
Profiling Chemobiological Connection between Natural Product and Target Space Based on Systematic Analysis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
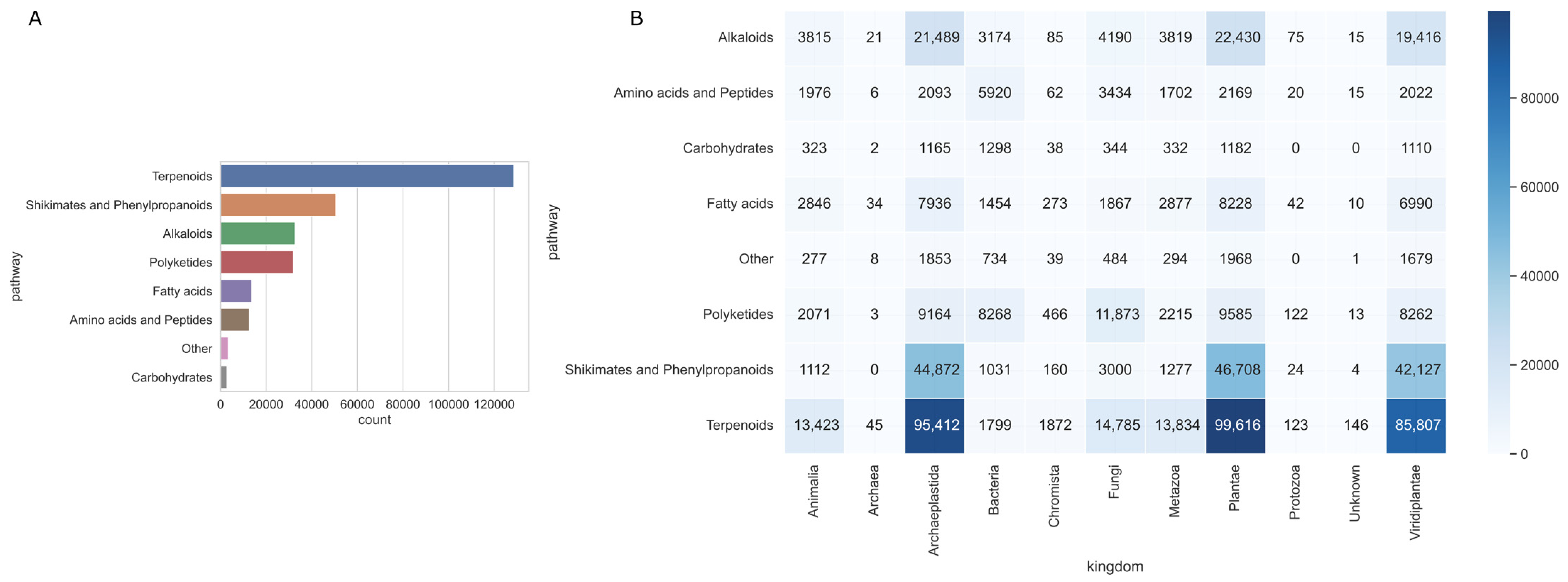
2.1. LOTUS Natural Products Analysis
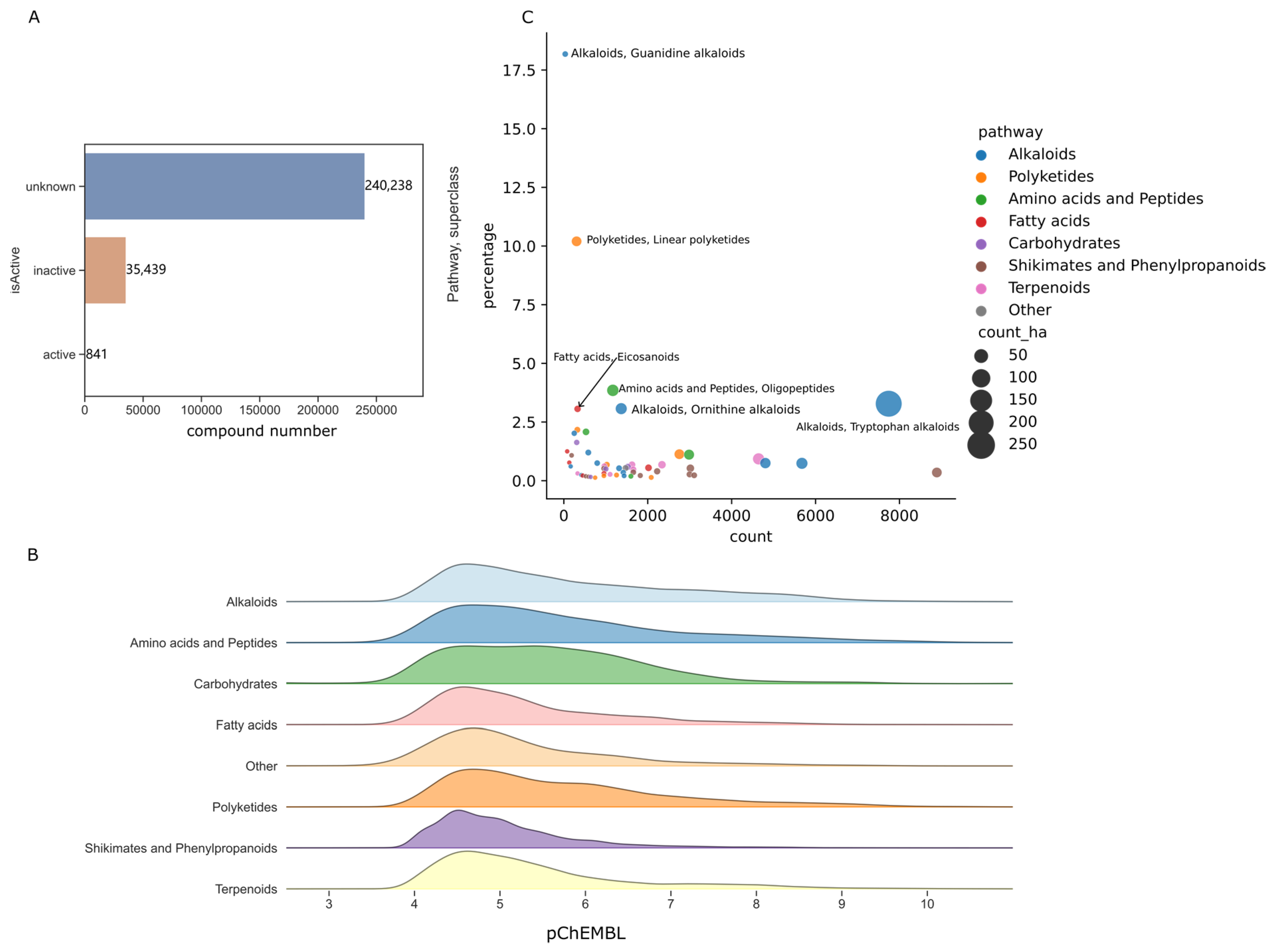
2.2. Activity Values Distribution of Natural Product
2.3. Target Space of Natural Products of High Hit Rate Superclass
2.4. Large-Scale Virtual Screening Based on the Deep Learning Model FusionDTA
3. Discussion
3.1. Biogenic Pathways of Natural Products and Bioresources Species Tendencies
3.2. Chemical Diversity and High Activities Hit Rates
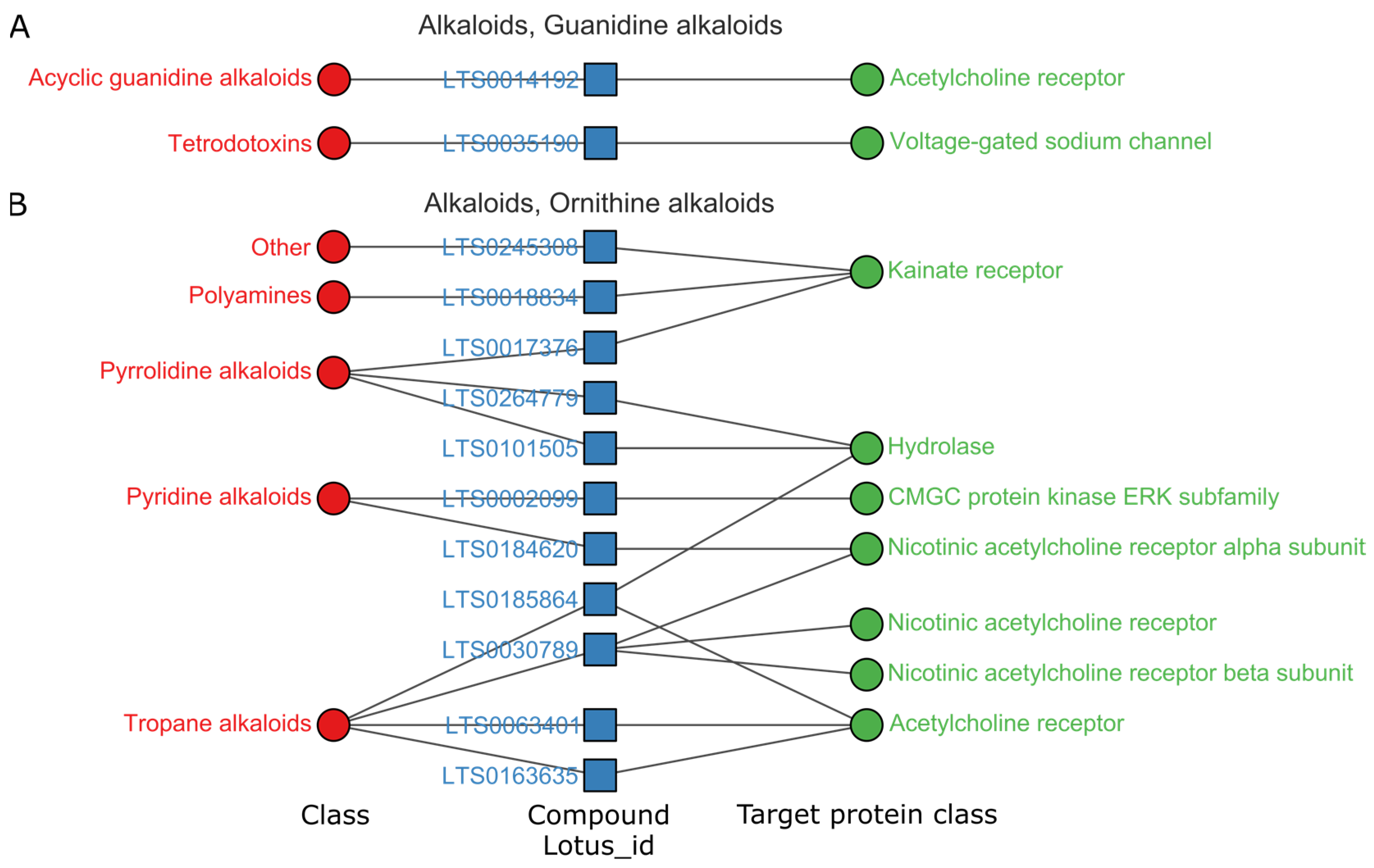
3.3. Privilege Targets of Natural Products in Superclass with High Hit Rates
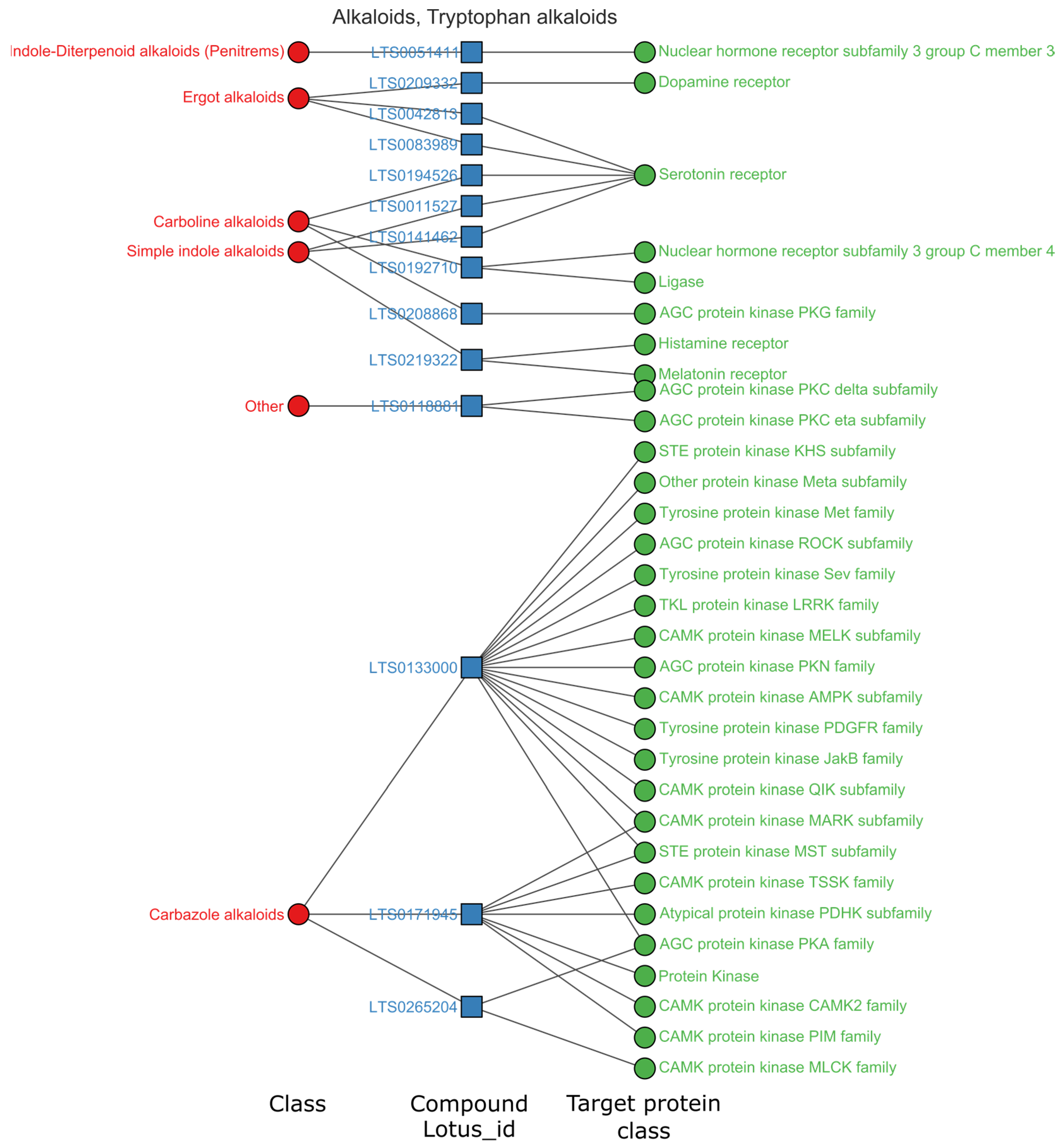
3.3.1. Alkaloids Mainly Regulate the Central Nervous System
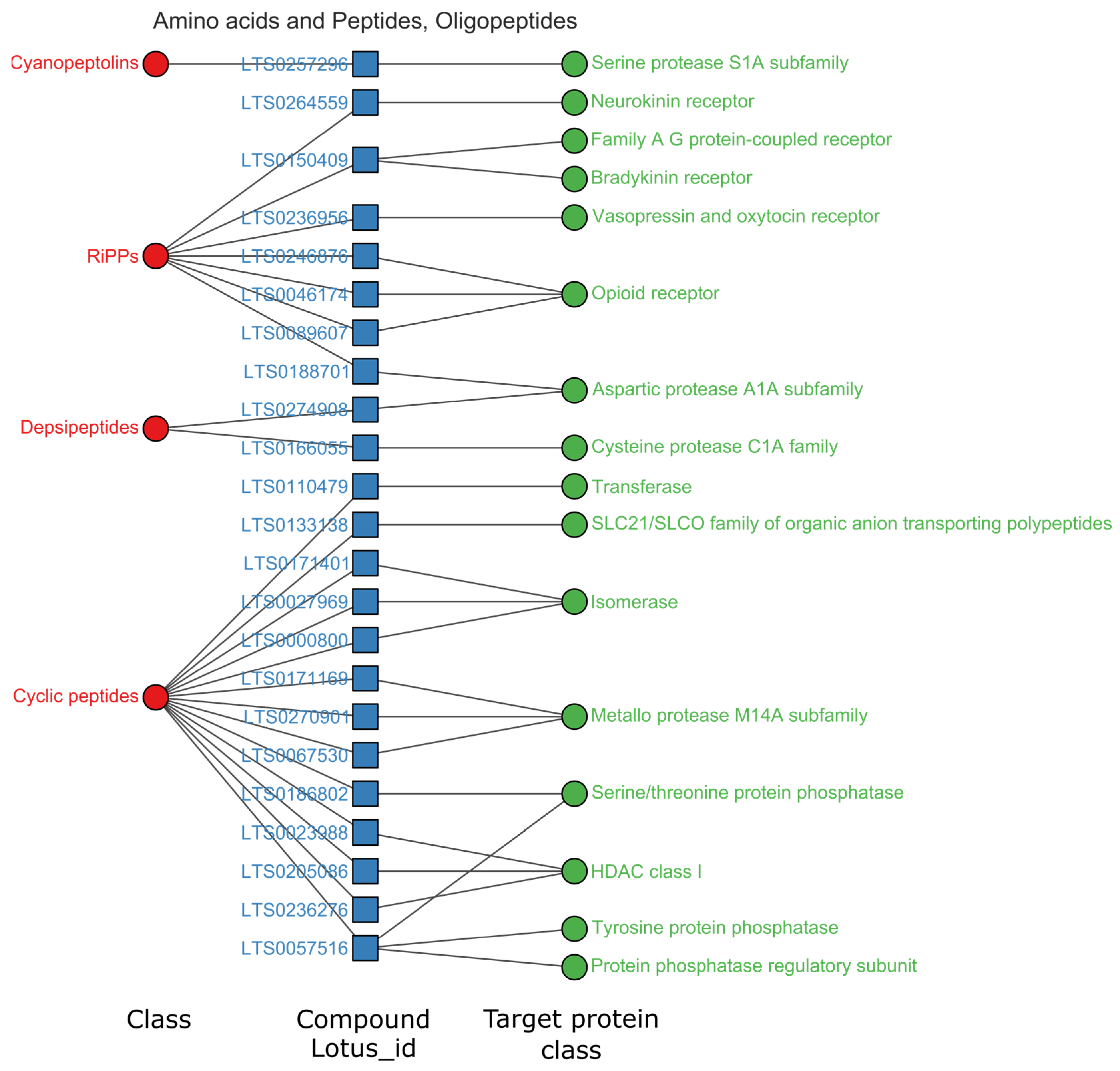
3.3.2. Oligopeptides and Linear Polyketides Targeted HDAC Class I
3.3.3. Eicosanoids Acted Primarily on Prostanoid and Leukotriene Receptors
3.4. Potential Targets of Natural Products Predicated by a Deep Learning Model, FusionDTA
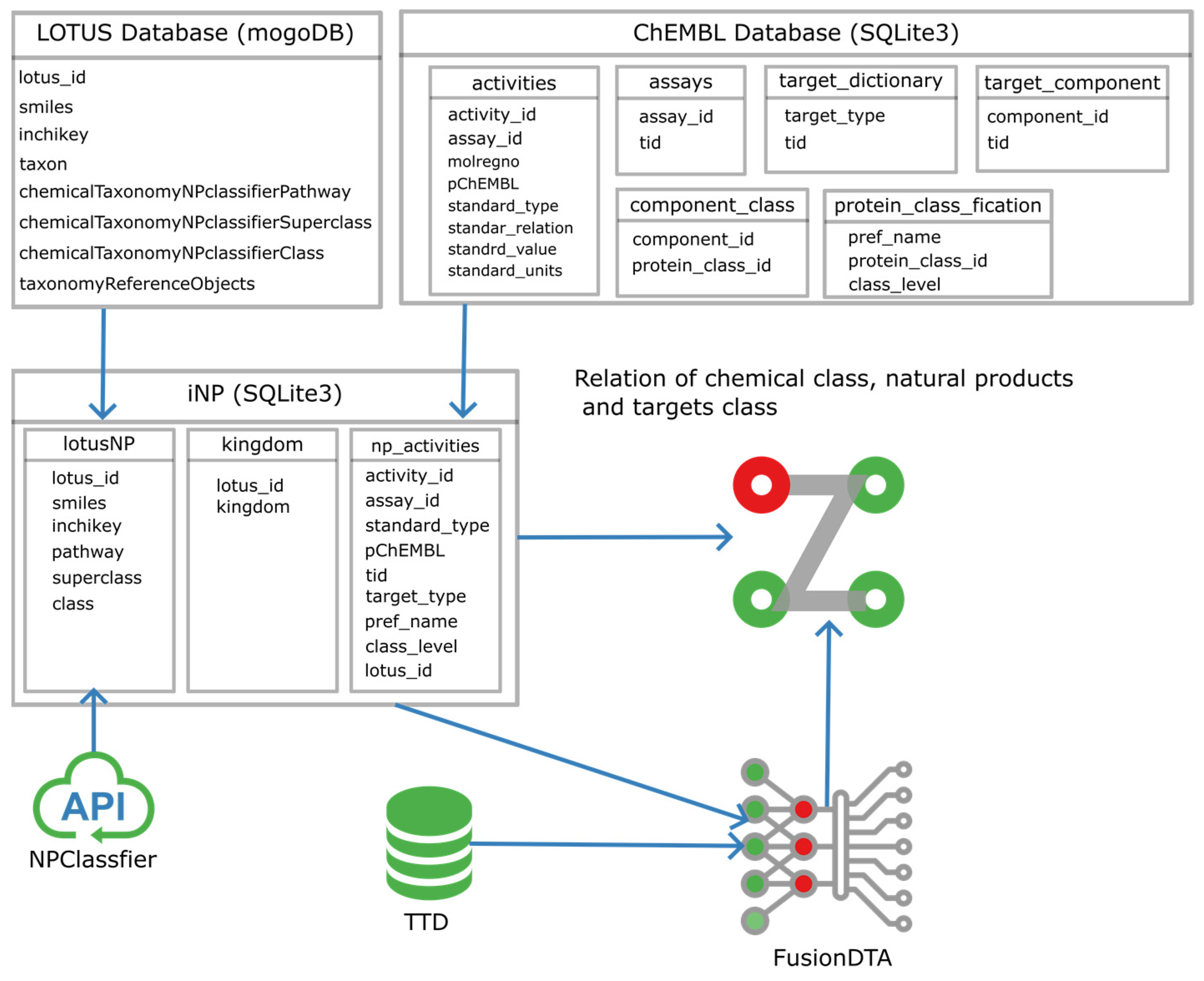
4. Materials and Methods
4.1. Database
4.2. Python Packages and Software
4.3. LOTUS Natural Product Data Preparation
4.4. Extract and Summary of LOTUS Taxon Information
4.5. Distribution Analysis of Natural Products Activity Data
4.6. Predicting Affinities of Natural Products and Drug Targets
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stone, S.; Newman, D.J.; Colletti, S.L.; Tan, D.S. Cheminformatic Analysis of Natural Product-Based Drugs and Chemical Probes. Nat. Prod. Rep. 2022, 39, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Supuran, C.T. Natural Products in Drug Discovery: Advances and Opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef] [PubMed]
- Zabolotna, Y.; Ertl, P.; Horvath, D.; Bonachera, F.; Marcou, G.; Varnek, A. NP Navigator: A New Look at the Natural Product Chemical Space. Mol. Inform. 2021, 40, 2100068. [Google Scholar] [CrossRef]
- Coley, C.W. Defining and Exploring Chemical Spaces. Trends Chem. 2021, 3, 133–145. [Google Scholar] [CrossRef]
- Sánchez-Ruiz, A.; Colmenarejo, G. Systematic Analysis and Prediction of the Target Space of Bioactive Food Compounds: Filling the Chemobiological Gaps. J. Chem. Inf. Model. 2022, 62, 3734–3751. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hsieh, C.-Y.; Wang, M.; Wang, X.; Wu, Z.; Jiang, D.; Liao, B.; Zhang, X.; Yang, B.; He, Q.; et al. Multi-Constraint Molecular Generation Based on Conditional Transformer, Knowledge Distillation and Reinforcement Learning. Nat. Mach. Intell. 2021, 3, 914–922. [Google Scholar] [CrossRef]
- Yang, Y.; Yao, K.; Repasky, M.P.; Leswing, K.; Abel, R.; Shoichet, B.K.; Jerome, S.V. Efficient Exploration of Chemical Space with Docking and Deep Learning. J. Chem. Theory Comput. 2021, 17, 7106–7119. [Google Scholar] [CrossRef]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards Direct Deposition of Bioassay Data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef]
- Rutz, A.; Sorokina, M.; Galgonek, J.; Mietchen, D.; Willighagen, E.; Gaudry, A.; Graham, J.G.; Stephan, R.; Page, R.; Vondrášek, J.; et al. The LOTUS Initiative for Open Knowledge Management in Natural Products Research. eLife 2022, 11, e70780. [Google Scholar] [CrossRef]
- Yuan, W.; Chen, G.; Chen, C.Y.-C. FusionDTA: Attention-Based Feature Polymerizer and Knowledge Distillation for Drug-Target Binding Affinity Prediction. Brief. Bioinform. 2022, 23, bbab506. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, Y.; Lian, X.; Li, F.; Wang, C.; Zhu, F.; Qiu, Y.; Chen, Y. Therapeutic Target Database Update 2022: Facilitating Drug Discovery with Enriched Comparative Data of Targeted Agents. Nucleic Acids Res. 2022, 50, D1398–D1407. [Google Scholar] [CrossRef]
- Kim, H.W.; Wang, M.; Leber, C.A.; Nothias, L.-F.; Reher, R.; Kang, K.B.; van der Hooft, J.J.J.; Dorrestein, P.C.; Gerwick, W.H.; Cottrell, G.W. NPClassifier: A Deep Neural Network-Based Structural Classification Tool for Natural Products. J. Nat. Prod. 2021, 84, 2795–2807. [Google Scholar] [CrossRef]
- Bonanni, D.; Pinzi, L.; Rastelli, G. Development of Machine Learning Classifiers to Predict Compound Activity on Prostate Cancer Cell Lines. J. Cheminform. 2022, 14, 77. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, J.V.; Pajouhesh, H.; Beckley, J.T.; Delwig, A.; Du Bois, J.; Hunter, J.C. Challenges and Opportunities for Therapeutics Targeting the Voltage-Gated Sodium Channel Isoform NaV1.7. J. Med. Chem. 2019, 62, 8695–8710. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Ogawa, A.K.; Du, X.; Houk, K.N.; Armstrong, R.W. A Model for Binding of Structurally Diverse Natural Product Inhibitors of Protein Phosphatases PP1 and PP2A. J. Med. Chem. 1997, 40, 3199–3206. [Google Scholar] [CrossRef] [PubMed]
- Gulledge, B.M.; Aggen, J.B.; Chamberlin, A.R. Linearized and Truncated Microcystin Analogues as Inhibitors of Protein Phosphatases 1 and 2A. Bioorg. Med. Chem. Lett. 2003, 13, 2903–2906. [Google Scholar] [CrossRef]
- Quinn, R.J.; Taylor, C.; Suganuma, M.; Fujiki, H. The Conserved Acid Binding Domain Model of Inhibitors of Protein Phosphatases 1 and 2A: Molecular Modelling Aspects. Bioorg. Med. Chem. Lett. 1993, 3, 1029–1034. [Google Scholar] [CrossRef]
- Hayashi, S.; Nakata, E.; Morita, A.; Mizuno, K.; Yamamura, K.; Kato, A.; Ohashi, K. Discovery of {1-[4-(2-{hexahydropyrrolo[3,4-c]Pyrrol-2(1H)-Yl}-1H-Benzimidazol-1-Yl)Piperidin-1-Yl]Cyclooctyl}methanol, Systemically Potent Novel Non-Peptide Agonist of Nociceptin/Orphanin FQ Receptor as Analgesic for the Treatment of Neuropathic Pain: Design, Synthesis, and Structure–Activity Relationships. Bioorg. Med. Chem. 2010, 18, 7675–7699. [Google Scholar] [CrossRef]
- Abram, T.S.; Böshagen, H.; Butler, J.E.; Cuthbert, N.J.; Francis, H.P.; Gardiner, P.J.; Hartwig, W.; Kluender, H.C.; Norman, P.; Meier, H.; et al. A New Structural Analogue Antagonist of Peptido-Leukotrienes. The Discovery of Bay X7195. Bioorg. Med. Chem. Lett. 1993, 3, 1517–1522. [Google Scholar] [CrossRef]
- Djoumbou Feunang, Y.; Eisner, R.; Knox, C.; Chepelev, L.; Hastings, J.; Owen, G.; Fahy, E.; Steinbeck, C.; Subramanian, S.; Bolton, E.; et al. ClassyFire: Automated Chemical Classification with a Comprehensive, Computable Taxonomy. J. Cheminform. 2016, 8, 61. [Google Scholar] [CrossRef] [PubMed]
- Lichman, B.R. The Scaffold-Forming Steps of Plant Alkaloid Biosynthesis. Nat. Prod. Rep. 2021, 38, 103–129. [Google Scholar] [CrossRef] [PubMed]
- Anwar, M.; Chen, L.; Xiao, Y.; Wu, J.; Zeng, L.; Li, H.; Wu, Q.; Hu, Z. Recent Advanced Metabolic and Genetic Engineering of Phenylpropanoid Biosynthetic Pathways. Int. J. Mol. Sci. 2021, 22, 9544. [Google Scholar] [CrossRef] [PubMed]
- Harms, V.; Kirschning, A.; Dickschat, J.S. Nature-Driven Approaches to Non-Natural Terpene Analogues. Nat. Prod. Rep. 2020, 37, 1080–1097. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A.; Lenaeus, M.J.; Gamal El-Din, T.M. Structure and Pharmacology of Voltage-Gated Sodium and Calcium Channels. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 133–154. [Google Scholar] [CrossRef]
- Papke, R.L.; Horenstein, N.A. Therapeutic Targeting of A7 Nicotinic Acetylcholine Receptors. Pharmacol. Rev. 2021, 73, 1118–1149. [Google Scholar] [CrossRef]
- Kałużna-Czaplińska, J.; Gątarek, P.; Chirumbolo, S.; Chartrand, M.S.; Bjørklund, G. How Important Is Tryptophan in Human Health? Crit. Rev. Food Sci. Nutr. 2019, 59, 72–88. [Google Scholar] [CrossRef]
- Quintero-Villegas, A.; Valdés-Ferrer, S.I. Central Nervous System Effects of 5-HT7 Receptors: A Potential Target for Neurodegenerative Diseases. Mol. Med. 2022, 28, 70. [Google Scholar] [CrossRef]
- Jones, L.A.; Sun, E.W.; Martin, A.M.; Keating, D.J. The Ever-Changing Roles of Serotonin. Int. J. Biochem. Cell Biol. 2020, 125, 105776. [Google Scholar] [CrossRef]
- Gannon, M.; Wang, Q. Complex Noradrenergic Dysfunction in Alzheimer’s Disease: Low Norepinephrine Input Is Not Always to Blame. Brain Res. 2019, 1702, 12–16. [Google Scholar] [CrossRef]
- Bekdash, R.A. The Cholinergic System, the Adrenergic System and the Neuropathology of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 1273. [Google Scholar] [CrossRef]
- Franco, R.; Reyes-Resina, I.; Navarro, G. Dopamine in Health and Disease: Much More Than a Neurotransmitter. Biomedicines 2021, 9, 109. [Google Scholar] [CrossRef] [PubMed]
- Cossu, G.; Rinaldi, R.; Colosimo, C. The Rise and Fall of Impulse Control Behavior Disorders. Park. Relat. Disord. 2018, 46, S24–S29. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Li, H. Structure-Based Inhibitor Discovery of Class I Histone Deacetylases (HDACs). Int. J. Mol. Sci. 2020, 21, 8828. [Google Scholar] [CrossRef] [PubMed]
- Obeng, S.; Hiranita, T.; León, F.; McMahon, L.R.; McCurdy, C.R. Novel Approaches, Drug Candidates, and Targets in Pain Drug Discovery. J. Med. Chem. 2021, 64, 6523–6548. [Google Scholar] [CrossRef] [PubMed]
- Biringer, R.G. A Review of Prostanoid Receptors: Expression, Characterization, Regulation, and Mechanism of Action. J. Cell Commun. Signal. 2021, 15, 155–184. [Google Scholar] [CrossRef]
- Chun, K.-S.; Lao, H.-C.; Trempus, C.S.; Okada, M.; Langenbach, R. The Prostaglandin Receptor EP2 Activates Multiple Signaling Pathways and β-Arrestin1 Complex Formation during Mouse Skin Papilloma Development. Carcinogenesis 2009, 30, 1620–1627. [Google Scholar] [CrossRef]
- Wallin, J.; Svenningsson, P. Potential Effects of Leukotriene Receptor Antagonist Montelukast in Treatment of Neuroinflammation in Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 5606. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Y.; Zhang, S.; Li, C.; Zhang, L. Modulation of Neuroinflammation by Cysteinyl Leukotriene 1 and 2 Receptors: Implications for Cerebral Ischemia and Neurodegenerative Diseases. Neurobiol. Aging 2020, 87, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rives, A.; Meier, J.; Sercu, T.; Goyal, S.; Lin, Z.; Liu, J.; Guo, D.; Ott, M.; Zitnick, C.L.; Ma, J.; et al. Biological Structure and Function Emerge from Scaling Unsupervised Learning to 250 Million Protein Sequences. Proc. Natl. Acad. Sci. USA 2021, 118, e2016239118. [Google Scholar] [CrossRef]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive Analysis of Kinase Inhibitor Selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, D.; Li, X.; Miao, Y.; Zhang, Q. Profiling Chemobiological Connection between Natural Product and Target Space Based on Systematic Analysis. Int. J. Mol. Sci. 2023, 24, 11265. https://doi.org/10.3390/ijms241411265
Wang D, Li X, Miao Y, Zhang Q. Profiling Chemobiological Connection between Natural Product and Target Space Based on Systematic Analysis. International Journal of Molecular Sciences. 2023; 24(14):11265. https://doi.org/10.3390/ijms241411265
Chicago/Turabian StyleWang, Disheng, Xue Li, Yicheng Miao, and Qiang Zhang. 2023. "Profiling Chemobiological Connection between Natural Product and Target Space Based on Systematic Analysis" International Journal of Molecular Sciences 24, no. 14: 11265. https://doi.org/10.3390/ijms241411265
APA StyleWang, D., Li, X., Miao, Y., & Zhang, Q. (2023). Profiling Chemobiological Connection between Natural Product and Target Space Based on Systematic Analysis. International Journal of Molecular Sciences, 24(14), 11265. https://doi.org/10.3390/ijms241411265