
Global Protein Profiling in Processed Immunohistochemistry Tissue Sections
 , ,
, ,  ,
,
Abstract
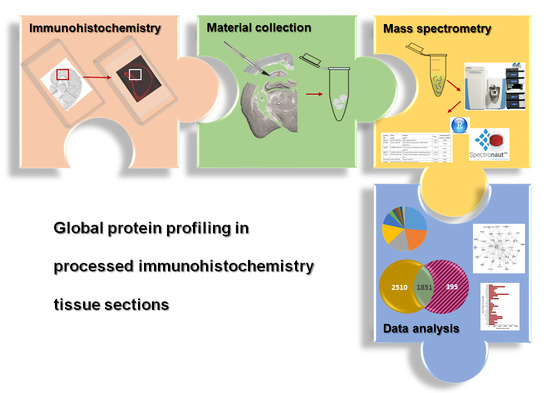
1. Introduction
2. Results
2.1. Protein Extraction Protocols for Proteomics
2.2. Protein Pattern of CA1 Region
2.3. Functional Enrichment Analysis of the Proteins Identified in the CA1 Region
2.4. Ingenuity Pathway Analysis
2.5. Comparison of Tissue-Derived CA1 Protein Patterns to Published Data Sets
3. Materials and Methods
3.1. Mice
3.2. Preparation of Embedded Slices
3.3. Preparation of CA1 Region Samples for Mass Spectrometry
3.4. Preparation of Whole Brain Samples for Mass Spectrometry
3.5. Mass Spectrometric Analysis
3.6. Bioinformatical Data Analysis and Statistics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wettersten, H.I.; Hakimi, A.A.; Morin, D.; Bianchi, C.; Johnstone, M.E.; Donohoe, D.R.; Trott, J.F.; Aboud, O.A.; Stirdivant, S.; Neri, B.; et al. Grade-Dependent Metabolic Reprogramming in Kidney Cancer Revealed by Combined Proteomics and Metabolomics Analysis. Cancer Res. 2015, 75, 2541–2552. [Google Scholar] [CrossRef] [PubMed]
- Zand, B.; Previs, R.A.; Zacharias, N.M.; Rupaimoole, R.; Mitamura, T.; Nagaraja, A.S.; Guindani, M.; Dalton, H.J.; Yang, L.; Baddour, J.; et al. Role of Increased n-acetylaspartate Levels in Cancer. J. Natl. Cancer Inst. 2016, 108, djv426. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Reznik, E.; Lee, H.J.; Gundem, G.; Jonsson, P.; Sarungbam, J.; Bialik, A.; Sanchez-Vega, F.; Creighton, C.J.; Hoekstra, J.; et al. Abnormal oxidative metabolism in a quiet genomic background underlies clear cell papillary renal cell carcinoma. eLife 2019, 8, e38986. [Google Scholar] [CrossRef] [PubMed]
- Griesser, E.; Wyatt, H.; Ten Have, S.; Stierstorfer, B.; Lenter, M.; Lamond, A.I. Quantitative Profiling of the Human Substantia Nigra Proteome from Laser-capture Microdissected FFPE Tissue. Mol. Cell Proteom. 2020, 19, 839–851. [Google Scholar] [CrossRef]
- Taverna, D.; Mignogna, C.; Santise, G.; Gaspari, M.; Cuda, G. On-Tissue Hydrogel-Mediated Nondestructive Proteomic Characterization: Application to fr/fr and FFPE Tissues and Insights for Quantitative Proteomics Using a Case of Cardiac Myxoma. Proteom. Clin. Appl. 2019, 13, e1700167. [Google Scholar] [CrossRef]
- Tanca, A.; Abbondio, M.; Pisanu, S.; Pagnozzi, D.; Uzzau, S.; Addis, M.F. Critical comparison of sample preparation strategies for shotgun proteomic analysis of formalin-fixed, paraffin-embedded samples: Insights from liver tissue. Clin. Proteom. 2014, 11, 28. [Google Scholar] [CrossRef]
- Morales-Betanzos, C.A.; Lee, H.; Gonzalez Ericsson, P.I.; Balko, J.M.; Johnson, D.B.; Zimmerman, L.J.; Liebler, D.C. Quantitative Mass Spectrometry Analysis of PD-L1 Protein Expression, N-glycosylation and Expression Stoichiometry with PD-1 and PD-L2 in Human Melanoma. Mol. Cell Proteom. 2017, 16, 1705–1717. [Google Scholar] [CrossRef]
- Balgley, B.M.; Guo, T.; Zhao, K.; Fang, X.; Tavassoli, F.A.; Lee, C.S. Evaluation of archival time on shotgun proteomics of formalin-fixed and paraffin-embedded tissues. J. Proteome Res. 2009, 8, 917–925. [Google Scholar] [CrossRef]
- Sprung, R.W.; Martinez, M.A.; Carpenter, K.L.; Ham, A.J.; Washington, M.K.; Arteaga, C.L.; Sanders, M.E.; Liebler, D.C. Precision of multiple reaction monitoring mass spectrometry analysis of formalin-fixed, paraffin-embedded tissue. J. Proteome Res. 2012, 11, 3498–3505. [Google Scholar] [CrossRef]
- Magaki, S.; Hojat, S.A.; Wei, B.; So, A.; Yong, W.H. An Introduction to the Performance of Immunohistochemistry. Methods Mol. Biol. 2019, 1897, 289–298. [Google Scholar] [CrossRef]
- Ramos-Vara, J.A. Principles and Methods of Immunohistochemistry. Methods Mol. Biol. 2017, 1641, 115–128. [Google Scholar] [CrossRef]
- Sholl, L.M.; Aisner, D.L.; Allen, T.C.; Beasley, M.B.; Borczuk, A.C.; Cagle, P.T.; Capelozzi, V.; Dacic, S.; Hariri, L.; Kerr, K.M.; et al. Programmed Death Ligand-1 Immunohistochemistry—A New Challenge for Pathologists: A Perspective From Members of the Pulmonary Pathology Society. Arch. Pathol. Lab Med. 2016, 140, 341–344. [Google Scholar] [CrossRef] [PubMed]
- Obi, E.N.; Tellock, D.A.; Thomas, G.J.; Veenstra, T.D. Biomarker Analysis of Formalin-Fixed Paraffin-Embedded Clinical Tissues Using Proteomics. Biomolecules 2023, 13, 96. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.S.; McConechy, M.K.; Cochrane, D.R.; Nazeran, T.; Karnezis, A.N.; Huntsman, D.G.; Morin, G.B. Quantitative Profiling of Single Formalin Fixed Tumour Sections: Proteomics for translational research. Sci. Rep. 2016, 6, 34949. [Google Scholar] [CrossRef]
- Neubert, H.; Palandra, J.; Fernandez Ocana, M. Quantification of biotherapeutic targets: New opportunities with immunoaffinity LC-MS/MS. Bioanalysis 2014, 6, 1731–1733. [Google Scholar] [CrossRef]
- Santockyte, R.; Zeng, J.; Zheng, N. Critical reagent screening and characterization: Benefits and approaches for protein biomarker assays by hybrid LC-MS. Bioanalysis 2019, 11, 785–795. [Google Scholar] [CrossRef]
- Zhao, Y.; Gu, H.; Zheng, N.; Zeng, J. Critical considerations for immunocapture enrichment LC-MS bioanalysis of protein therapeutics and biomarkers. Bioanalysis 2018, 10, 987–995. [Google Scholar] [CrossRef]
- Zheng, N.; Catlett, I.M.; Taylor, K.; Gu, H.; Pattoli, M.A.; Neely, R.J.; Li, W.; Allentoff, A.; Yuan, X.; Ciccimaro, E.; et al. Determination of Real Time in Vivo Drug Receptor Occupancy for a Covalent Binding Drug as a Clinical Pharmacodynamic Biomarker by Immunocapture-LC-MS/MS. Anal. Chem. 2019, 91, 8443–8452. [Google Scholar] [CrossRef] [PubMed]
- Emmert-Buck, M.R.; Bonner, R.F.; Smith, P.D.; Chuaqui, R.F.; Zhuang, Z.; Goldstein, S.R.; Weiss, R.A.; Liotta, L.A. Laser capture microdissection. Science 1996, 274, 998–1001. [Google Scholar] [CrossRef]
- Orba, Y.; Tanaka, S.; Nishihara, H.; Kawamura, N.; Itoh, T.; Shimizu, M.; Sawa, H.; Nagashima, K. Application of laser capture microdissection to cytologic specimens for the detection of immunoglobulin heavy chain gene rearrangement in patients with malignant lymphoma. Cancer 2003, 99, 198–204. [Google Scholar] [CrossRef]
- Kihara, A.H.; Moriscot, A.S.; Ferreira, P.J.; Hamassaki, D.E. Protecting RNA in fixed tissue: An alternative method for LCM users. J. Neurosci. Methods 2005, 148, 103–107. [Google Scholar] [CrossRef]
- Gallagher, R.I.; Blakely, S.R.; Liotta, L.A.; Espina, V. Laser capture microdissection: Arcturus(XT) infrared capture and UV cutting methods. Methods Mol. Biol. 2012, 823, 157–178. [Google Scholar] [CrossRef]
- Korovesi, A.G.; Anagnostopoulos, A.K.; Pierros, V.; Stravopodis, D.J.; Tsangaris, G.T. Normal Mouse Brain Proteome II: Analysis of Brain Regions by High-resolution Mass Spectrometry. Cancer Genom. Proteom. 2020, 17, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef]
- Blake, J.A.; Baldarelli, R.; Kadin, J.A.; Richardson, J.E.; Smith, C.L.; Bult, C.J.; Mouse Genome Database, G. Mouse Genome Database (MGD): Knowledgebase for mouse-human comparative biology. Nucleic Acids Res. 2021, 49, D981–D987. [Google Scholar] [CrossRef] [PubMed]
- Gerber, K.J.; Dammer, E.B.; Duong, D.M.; Deng, Q.; Dudek, S.M.; Seyfried, N.T.; Hepler, J.R. Specific Proteomes of Hippocampal Regions CA2 and CA1 Reveal Proteins Linked to the Unique Physiology of Area CA2. J. Proteome Res. 2019, 18, 2571–2584. [Google Scholar] [CrossRef]
- Blankenburg, S.; Hentschker, C.; Nagel, A.; Hildebrandt, P.; Michalik, S.; Dittmar, D.; Surmann, K.; Voelker, U. Improving Proteome Coverage for Small Sample Amounts: An Advanced Method for Proteomics Approaches with Low Bacterial Cell Numbers. Proteomics 2019, 19, 1900192. [Google Scholar] [CrossRef]
- Oliveros, J.C. Venny. An Interactive Tool for Comparing Lists with Venn’s Diagrams. Available online: https://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 13 April 2023).
- Wisniewski, J.R.; Dus, K.; Mann, M. Proteomic workflow for analysis of archival formalin-fixed and paraffin-embedded clinical samples to a depth of 10 000 proteins. Proteom. Clin. Appl. 2013, 7, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Muller, T.; Kalxdorf, M.; Longuespee, R.; Kazdal, D.N.; Stenzinger, A.; Krijgsveld, J. Automated sample preparation with SP3 for low-input clinical proteomics. Mol. Syst. Biol. 2020, 16, e9111. [Google Scholar] [CrossRef]
- Fujii, K.; Miyata, Y.; Takahashi, I.; Koizumi, H.; Saji, H.; Hoshikawa, M.; Takagi, M.; Nishimura, T.; Nakamura, H. Differential Proteomic Analysis between Small Cell Lung Carcinoma (SCLC) and Pulmonary Carcinoid Tumors Reveals Molecular Signatures for Malignancy in Lung Cancer. Proteom. Clin. Appl. 2018, 12, e1800015. [Google Scholar] [CrossRef]
- Bohnenberger, H.; Kaderali, L.; Strobel, P.; Yepes, D.; Plessmann, U.; Dharia, N.V.; Yao, S.; Heydt, C.; Merkelbach-Bruse, S.; Emmert, A.; et al. Comparative proteomics reveals a diagnostic signature for pulmonary head-and-neck cancer metastasis. EMBO Mol. Med. 2018, 10, e8428. [Google Scholar] [CrossRef] [PubMed]
- Kuras, M.; Woldmar, N.; Kim, Y.; Hefner, M.; Malm, J.; Moldvay, J.; Dome, B.; Fillinger, J.; Pizzatti, L.; Gil, J.; et al. Proteomic Workflows for High-Quality Quantitative Proteome and Post-Translational Modification Analysis of Clinically Relevant Samples from Formalin-Fixed Paraffin-Embedded Archives. J. Proteome Res. 2021, 20, 1027–1039. [Google Scholar] [CrossRef] [PubMed]
- Arima, K.; Lau, M.C.; Zhao, M.; Haruki, K.; Kosumi, K.; Mima, K.; Gu, M.; Vayrynen, J.P.; Twombly, T.S.; Baba, Y.; et al. Metabolic Profiling of Formalin-Fixed Paraffin-Embedded Tissues Discriminates Normal Colon from Colorectal Cancer. Mol. Cancer Res. 2020, 18, 883–890. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry Name | Protein Name | Gene | Function/ Involvement in * | Mean Intensity; Range 180–9,700,000 |
|---|---|---|---|---|
| APOH_MOUSE | Beta-2-glycoprotein 1 | Apoh | cognitive decline | 13,058 |
| CD38_MOUSE | ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 | Cd38 | dendritic organization long-term synaptic depression | 59,130 |
| CRHBP_MOUSE | Corticotropin-releasing factor-binding protein | Crhbp | stress susceptibility | 30,011 |
| CSMD3_MOUSE | CUB and sushi domain-containing protein 3 | Csmd3 | dendrite development | 21,276 |
| DOC2B_MOUSE | Double C2-like domain-containing protein beta | Doc2b | neuronal activity synaptic release | 18,311 |
| DOP2_MOUSE | Protein dopey-2 | Dop1b | mental retardation | 24,725 |
| EPHA5_MOUSE | Ephrin type-A receptor 5 | Epha5 | synaptic plasticity | 72,835 |
| FGF13_MOUSE | Fibroblast growth factor 13 | Fgf13 | synaptic excitatory-inhibitory imbalance | 32,178 |
| FBCD1_MOUSE | Fibrinogen C domain-containing protein 1 | Fibcd1 | neurodevelopmental disorder | 14,255 |
| FLRT2_MOUSE | Leucine-rich repeat transmembrane protein FLRT2 | Flrt2 | synaptic plasticity spatial memory | 35,696 |
| FUT8_MOUSE | Alpha-(1,6)-fucosyltransferase | Fut8 | hippocampal Long Term Potentiation | 5973 |
| KIRR3_MOUSE | Kin of IRRE-like protein 3 | Kirrel3 | intellectual disability | 5782 |
| LEG3_MOUSE | Galectin-3 | Lgals3 | hippocampal formation | 13,470 |
| LRFN2_MOUSE | Leucine-rich repeat and fibronectin type-III domain-containing protein 2 | Lrfn2 | synaptic adhesion synaptic plasticity | 18,303 |
| MDGA1_MOUSE | MAM domain-containing glycosyl-phosphatidylinositol anchor protein 1 | Mdga1 | synapse inhibition | 33,520 |
| MK_MOUSE | Midkine | Mdk | memory | 14,722 |
| MT3_MOUSE | Metallothionein-3 | Mt3 | apoptosis of neurons | 13,294 |
| MYD88_MOUSE | Myeloid differentiation primary response protein MyD88 | Myd88 | signal transduction neuronal activity | 10,861 |
| NTNG2_MOUSE | Netrin-G2 | Ntng2 | axon differentiation | 25,387 |
| PK3CG_MOUSE | Phosphatidylinositol 4 | Pik3cg | long-term depression cognitive impairments | 7427 |
| KS6B1_MOUSE | Ribosomal protein S6 kinase b1 | Rps6kb1 | developing and mature of neuronal cells | 29,356 |
| RTN4R_MOUSE | Reticulon-4 receptor | Rtn4r | aging and cognitive decline | 48,793 |
| SORC3_MOUSE | VPS10 domain-containing receptor SorCS3 | Sorcs3 | postsynaptic modulation of synaptic depression | 49,851 |
| TAFA5_MOUSE | Chemokine-like protein TAFA-5 | Tafa5 | spatial memory | 31,477 |
| ITF2_MOUSE | Transcription factor 4 | Tcf4 | dendritic spine density | 19,181 |
| TRPC4_MOUSE | Short transient receptor potential channel 4 | Trpc4 | hippocampal synaptic transmission | 30,792 |
| ZEB2_MOUSE | Zinc finger E-box-binding homeobox 2 | Zeb2 | neuronal development | 21,158 |
| Canonical Pathway | p-Value | Overlap Molecules in Dataset/Number of Molecules Whole Pathway | Rich Factor | Molecules in Dataset |
|---|---|---|---|---|
| G Beta Gamma Signaling | 3.90 × 10−5 | 12/127 | 0.094 | ARHGEF6, CACNB2, CACNG4, GNA14, GNB3, GNG5, GNG10, KCNJ6, KCNJ9, PIK3CG, PRKCI, PRKCZ |
| Opioid Signaling Pathway | 8.09 × 10−5 | 18/276 | 0.065 | CACNB2, CACNG4, GNA14, GNB3, GNG5, GNG10, GRK3, KCNJ6, KCNJ9, PIK3CG, PRKCI, PRKCZ, RAC3, RGS17, RPS6KB1, RYR1, RYR3, TCF4 |
| Axonal Guidance Signaling | 3.91 × 10−4 | 24/283 | 0.085 | ARHGEF6, EPHA5, EPHA6, EPHA7, EPHB1, EPHB6, FZD7, GNA14, GNB3, GNG5, GNG10, HERC2, MMP17, NRP2, NTNG1, NTNG2, PIK3CG, PRKCI, PRKCZ, RAC3, ROBO1, RTN4R, SLIT1, UNC5C |
| ID | Molecules in Network | Score | Focus Molecules | Top Diseases and Functions |
|---|---|---|---|---|
| 1 | ADGRF5, ADRM1, APOL2, APP, CERK, CNRIP1, EPHB6, FGF13, GOLIM4, HDAC4, ITGB1, JUN, KCNAB3, KCNJ6, KCNJ9, LAMB1, Map3k7, MECP2, NACC2, NCOR1, NEXN, NF2, NYAP1, PDGFRB, PREX2, RAC1, ROBO1, SH3RF1, SIDT1, SLC35F6, TBC1D2B, TBC1D9B, TCF20, TSPAN9, UBQLN2 | 17 | 16 | Cell Death and Survival, Neurological Disease, Organismal Injury and Abnormalities |
| 2 | Abcb1b, ACTB, ACTG1, APP, ATP12A, Atp5e, BACE1, CAT, DPYSL2, EIF5A2, EOMES, FZD7, GAP43, GFAP, GRM5, H2AC7, HECW1, Ly6a (includes others), MAPK14, MEF2C, MRTFA, Mt3, NTRK2, PLA2G4A, PSEN1, PTGER2, PURA, RELA, SH3PXD2A, SLC1A2, SMPD2, SNAP25, SNAPIN, TCF4, VAMP2 | 15 | 15 | Cell Death and Survival, Neurological Disease, Organismal Injury and Abnormalities |
| 3 | ARR3, AURKA, BAD, BCL2L1, CAST, CDK5, DLGAP3, FTH1, GABRD, GNAS, GNG10, GNG5, GRK3, Gstm6, HK2, HMOX1, HSPA5, MAPK14, MIB1, Nefm, OPRK1, P2RX7, PDPK1, PRKCI, RPS6KB1, RYR1, SLC11A2, SLC40A1, SOD1, SRXN1, TFRC, THRB, Tmsb4x (includes others), UTS2, VIP | 15 | 15 | Cell Death and Survival, Free Radical Scavenging, Organismal Injury and Abnormalities |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Venz, S.; von Bohlen und Halbach, V.; Hentschker, C.; Junker, H.; Kuss, A.W.; Sura, T.; Krüger, E.; Völker, U.; von Bohlen und Halbach, O.; Jensen, L.R.; et al. Global Protein Profiling in Processed Immunohistochemistry Tissue Sections. Int. J. Mol. Sci. 2023, 24, 11308. https://doi.org/10.3390/ijms241411308
Venz S, von Bohlen und Halbach V, Hentschker C, Junker H, Kuss AW, Sura T, Krüger E, Völker U, von Bohlen und Halbach O, Jensen LR, et al. Global Protein Profiling in Processed Immunohistochemistry Tissue Sections. International Journal of Molecular Sciences. 2023; 24(14):11308. https://doi.org/10.3390/ijms241411308
Chicago/Turabian StyleVenz, Simone, Viola von Bohlen und Halbach, Christian Hentschker, Heike Junker, Andreas Walter Kuss, Thomas Sura, Elke Krüger, Uwe Völker, Oliver von Bohlen und Halbach, Lars Riff Jensen, and et al. 2023. "Global Protein Profiling in Processed Immunohistochemistry Tissue Sections" International Journal of Molecular Sciences 24, no. 14: 11308. https://doi.org/10.3390/ijms241411308
APA StyleVenz, S., von Bohlen und Halbach, V., Hentschker, C., Junker, H., Kuss, A. W., Sura, T., Krüger, E., Völker, U., von Bohlen und Halbach, O., Jensen, L. R., & Hammer, E. (2023). Global Protein Profiling in Processed Immunohistochemistry Tissue Sections. International Journal of Molecular Sciences, 24(14), 11308. https://doi.org/10.3390/ijms241411308