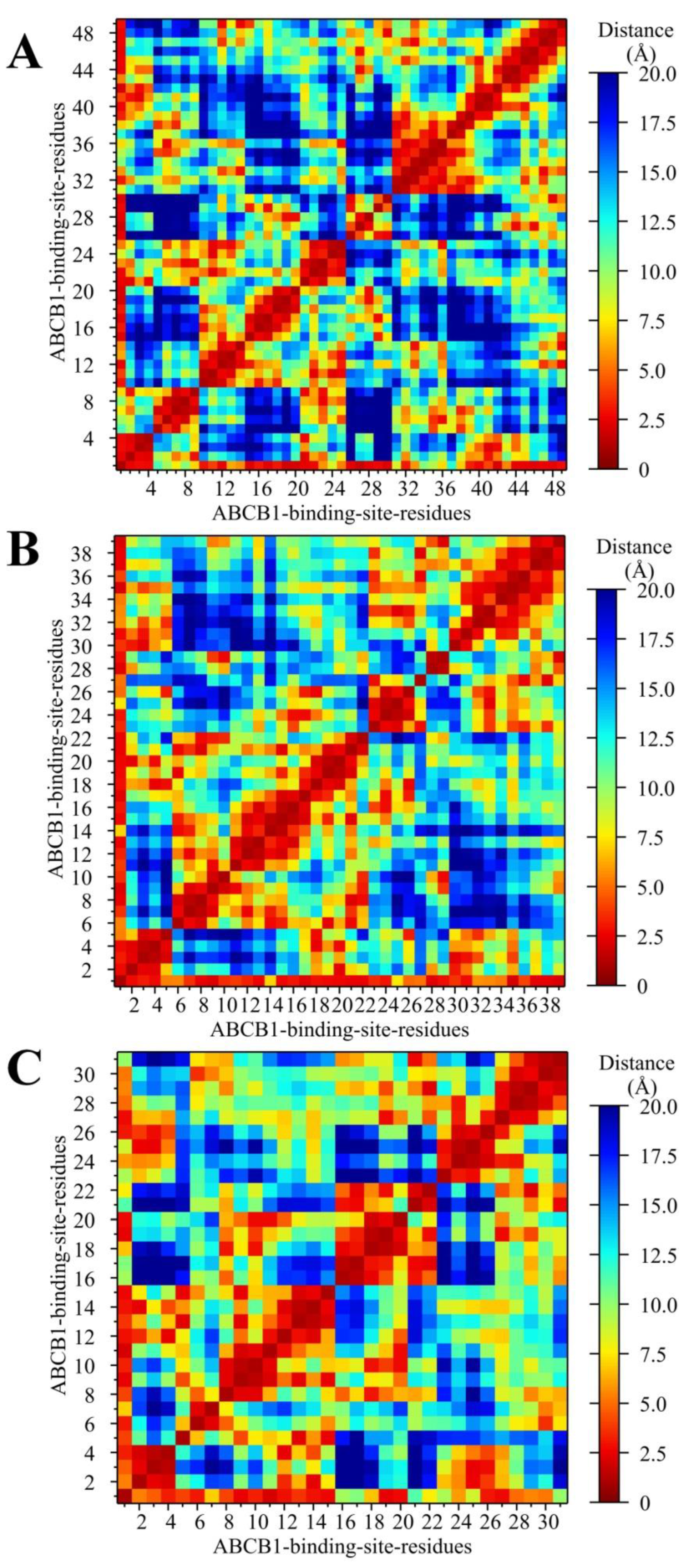
2.5.2. Analysis of the Conformation Changes in the NBDs and TMDs of Ligand–ABCB1 Complexes
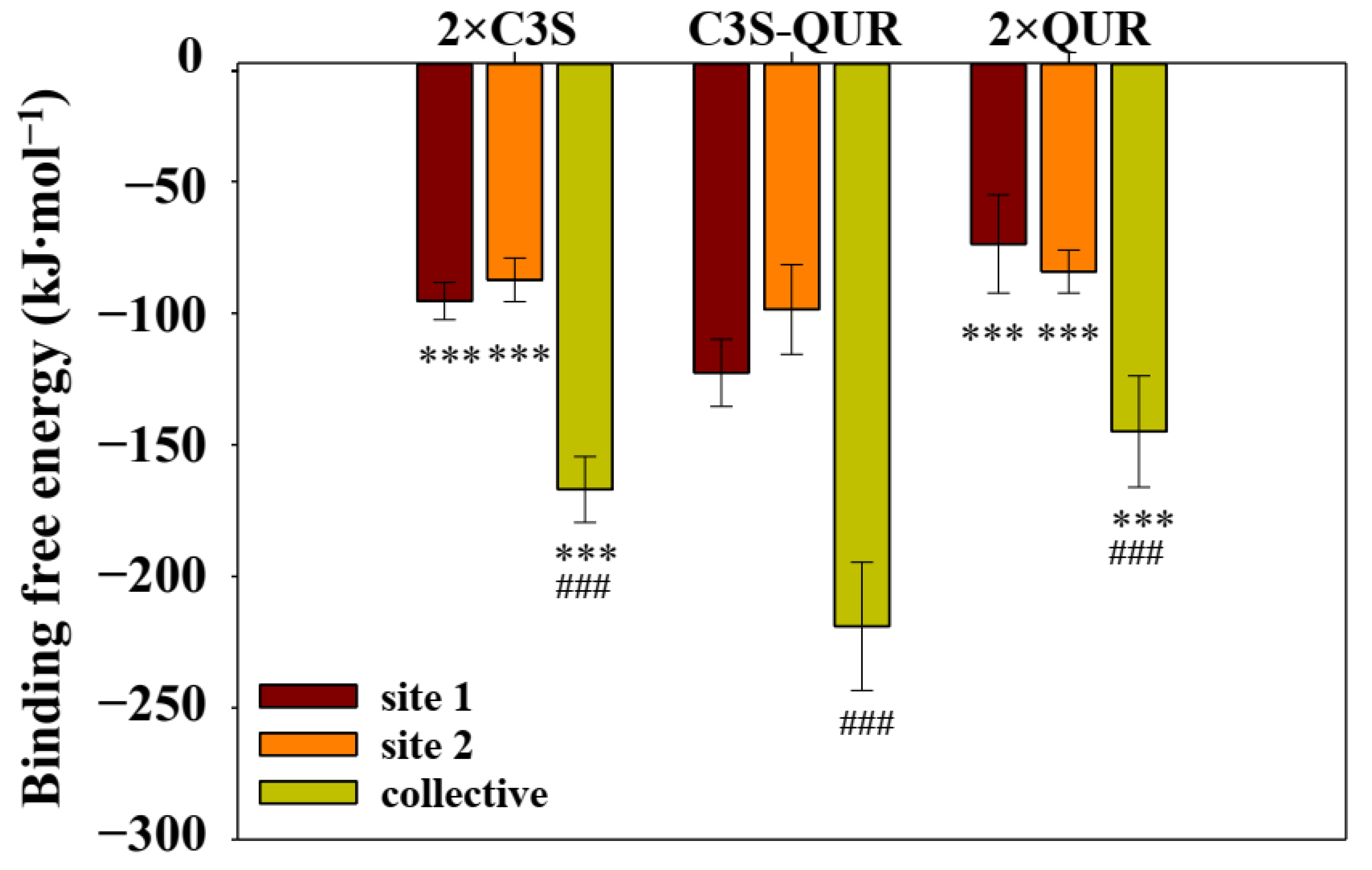
2.5.3. Binding Free Energy of the Different Ligand–ABCB1 Complexes
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culturing
4.3. Transport Activity Measurements
4.4. Crude Membrane Preparation for ATPase Activity Measurements
4.5. ATPase Activity Measurements
4.6. UIC2 Reactivity Assay
4.7. Flow Cytometry
4.8. Statistical Analysis
4.9. Molecular Docking
4.10. Molecular Dynamics
4.11. MD Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sarkadi, B.; Szakács, G. Understanding transport through pharmacological barriers—Are we there yet? Nat. Rev. Drug Discov. 2010, 9, 897–898. [Google Scholar] [CrossRef]
- Szakács, G.; Váradi, A.; Özvegy-Laczka, C.; Sarkadi, B. The role of ABC transporters in drug absorption, distribution, metabolism, excretion and toxicity (ADME–Tox). Drug Discov. Today 2008, 13, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Sarkadi, B.; Homolya, L.; Szakács, G.; Váradi, A. Human multidrug resistance ABCB and ABCG transporters: Participation in a chemoimmunity defense system. Physiol. Rev. 2006, 86, 1179–1236. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP–dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Stockner, T.; Gradisch, R.; Schmitt, L. The role of the degenerate nucleotide binding site in type I ABC exporters. FEBS Lett. 2020, 594, 3815–3838. [Google Scholar] [CrossRef]
- Aller, S.G.; Yu, J.; Ward, A.; Weng, Y.; Chittaboina, S.; Zhuo, R.; Harrell, P.M.; Trinh, Y.T.; Zhang, Q.; Urbatsch, I.L.; et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 2009, 323, 1718–1722. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Chen, J. Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science 2018, 359, 915–919. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Pastan, I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu. Rev. Biochem. 1993, 62, 385–427. [Google Scholar] [CrossRef]
- Alam, A.; Kowal, J.; Broude, E.; Roninson, I.; Locher, K.P. Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science 2019, 363, 753–756. [Google Scholar] [CrossRef]
- Nosol, K.; Romane, K.; Irobalieva, R.N.; Alam, A.; Kowal, J.; Fujita, N.; Locher, K.P. Cryo-EM structures reveal distinct mechanisms of inhibition of the human multidrug transporter ABCB1. Proc. Natl. Acad. Sci. USA 2020, 117, 26245–26253. [Google Scholar] [CrossRef]
- Dey, S.; Ramachandra, M.; Pastan, I.; Gottesman, M.M.; Ambudkar, S.V. Evidence for two nonidentical drug-interaction sites in the human P-glycoprotein. Proc. Natl. Acad. Sci. USA 1997, 94, 10594–10599. [Google Scholar] [CrossRef]
- Klepsch, F.; Chiba, P.; Ecker, G.F. Exhaustive sampling of docking poses reveals binding hypotheses for propafenone type inhibitors of P-glycoprotein. PLoS Comput. Biol. 2011, 7, e1002036. [Google Scholar] [CrossRef]
- Shapiro, A.B.; Fox, K.; Lam, P.; Ling, V. Stimulation of P-glycoprotein-mediated drug transport by prazosin and progesterone. Evidence for a third drug-binding site. Eur. J. Biochem. 1999, 259, 841–850. [Google Scholar] [CrossRef]
- Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Simultaneous binding of two different drugs in the binding pocket of the human multidrug resistance P-glycoprotein. J. Biol. Chem. 2003, 278, 39706–39710. [Google Scholar] [CrossRef]
- Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Substrate-induced conformational changes in the transmembrane segments of human P-glycoprotein. Direct evidence for the substrate-induced fit mechanism for drug binding. J. Biol. Chem. 2003, 278, 13603–13606. [Google Scholar] [CrossRef]
- Alam, A.; Küng, R.; Kowal, J.; McLeod, R.A.; Tremp, N.; Broude, E.V.; Roninson, I.B.; Stahlberg, H.; Locher, K.P. Structure of a zosuquidar and UIC2-bound human-mouse chimeric ABCB1. Proc. Natl. Acad. Sci. USA 2018, 115, E1973–E1982. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Drug binding in human P-glycoprotein causes conformational changes in both nucleotide-binding domains. J. Biol. Chem. 2003, 278, 1575–1578. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.B.; Ling, V. Effect of quercetin on Hoechst 33342 transport by purified and reconstituted P-glycoprotein. Biochem. Pharmacol. 1997, 53, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Tarapcsák, S.; Gyöngy, Z.; Ritter, Z.; Batta, G.; Bosire, R.; Remenyik, J.; Goda, K. Effects of Polyphenols on P-Glycoprotein (ABCB1) Activity. Pharmaceutics 2021, 13, 2062. [Google Scholar] [CrossRef]
- Goda, K.; Fenyvesi, F.; Bacsó, Z.; Nagy, H.; Márián, T.; Megyeri, A.; Krasznai, Z.; Juhász, I.; Vecsernyés, M.; Szabó, G., Jr. Complete inhibition of P-glycoprotein by simultaneous treatment with a distinct class of modulators and the UIC2 monoclonal antibody. J. Pharmacol. Exp. Ther. 2007, 320, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Muzi, M.; Mankoff, D.A.; Link, J.M.; Shoner, S.; Collier, A.C.; Sasongko, L.; Unadkat, J.D. Imaging of cyclosporine inhibition of P-glycoprotein activity using 11C-verapamil in the brain: Studies of healthy humans. J. Nucl. Med. 2009, 50, 1267–1275. [Google Scholar] [CrossRef]
- Clouser, A.F.; Atkins, W.M. Long Range Communication between the Drug-Binding Sites and Nucleotide Binding Domains of the Efflux Transporter ABCB1. Biochemistry 2022, 61, 730–740. [Google Scholar] [CrossRef]
- Nagy, H.; Goda, K.; Fenyvesi, F.; Bacsó, Z.; Szilasi, M.; Kappelmayer, J.; Lustyik, G.; Cianfriglia, M.; Szabó, G., Jr. Distinct groups of multidrug resistance modulating agents are distinguished by competition of P-glycoprotein-specific antibodies. Biochem. Biophys. Res. Commun. 2004, 315, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Tuffery, P.; Derreumaux, P. Flexibility and binding affinity in protein–ligand, protein–protein and multi-component protein interactions: Limitations of current computational approaches. J. R. Soc. Interface 2012, 9, 20–33. [Google Scholar] [CrossRef]
- Mora Lagares, L.; Minovski, N.; Caballero Alfonso, A.Y.; Benfenati, E.; Wellens, S.; Culot, M.; Gosselet, F.; Novič, M. Homology Modeling of the Human P-glycoprotein (ABCB1) and Insights into Ligand Binding through Molecular Docking Studies. Int. J. Mol. Sci. 2020, 21, 4058. [Google Scholar] [CrossRef]
- Chufan, E.E.; Kapoor, K.; Ambudkar, S.V. Drug–protein hydrogen bonds govern the inhibition of the ATP hydrolysis of the multidrug transporter P-glycoprotein. Biochem. Pharmacol. 2016, 101, 40–53. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Hoag, H.; Wu, C.; Liu, H.; Yin, H.; Dong, J.; Qian, Z.; Miao, F.; Liu, M.; Miao, J. Experimental and Simulation Identification of Xanthohumol as an Inhibitor and Substrate of ABCB1. Appl. Sci. 2018, 8, 681. [Google Scholar] [CrossRef]
- Mora Lagares, L.; Pérez-Castillo, Y.; Minovski, N.; Novič, M. Structure–Function Relationships in the Human P-Glycoprotein (ABCB1): Insights from Molecular Dynamics Simulations. Int. J. Mol. Sci. 2022, 23, 362. [Google Scholar] [CrossRef]
- Litman, T.; Nielsen, D.; Skovsgaard, T.; Zeuthen, T.; Stein, W.D. ATPase activity of P-glycoprotein related to emergence of drug resistance in Ehrlich ascites tumor cell lines. Biochim. Biophys. Acta 1997, 1361, 147–158. [Google Scholar] [CrossRef]
- Bankstahl, J.P.; Bankstahl, M.; Römermann, K.; Wanek, T.; Stanek, J.; Windhorst, A.D.; Fedrowitz, M.; Erker, T.; Müller, M.; Löscher, W.; et al. Tariquidar and elacridar are dose-dependently transported by P-glycoprotein and Bcrp at the blood-brain barrier: A small-animal positron emission tomography and in vitro study. Drug Metab. Dispos. 2013, 41, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Bohn, K.; Lange, A.; Chmielewski, J.; Hrycyna, C.A. Dual Modulation of Human P-Glycoprotein and ABCG2 with Prodrug Dimers of the Atypical Antipsychotic Agent Paliperidone in a Model of the Blood–Brain Barrier. Mol. Pharm. 2017, 14, 1107–1119. [Google Scholar] [CrossRef] [PubMed]
- Namanja-Magliano, H.A.; Bohn, K.; Agrawal, N.; Willoughby, M.E.; Hrycyna, C.A.; Chmielewski, J. Dual inhibitors of the human blood-brain barrier drug efflux transporters P-glycoprotein and ABCG2 based on the antiviral azidothymidine. Bioorganic Med. Chem. 2017, 25, 5128–5132. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.-F.; Zhao, Y.; Chow, T.W.S.; Yan, C.S.W.; Ma, D.L.; Burkett, B.A.; Wong, I.L.K.; Chow, L.M.C.; Chan, T.H. Flavonoid dimers as bivalent modulators for p-glycoprotein-based multidrug resistance: Structure-activity relationships. ChemMedChem 2009, 4, 594–614. [Google Scholar] [CrossRef]
- Nagy, H.; Goda, K.; Arceci, R.; Cianfriglia, M.; Mechetner, E.; Szabó, G., Jr. P-Glycoprotein conformational changes detected by antibody competition. Eur. J. Biochem. 2001, 268, 2416–2420. [Google Scholar] [CrossRef]
- Farman, S.; Javed, A.; Arshia; Khan, K.M.; Nasir, A.; Khan, A.U.; Lodhi, M.A.; Gul, H.; Khan, F.; Asad, M.; et al. Benzophenone Sulfonamide Derivatives as Interacting Partners and Inhibitors of Human P-glycoprotein. Anti-Cancer Agents Med. Chem. 2020, 20, 1739–1751. [Google Scholar] [CrossRef]
- Chufan, E.E.; Sim, H.-M.; Ambudkar, S.V. Chapter Three—Molecular Basis of the Polyspecificity of P-Glycoprotein (ABCB1): Recent Biochemical and Structural Studies. In Advances in Cancer Research; Schuetz, J.D., Ishikawa, T., Eds.; Academic Press: Cambridge, MA, USA, 2015; Volume 125, pp. 71–96. [Google Scholar]
- Szöllósi, J.; Horejsí, V.; Bene, L.; Angelisová, P.; Damjanovich, S. Supramolecular complexes of MHC class I, MHC class II, CD20, and tetraspan molecules (CD53, CD81, and CD82) at the surface of a B cell line JY. J. Immunol. 1996, 157, 2939–2946. [Google Scholar] [CrossRef]
- Bruggemann, E.P.; Currier, S.J.; Gottesman, M.M.; Pastan, I. Characterization of the azidopine and vinblastine binding site of P-glycoprotein. J. Biol. Chem. 1992, 267, 21020–21026. [Google Scholar] [CrossRef]
- Holló, Z.; Homolya, L.; Davis, C.W.; Sarkadi, B. Calcein accumulation as a fluorometric functional assay of the multidrug transporter. Biochim. Biophys. Acta (BBA)-Biomembr. 1994, 1191, 384–388. [Google Scholar] [CrossRef]
- Goda, K.; Balkay, L.; Márián, T.; Trón, L.; Aszalós, A.; Szabó, G. Intracellular pH does not affect drug extrusion by P-glycoprotein. J. Photochem. Photobiol. B Biol. 1996, 34, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Sarkadi, B.; Price, E.M.; Boucher, R.C.; Germann, U.A.; Scarborough, G.A. Expression of the human multidrug resistance cDNA in insect cells generates a high activity drug-stimulated membrane ATPase. J. Biol. Chem. 1992, 267, 4854–4858. [Google Scholar] [CrossRef]
- Tarapcsák, S.; Szalóki, G.; Telbisz, Á.; Gyöngy, Z.; Matúz, K.; Csősz, É.; Nagy, P.; Holb, I.J.; Rühl, R.; Nagy, L.; et al. Interactions of retinoids with the ABC transporters P-glycoprotein and Breast Cancer Resistance Protein. Sci. Rep. 2017, 7, 41376. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Rackers, J.A.; Wang, Z.; Lu, C.; Laury, M.L.; Lagardère, L.; Schnieders, M.J.; Piquemal, J.P.; Ren, P.; Ponder, J.W. Tinker 8: Software Tools for Molecular Design. J. Chem. Theory Comput. 2018, 14, 5273–5289. [Google Scholar] [CrossRef]
- Søndergaard, C.R.; Olsson, M.H.M.; Rostkowski, M.; Jensen, J.H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Jo, S.; Lim, J.B.; Klauda, J.B.; Im, W. CHARMM-GUI Membrane Builder for mixed bilayers and its application to yeast membranes. Biophys. J. 2009, 97, 50–58. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; MacKerell, A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) I: Bond perception and atom typing. J. Chem. Inf. Model. 2012, 52, 3144–3154. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2012, 40, D370–D376. [Google Scholar] [CrossRef] [PubMed]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef]
- Petersen, H.G. Accuracy and efficiency of the particle mesh Ewald method. J. Chem. Phys. 1995, 103, 3668–3679. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 2006, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A Gen. Phys. 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Open Source Drug Discovery Consortium; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| Hydrogen Bonding Partners in the Central Binding Site (Site 1) | Occupancy (%) | Hydrogen Bonding Partners in the “Access Tunnel” (Site 2) | Occupancy (%) |
| C3S(H63)—Gln838 * (OE1) ** | 19.1 | C3S(H63)—Gln725(OE1) | 35.5 |
| C3S(H63)—Asn721(O) | 40.4 | C3S(H72)—Glu875(OE2) | 18.9 |
| C3S(H60)—Gln725(OE1) | 99.1 | C3S(H72)—Glu875(OE1) | 23.4 |
| Gln838(E21)—C3S(O10) | 42.7 | Tyr953(HH)—C3S(O13) | 92.9 |
| Gln838(E21)—C3S(O16) | 30.4 | Gln725(E21)—C3S(O10) | 35.4 |
| Gln838(E21)—C3S(O15) | 23.3 | Tyr310(HH)—C3S(O8) | 75.5 |
| Tyr310(HH)—C3S(O9) | 43.5 | ||
| Tyr307(HH)—C3S(O8) | 97.3 | ||
| Trp232(HE1)—C3S(O13) | 37.2 | ||
| Ser228(HG1)—C3S(O14) | 22.3 | ||
 | |||
|---|---|---|---|
| Hydrogen Bonding Partners in the Central Binding Site (Site 1) | Occupancy (%) | Hydrogen Bonding Partners in the “Access tunnel” (Site 2) | Occupancy (%) |
| C3S(H32)—Gln990 * (OE1) ** | 52.0 | Gln347(E21)—QUR(O3) | 33.9 |
| C3S(H30)—Phe983(O) | 88.6 | Try310(HH)—QUR(O7) | 58.8 |
| C3S(H15)—Glu875(OE2) | 27.2 | ||
| C3S(H15)—Glu875(OE1) | 38.6 | ||
| Gln990(E21)—C3S(O2) | 76.3 | ||
| Trp232(HE1)—C3S(O8) | 44.5 | ||
| Trp232(HE1)—C3S(O9) | 44.6 | ||
| Tyr953(HH)—C3S(O14) | 38.0 | ||
 | |||
|---|---|---|---|
| Hydrogen Bonding Partners in the Central Binding Site (Site 1) | Occupancy (%) | Hydrogen Bonding Partners in the “Access Tunnel” (Site 2) | Occupancy (%) |
| Tyr953(HH)—QUR *(O6) ** | 17.7 | Tyr310(HH)—QUR(O7) | 97.5 |
| Gln725(E21)—QUR(O2) | 32.1 | Ser228(HG1)—QUR(O5) | 93.4 |
| Gln725(E21)—QUR(O4) | 40.9 | ||
| System | Van Der Waals Energy (kJ∙mol−1) | Electrostatic Energy (kJ∙mol−1) | Polar Solvation Energy (kJ∙mol−1) | SASA Energy (kJ∙mol−1) | Binding Free Energy (kJ∙mol−1) |
|---|---|---|---|---|---|
| 2×C3S–ABCB1 complex | |||||
| site 1 | −133.84 (7.85) | −10.00 (2.40) | 62.33 (4.04) | −13.89 (0.82) | −95.41 (7.09) |
| site 2 | −133.04 (9.05) | −13.28 (2.64) | 73.21 (4.99) | −14.29 (0.70) | −87.37 (8.31) |
| total | −266.87 (12.72) | −23.30 (3.56) | 149.41 (7.33) | −26.29 (1.16) | −166.99 (12.53) |
| C3S-QUR–ABCB1 complex | |||||
| site 1 (C3S) | −231.98 (14.12) | −35.86 (6.29) | 171.81 (7.89) | −26.61 (0.98) | −122.66 (12.74) |
| site 2 (QUR) | −219.58 (15.04) | −34.88 (8.21) | 181.38 (13.72) | −25.52 (1.00) | −98.60 (1.70) |
| total | −451.55 (21.97) | −70.75 (8.99) | 351.84 (17.06) | −48.40 (1.48) | −218.96 (24.39) |
| 2×QUR–ABCB1 complex | |||||
| site 1 | −201.51 (15.56) | −39.57 (11.11) | 193.47 (19.26) | −26.07 (1.16) | −73.70 (18.64) |
| site 2 | −119.67 (8.89) | −11.01 (2.98) | 60.21 (6.35) | −13.79 (0.82) | −84.17 (8.17) |
| total | −321.26 (19.26) | −50.60 (11.35) | 263.44 (19.46) | −36.61 (1.70) | −144.93 (21.17) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, K.; Patil, R.B.; Patel, V.; Remenyik, J.; Hegedűs, T.; Goda, K. Synergistic Inhibitory Effect of Quercetin and Cyanidin-3O-Sophoroside on ABCB1. Int. J. Mol. Sci. 2023, 24, 11341. https://doi.org/10.3390/ijms241411341
Singh K, Patil RB, Patel V, Remenyik J, Hegedűs T, Goda K. Synergistic Inhibitory Effect of Quercetin and Cyanidin-3O-Sophoroside on ABCB1. International Journal of Molecular Sciences. 2023; 24(14):11341. https://doi.org/10.3390/ijms241411341
Chicago/Turabian StyleSingh, Kuljeet, Rajesh B. Patil, Vikas Patel, Judit Remenyik, Tamás Hegedűs, and Katalin Goda. 2023. "Synergistic Inhibitory Effect of Quercetin and Cyanidin-3O-Sophoroside on ABCB1" International Journal of Molecular Sciences 24, no. 14: 11341. https://doi.org/10.3390/ijms241411341
APA StyleSingh, K., Patil, R. B., Patel, V., Remenyik, J., Hegedűs, T., & Goda, K. (2023). Synergistic Inhibitory Effect of Quercetin and Cyanidin-3O-Sophoroside on ABCB1. International Journal of Molecular Sciences, 24(14), 11341. https://doi.org/10.3390/ijms241411341