

Targeting Prostate Cancer, the ‘Tousled Way’


Abstract
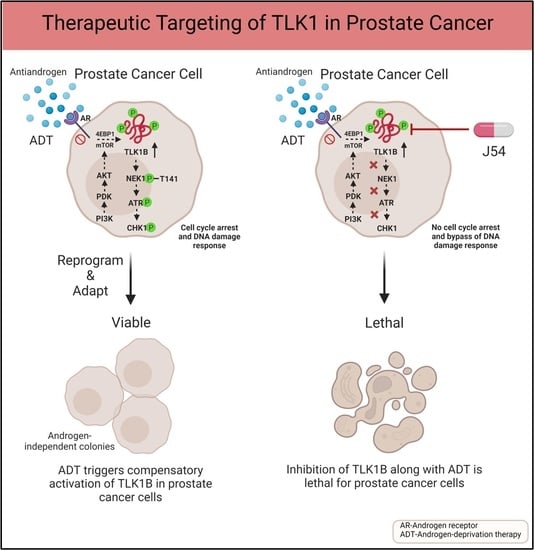
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction

2. Progression of PCa to AR-Negative Lethal Disease: Understanding the Implications
3. Clinical Signs of AR-Negative Disease
4. Human TLKs, Their Substrate Interaction, and Functional Significance
5. TLK1 in PCa Progression
6. Phenothiazines (PTH) as TLK1 Inhibitors
7. Discussion
8. Factual Considerations and Alternative Modalities
9. Bicalutamide vs. Enzalutamide: Potential Therapeutic Competition and Comparative Analysis

10. Perspective and Future Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Heinlein, C.A.; Chang, C. Androgen receptor in prostate cancer. Endocr. Rev. 2004, 25, 276–308. [Google Scholar] [CrossRef]
- Harris, W.P.; Mostaghel, E.A.; Nelson, P.S.; Montgomery, B. Androgen deprivation therapy: Progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat. Clin. Pract. Urol. 2009, 6, 76–85. [Google Scholar] [CrossRef]
- Merseburger, A.S.; Haas, G.P.; von Klot, C.A. An update on enzalutamide in the treatment of prostate cancer. Ther. Adv. Urol. 2015, 7, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.; Jazayeri, S.B.; Gautam, S.; Norez, D.; Alam, M.U.; Tanneru, K.; Bazargani, S.; Costa, J.; Bandyk, M.; Ganapathi, H.P.; et al. Comparative efficacy of apalutamide darolutamide and enzalutamide for treatment of non-metastatic castrate-resistant prostate cancer: A systematic review and network meta-analysis. Urol. Oncol. 2020, 38, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Karantanos, T.; Corn, P.G.; Thompson, T.C. Prostate cancer progression after androgen deprivation therapy: Mechanisms of castrate resistance and novel therapeutic approaches. Oncogene 2013, 32, 5501–5511. [Google Scholar] [CrossRef]
- Imamura, Y.; Sadar, M.D. Androgen receptor targeted therapies in castration-resistant prostate cancer: Bench to clinic. Int. J. Urol. 2016, 23, 654–665. [Google Scholar] [CrossRef]
- Zhu, M.L.; Kyprianou, N. Androgen receptor and growth factor signaling cross-talk in prostate cancer cells. Endocr. Relat. Cancer 2008, 15, 841–849. [Google Scholar] [CrossRef]
- Craft, N.; Shostak, Y.; Carey, M.; Sawyers, C.L. A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the HER-2/neu tyrosine kinase. Nat. Med. 1999, 5, 280–285. [Google Scholar] [CrossRef]
- Seruga, B.; Ocana, A.; Tannock, I.F. Drug resistance in metastatic castration-resistant prostate cancer. Nat. Rev. Clin. Oncol. 2011, 8, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Fizazi, K.; Saad, F.; Rathenborg, P.; Shore, N.; Ferreira, U.; Ivashchenko, P.; Demirhan, E.; Modelska, K.; Phung, D.; et al. Enzalutamide in Men with Nonmetastatic, Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2018, 378, 2465–2474. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; van Gent, D.C.; Incrocci, L.; van Weerden, W.M.; Nonnekens, J. Role of the DNA damage response in prostate cancer formation, progression and treatment. Prostate Cancer Prostatic Dis. 2020, 23, 24–37. [Google Scholar] [CrossRef]
- Goodwin, J.F.; Schiewer, M.J.; Dean, J.L.; Schrecengost, R.S.; de Leeuw, R.; Han, S.; Ma, T.; Den, R.B.; Dicker, A.P.; Feng, F.Y.; et al. A hormone-DNA repair circuit governs the response to genotoxic insult. Cancer Discov. 2013, 3, 1254–1271. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Karanika, S.; Yang, G.; Wang, J.; Park, S.; Broom, B.M.; Manyam, G.C.; Wu, W.; Luo, Y.; Basourakos, S.; et al. Androgen receptor inhibitor-induced “BRCAness” and PARP inhibition are synthetically lethal for castration-resistant prostate cancer. Sci. Signal. 2017, 10, eaam7479. [Google Scholar] [CrossRef]
- Polkinghorn, W.R.; Parker, J.S.; Lee, M.X.; Kass, E.M.; Spratt, D.E.; Iaquinta, P.J.; Arora, V.K.; Yen, W.-F.; Cai, L.; Zheng, D.; et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov. 2013, 3, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekar, T.; Yang, J.C.; Gao, A.C.; Evans, C.P. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl. Androl. Urol. 2015, 4, 365–380. [Google Scholar]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef]
- Groner, A.C.; Cato, L.; de Tribolet-Hardy, J.; Bernasocchi, T.; Janouskova, H.; Melchers, D.; Houtman, R.; Cato, A.C.; Tschopp, P.; Gu, L.; et al. TRIM24 Is an Oncogenic Transcriptional Activator in Prostate Cancer. Cancer Cell 2016, 29, 846–858. [Google Scholar] [CrossRef]
- Montgomery, R.B.; Mostaghel, E.A.; Vessella, R.; Hess, D.L.; Kalhorn, T.F.; Higano, C.S.; True, L.D.; Nelson, P.S. Maintenance of intratumoral androgens in metastatic prostate cancer: A mechanism for castration-resistant tumor growth. Cancer Res. 2008, 68, 4447–4454. [Google Scholar] [CrossRef]
- Singh, V.; Connelly, Z.M.; Shen, X.; De Benedetti, A. Identification of the proteome complement of humanTLK1 reveals it binds and phosphorylates NEK1 regulating its activity. Cell Cycle 2017, 16, 915–926. [Google Scholar] [CrossRef]
- Polci, R.; Peng, A.; Chen, P.L.; Riley, D.J.; Chen, Y. NIMA-related protein kinase 1 is involved early in the ionizing radiation-induced DNA damage response. Cancer Res. 2004, 64, 8800–8803. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, P.L.; Chen, C.F.; Jiang, X.; Riley, D.J. Never-in-mitosis related kinase 1 functions in DNA damage response and checkpoint control. Cell Cycle 2008, 7, 3194–3201. [Google Scholar] [CrossRef]
- Liu, S.; Ho, C.K.; Ouyang, J.; Zou, L. Nek1 kinase associates with ATR-ATRIP and primes ATR for efficient DNA damage signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 2175–2180. [Google Scholar] [CrossRef] [PubMed]
- Khalil, M.I.; Ghosh, I.; Singh, V.; Chen, J.; Zhu, H.; De Benedetti, A. NEK1 Phosphorylation of YAP Promotes Its Stabilization and Transcriptional Output. Cancers 2020, 12, 3666. [Google Scholar] [CrossRef] [PubMed]
- Kuser-Abali, G.; Alptekin, A.; Lewis, M.; Garraway, I.P.; Cinar, B. YAP1 and AR interactions contribute to the switch from androgen-dependent to castration-resistant growth in prostate cancer. Nat. Commun. 2015, 6, 8126. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Khalil, M.I.; De Benedetti, A. The TLK1/Nek1 axis contributes to mitochondrial integrity and apoptosis prevention via phosphorylation of VDAC1. Cell Cycle 2020, 19, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Khalil, M.I.; Madere, C.; Ghosh, I.; Adam, R.M.; De Benedetti, A. Interaction of TLK1 and AKTIP as a Potential Regulator of AKT Activation in Castration-Resistant Prostate Cancer Progression. Pathophysiology 2021, 28, 339–354. [Google Scholar] [CrossRef]
- Bluemn, E.G.; Coleman, I.M.; Lucas, J.M.; Coleman, R.T.; Hernandez-Lopez, S.; Tharakan, R.; Bianchi-Frias, D.; Dumpit, R.F.; Kaipainen, A.; Corella, A.N.; et al. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell 2017, 32, 474–489.e476. [Google Scholar] [CrossRef]
- Kregel, S.; Wang, C.; Han, X.; Xiao, L.; Fernandez-Salas, E.; Bawa, P.; McCollum, B.L.; Wilder-Romans, K.; Apel, I.J.; Cao, X.; et al. Androgen receptor degraders overcome common resistance mechanisms developed during prostate cancer treatment. Neoplasia 2020, 22, 111–119. [Google Scholar] [CrossRef]
- Flanagan, J.J.; Neklesa, T.K. Targeting Nuclear Receptors with PROTAC degraders. Mol. Cell Endocrinol. 2019, 493, 110452. [Google Scholar] [CrossRef]
- Neklesa, T.; Snyder, L.B.; Willard, R.R.; Vitale, N.; Pizzano, J.; A Gordon, D.; Bookbinder, M.; Macaluso, J.; Dong, H.; Ferraro, C.; et al. ARV-110, An oral androgen receptor PROTAC degrader for prostate cancer. J. Clin. Oncol. 2019, 37, 259. [Google Scholar] [CrossRef]
- Fléchon, A.; Pouessel, D.; Ferlay, C.; Perol, D.; Beuzeboc, P.; Gravis, G.; Joly, F.; Oudard, S.; Deplanque, G.; Zanetta, S.; et al. Phase II study of carboplatin and etoposide in patients with anaplastic progressive metastatic castration-resistant prostate cancer (mCRPC) with or without neuroendocrine differentiation: Results of the French Genito-Urinary Tumor Group (GETUG) P01 trial. Ann. Oncol. 2011, 22, 2476–2481. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, A.M.; Harzstark, A.L.; Corn, P.G.; Wen, S.; Araujo, J.C.; Tu, S.-M.; Pagliaro, L.C.; Kim, J.; Millikan, R.E.; Ryan, C.; et al. Platinum-based chemotherapy for variant castrate-resistant prostate cancer. Clin. Cancer Res. 2013, 19, 3621–3630. [Google Scholar] [CrossRef] [PubMed]
- Loriot, Y.; Massard, C.; Gross-Goupil, M.; Di Palma, M.; Escudier, B.; Bossi, A.; Fizazi, K. Combining carboplatin and etoposide in docetaxel-pretreated patients with castration-resistant prostate cancer: A prospective study evaluating also neuroendocrine features. Ann. Oncol. 2009, 20, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Metzger, A.L.; Abel, S.; Wegner, R.E.; Fuhrer, R.; Mao, S.; Miller, R.; Beriwal, S.; Horne, Z.D. Patterns of care and outcomes in small cell carcinoma of the prostate: A national cancer database analysis. Prostate 2019, 79, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Conteduca, V.; Oromendia, C.; Eng, K.W.; Bareja, R.; Sigouros, M.; Molina, A.; Faltas, B.M.; Sboner, A.; Mosquera, J.M.; Elemento, O.; et al. Clinical features of neuroendocrine prostate cancer. Eur. J. Cancer 2019, 121, 7–18. [Google Scholar] [CrossRef]
- Shah, R.B.; Mehra, R.; Chinnaiyan, A.M.; Shen, R.; Ghosh, D.; Zhou, M.; MacVicar, G.R.; Varambally, S.; Harwood, J.; Bismar, T.A.; et al. Androgen-independent prostate cancer is a heterogeneous group of diseases: Lessons from a rapid autopsy program. Cancer Res. 2004, 64, 9209–9216. [Google Scholar] [CrossRef]
- Beltran, H.; Tomlins, S.; Aparicio, A.; Arora, V.; Rickman, D.; Ayala, G.; Huang, J.; True, L.; Gleave, M.E.; Soule, H.; et al. Aggressive variants of castration-resistant prostate cancer. Clin. Cancer Res. 2014, 20, 2846–2850. [Google Scholar] [CrossRef]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef]
- Epstein, J.I.; Amin, M.B.; Beltran, H.; Lotan, T.L.; Mosquera, J.-M.; Reuter, V.E.; Robinson, B.D.; Troncoso, P.; Rubin, M.A. Proposed morphologic classification of prostate cancer with neuroendocrine differentiation. Am. J. Surg. Pathol. 2014, 38, 756–767. [Google Scholar] [CrossRef]
- Randolph, T.L.; Amin, M.B.; Ro, J.Y.; Ayala, A.G. Histologic variants of adenocarcinoma and other carcinomas of prostate: Pathologic criteria and clinical significance. Mod. Pathol. 1997, 10, 612–629. [Google Scholar] [PubMed]
- Zou, M.; Toivanen, R.; Mitrofanova, A.; Floch, N.; Hayati, S.; Sun, Y.; Le Magnen, C.; Chester, D.; Mostaghel, E.A.; Califano, A.; et al. Transdifferentiation as a Mechanism of Treatment Resistance in a Mouse Model of Castration-Resistant Prostate Cancer. Cancer Discov. 2017, 7, 736–749. [Google Scholar] [CrossRef]
- Zhang, X.Q.; Kondrikov, D.; Yuan, T.C.; Lin, F.F.; Hansen, J.; Lin, M.F. Receptor protein tyrosine phosphatase alpha signaling is involved in androgen depletion-induced neuroendocrine differentiation of androgen-sensitive LNCaP human prostate cancer cells. Oncogene 2003, 22, 6704–6716. [Google Scholar] [CrossRef][Green Version]
- Bishop, J.L.; Thaper, D.; Vahid, S.; Davies, A.; Ketola, K.; Kuruma, H.; Jama, R.; Nip, K.M.; Angeles, A.; Johnson, F.; et al. The Master Neural Transcription Factor BRN2 Is an Androgen Receptor-Suppressed Driver of Neuroendocrine Differentiation in Prostate Cancer. Cancer Discov. 2017, 7, 54–71. [Google Scholar] [CrossRef]
- Lotan, T.L.; Gupta, N.S.; Wang, W.; Toubaji, A.; Haffner, M.C.; Chaux, A.; Hicks, J.L.; Meeker, A.K.; Bieberich, C.J.; De Marzo, A.M.; et al. ERG gene rearrangements are common in prostatic small cell carcinomas. Mod. Pathol. 2011, 24, 820–828. [Google Scholar] [CrossRef]
- Beltran, H.; Rickman, D.S.; Park, K.; Chae, S.S.; Sboner, A.; MacDonald, T.Y.; Wang, Y.; Sheikh, K.L.; Terry, S.; Tagawa, S.T.; et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011, 1, 487–495. [Google Scholar] [CrossRef]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Tretiakova, M.S.; Silvis, M.R.; Lucas, J.; Klezovitch, O.; Coleman, I.; Bolouri, H.; Kutyavin, V.I.; Morrissey, C.; True, L.D.; et al. ERG Activates the YAP1 Transcriptional Program and Induces the Development of Age-Related Prostate Tumors. Cancer Cell 2015, 27, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, I.; Khalil, M.I.; Mirza, R.; King, J.; Olatunde, D.; De Benedetti, A. NEK1-Mediated Phosphorylation of YAP1 Is Key to Prostate Cancer Progression. Biomedicines 2023, 11, 743. [Google Scholar] [CrossRef]
- Yu, X.; Wang, Y.; Jiang, M.; Bierie, B.; Roy-Burman, P.; Shen, M.M.; Taketo, M.M.; Wills, M.; Matusik, R.J. Activation of beta-Catenin in mouse prostate causes HGPIN and continuous prostate growth after castration. Prostate 2009, 69, 249–262. [Google Scholar] [CrossRef]
- Kuwahara, A.; Hirabayashi, Y.; Knoepfler, P.S.; Taketo, M.M.; Sakai, J.; Kodama, T.; Gotoh, Y. Wnt signaling and its downstream target N-myc regulate basal progenitors in the developing neocortex. Development 2010, 137, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.; Brady, N.J.; Bareja, R.; Robinson, B.; Conteduca, V.; Augello, M.A.; Puca, L.; Ahmed, A.; Dardenne, E.; Lu, X.; et al. N-Myc-mediated epigenetic reprogramming drives lineage plasticity in advanced prostate cancer. J. Clin. Investig. 2019, 129, 3924–3940. [Google Scholar] [CrossRef] [PubMed]
- Dardenne, E.; Beltran, H.; Benelli, M.; Gayvert, K.; Berger, A.; Puca, L.; Cyrta, J.; Sboner, A.; Noorzad, Z.; MacDonald, T.; et al. N-Myc Induces an EZH2-Mediated Transcriptional Program Driving Neuroendocrine Prostate Cancer. Cancer Cell 2016, 30, 563–577. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Lee, J.K.; Witte, O.N.; Huang, J. FOXA2 is a sensitive and specific marker for small cell neuroendocrine carcinoma of the prostate. Mod. Pathol. 2017, 30, 1262–1272. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Chen, M.-W.; Terry, S.; Vacherot, F.; Chopin, D.K.; Bemis, D.L.; Kitajewski, J.; Benson, M.C.; Guo, Y.; Buttyan, R. A human- and male-specific protocadherin that acts through the wnt signaling pathway to induce neuroendocrine transdifferentiation of prostate cancer cells. Cancer Res. 2005, 65, 5263–5271. [Google Scholar] [CrossRef] [PubMed]
- Uysal-Onganer, P.; Kawano, Y.; Caro, M.; Walker, M.M.; Diez, S.; Darrington, R.S.; Waxman, J.; Kypta, R.M. Wnt-11 promotes neuroendocrine-like differentiation, survival and migration of prostate cancer cells. Mol. Cancer 2010, 9, 55. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, C.; Li, X.; Barnes, S.; Deng, S.; Hoover, E.; Chen, C.-C.; Lee, Y.S.; Zhang, Y.; Wang, C.; et al. Loss of CHD1 Promotes Heterogeneous Mechanisms of Resistance to AR-Targeted Therapy via Chromatin Dysregulation. Cancer Cell 2020, 37, 584–598.e511. [Google Scholar] [CrossRef]
- Labrecque, M.; Coleman, I.M.; Brown, L.G.; True, L.D.; Kollath, L.; Lakely, B.; Nguyen, H.M.; Yang, Y.C.; Gil Da Costa, R.M.; Kaipainen, A.; et al. Molecular profiling stratifies diverse phenotypes of treatment-refractory metastatic castration-resistant prostate cancer. J. Clin. Investig. 2019, 129, 4492–4505. [Google Scholar] [CrossRef]
- Beltran, H.; Hruszkewycz, A.; Scher, H.I.; Hildesheim, J.; Isaacs, J.; Yu, E.Y.; Kelly, K.; Lin, D.; Dicker, A.; Arnold, J.; et al. The Role of Lineage Plasticity in Prostate Cancer Therapy Resistance. Clin. Cancer Res. 2019, 25, 6916–6924. [Google Scholar] [CrossRef]
- Aggarwal, R.; Huang, J.; Alumkal, J.J.; Zhang, L.; Feng, F.Y.; Thomas, G.V.; Weinstein, A.S.; Friedl, V.; Zhang, C.; Witte, O.N.; et al. Clinical and Genomic Characterization of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer: A Multi-institutional Prospective Study. J. Clin. Oncol. 2018, 36, 2492–2503. [Google Scholar] [CrossRef]
- Alumkal, J.J.; Sun, D.; Lu, E.; Beer, T.M.; Thomas, G.V.; Latour, E.; Aggarwal, R.; Cetnar, J.; Ryan, C.J.; Tabatabaei, S.; et al. Transcriptional profiling identifies an androgen receptor activity-low, stemness program associated with enzalutamide resistance. Proc. Natl. Acad. Sci. USA 2020, 117, 12315–12323. [Google Scholar] [CrossRef]
- Sillje, H.H.; Takahashi, K.; Tanaka, K.; Van Houwe, G.; Nigg, E.A. Mammalian homologues of the plant Tousled gene code for cell-cycle-regulated kinases with maximal activities linked to ongoing DNA replication. EMBO J. 1999, 18, 5691–5702. [Google Scholar] [CrossRef] [PubMed]
- De Benedetti, A. The Tousled-Like Kinases as Guardians of Genome Integrity. ISRN Mol. Biol. 2012, 2012, 627596. [Google Scholar] [CrossRef] [PubMed]
- Segura-Bayona, S.; Stracker, T.H. The Tousled-like kinases regulate genome and epigenome stability: Implications in development and disease. Cell Mol. Life Sci. 2019, 76, 3827–3841. [Google Scholar] [CrossRef]
- Sunavala-Dossabhoy, G. Preserving salivary gland physiology against genotoxic damage—The Tousled way. Oral. Dis. 2018, 24, 1390–1398. [Google Scholar] [CrossRef]
- Li, Y.; DeFatta, R.; Anthony, C.; Sunavala, G.; De Benedetti, A. A translationally regulated Tousled kinase phosphorylates histone H3 and confers radioresistance when overexpressed. Oncogene 2001, 20, 726–738. [Google Scholar] [CrossRef]
- Mortuza, G.B.; Hermida, D.; Pedersen, A.-K.; Segura-Bayona, S.; López-Méndez, B.; Redondo, P.; Rüther, P.; Pozdnyakova, I.; Garrote, A.M.; Muñoz, I.G.; et al. Molecular basis of Tousled-Like Kinase 2 activation. Nat. Commun. 2018, 9, 2535. [Google Scholar] [CrossRef]
- Sunavala-Dossabhoy, G.; Fowler, M.; De Benedetti, A. Translation of the radioresistance kinase TLK1B is induced by gamma-irradiation through activation of mTOR and phosphorylation of 4E-BP1. BMC Mol. Biol. 2004, 5, 1. [Google Scholar] [CrossRef]
- Norton, K.S.; McClusky, D.; Sen, S.; Yu, H.; Meschonat, C.; Debenedetti, A.; Li, B.D. TLK1B is elevated with eIF4E overexpression in breast cancer. J. Surg. Res. 2004, 116, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Sunavala-Dossabhoy, G.; Li, Y.; Williams, B.; De Benedetti, A. A dominant negative mutant of TLK1 causes chromosome missegregation and aneuploidy in normal breast epithelial cells. BMC Cell Biol. 2003, 4, 16. [Google Scholar] [CrossRef]
- Klimovskaia, I.M.; Young, C.; Strømme, C.B.; Menard, P.; Jasencakova, Z.; Mejlvang, J.; Ask, K.; Ploug, M.; Nielsen, M.L.; Jensen, O.N.; et al. Tousled-like kinases phosphorylate Asf1 to promote histone supply during DNA replication. Nat. Commun. 2014, 5, 3394. [Google Scholar] [CrossRef] [PubMed]
- Sunavala-Dossabhoy, G.; Balakrishnan, S.K.; Sen, S.; Nuthalapaty, S.; De Benedetti, A. The radioresistance kinase TLK1B protects the cells by promoting repair of double strand breaks. BMC Mol. Biol. 2005, 6, 19. [Google Scholar] [CrossRef] [PubMed]
- Sunavala-Dossabhoy, G.; De Benedetti, A. Tousled homolog, TLK1, binds and phosphorylates Rad9; TLK1 acts as a molecular chaperone in DNA repair. DNA Repair 2009, 8, 87–102. [Google Scholar] [CrossRef] [PubMed]
- Awate, S.; De Benedetti, A. TLK1B mediated phosphorylation of Rad9 regulates its nuclear/cytoplasmic localization and cell cycle checkpoint. BMC Mol. Biol. 2016, 17, 3. [Google Scholar] [CrossRef] [PubMed]
- Canfield, C.; Rains, J.; De Benedetti, A. TLK1B promotes repair of DSBs via its interaction with Rad9 and Asf1. BMC Mol. Biol. 2009, 10, 110. [Google Scholar] [CrossRef]
- Feige, E.; Shalom, O.; Tsuriel, S.; Yissachar, N.; Motro, B. Nek1 shares structural and functional similarities with NIMA kinase. Biochim. Biophys. Acta 2006, 1763, 272–281. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, C.F.; Riley, D.J.; Chen, P.L. Nek1 kinase functions in DNA damage response and checkpoint control through a pathway independent of ATM and ATR. Cell Cycle 2011, 10, 655–663. [Google Scholar] [CrossRef]
- Singh, V.; Jaiswal, P.K.; Ghosh, I.; Koul, H.K.; Yu, X.; De Benedetti, A. Targeting the TLK1/NEK1 DDR axis with Thioridazine suppresses outgrowth of androgen independent prostate tumors. Int. J. Cancer 2019, 145, 1055–1067. [Google Scholar] [CrossRef]
- Ronald, S.; Awate, S.; Rath, A.; Carroll, J.; Galiano, F.; Dwyer, D.; Kleiner-Hancock, H.; Mathis, J.M.; Vigod, S.; De Benedetti, A. Phenothiazine Inhibitors of TLKs Affect Double-Strand Break Repair and DNA Damage Response Recovery and Potentiate Tumor Killing with Radiomimetic Therapy. Genes Cancer 2013, 4, 39–53. [Google Scholar] [CrossRef]
- Khalil, M.I.; De Benedetti, A. Tousled-like kinase 1, a novel factor with multifaceted role in mCRPC progression and development of therapy resistance. Cancer Drug Resist. 2022, 5, 93–101. [Google Scholar] [CrossRef]
- Shen, J.; Ma, B.; Zhang, X.; Sun, X.; Han, J.; Wang, Y.; Chu, L.; Xu, H.; Yang, Y. Thioridazine has potent antitumor effects on lung cancer stem-like cells. Oncol. Lett. 2017, 13, 1563–1568. [Google Scholar] [CrossRef]
- Spengler, G.; Csonka, A.; Molnar, J.; Amaral, L. The Anticancer Activity of the Old Neuroleptic Phenothiazine-type Drug Thioridazine. Anticancer Res. 2016, 36, 5701–5706. [Google Scholar] [CrossRef]
- Seo, S.U.; Cho, H.K.; Min, K.-J.; Woo, S.M.; Kim, S.; Park, J.-W.; Kim, S.H.; Choi, Y.H.; Keum, Y.S.; Hyun, J.W.; et al. Thioridazine enhances sensitivity to carboplatin in human head and neck cancer cells through downregulation of c-FLIP and Mcl-1 expression. Cell Death Dis. 2017, 8, e2599. [Google Scholar] [CrossRef]
- Luo, L.; Jin, X.; Zou, B.; Zhong, C.; Zhang, P.; Cheng, H.; Guo, Y.; Gou, M. Codelivery of thioridazine and doxorubicin using nanoparticles for effective breast cancer therapy. Int. J. Nanomed. 2016, 11, 4545–4552. [Google Scholar] [CrossRef]
- Torrey, E.F. Prostate cancer and schizophrenia. Urology 2006, 68, 1280–1283. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, P.B. The incidence of cancer in schizophrenic patients. J. Epidemiol. Community Health 1989, 43, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, P.B. Neuroleptic medication and reduced risk of prostate cancer in schizophrenic patients. Acta Psychiatr. Scand. 1992, 85, 390–393. [Google Scholar] [CrossRef] [PubMed]
- Hoeh, B.; Würnschimmel, C.; Flammia, R.S.; Horlemann, B.; Sorce, G.; Chierigo, F.; Tian, Z.; Saad, F.; Graefen, M.; Gallucci, M.; et al. Effect of Chemotherapy on Overall Survival in Contemporary Metastatic Prostate Cancer Patients. Front. Oncol. 2021, 11, 778858. [Google Scholar] [CrossRef] [PubMed]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef]
- Magee, J.A.; Chang, L.W.; Stormo, G.D.; Milbrandt, J. Direct, androgen receptor-mediated regulation of the FKBP5 gene via a distal enhancer element. Endocrinology 2006, 147, 590–598. [Google Scholar] [CrossRef]
- Chiu, Y.T.; Liu, J.; Tang, K.; Wong, Y.C.; Khanna, K.K.; Ling, M.T. Inactivation of ATM/ATR DNA damage checkpoint promotes androgen induced chromosomal instability in prostate epithelial cells. PLoS ONE 2012, 7, e51108. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.; Wu, M.; Ciavattone, N.; McKenty, N.; Menon, M.; Barrack, E.R.; Reddy, G.P.V.; Kim, S.H. ATM Inhibition Potentiates Death of Androgen Receptor-inactivated Prostate Cancer Cells with Telomere Dysfunction. J. Biol. Chem. 2015, 290, 25522–25533. [Google Scholar] [CrossRef] [PubMed]
- Karanika, S.; Karantanos, T.; Li, L.; Corn, P.G.; Thompson, T.C. DNA damage response and prostate cancer: Defects, regulation and therapeutic implications. Oncogene 2015, 34, 2815–2822. [Google Scholar] [CrossRef]
- Yu, P.; Duan, X.; Cheng, Y.; Liu, C.; Chen, Y.; Liu, W.; Yin, B.; Wang, X.; Tao, Z. Androgen-independent LNCaP cells are a subline of LNCaP cells with a more aggressive phenotype and androgen suppresses their growth by inducing cell cycle arrest at the G1 phase. Int. J. Mol. Med. 2017, 40, 1426–1434. [Google Scholar] [CrossRef]
- Singh, V.; Jaiswal, P.K.; Ghosh, I.; Koul, H.K.; Yu, X.; De Benedetti, A. The TLK1-Nek1 axis promotes prostate cancer progression. Cancer Lett. 2019, 453, 131–141. [Google Scholar] [CrossRef]
- Loberg, R.D.; St John, L.N.; Day, L.L.; Neeley, C.K.; Pienta, K.J. Development of the VCaP androgen-independent model of prostate cancer. Urol. Oncol. 2006, 24, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Jia, P.; Zhao, Z.; Shen, B. Key regulators in prostate cancer identified by co-expression module analysis. BMC Genom. 2014, 15, 1015. [Google Scholar] [CrossRef]
- Melo-Hanchuk, T.D.; Slepicka, P.F.; Meirelles, G.V.; Basei, F.L.; Lovato, D.V.; Granato, D.C.; Pauletti, B.A.; Domingues, R.R.; Leme, A.F.P.; Pelegrini, A.L.; et al. NEK1 kinase domain structure and its dynamic protein interactome after exposure to Cisplatin. Sci. Rep. 2017, 7, 5445. [Google Scholar] [CrossRef] [PubMed]
- Salem, O.; Hansen, C.G. The Hippo Pathway in Prostate Cancer. Cells 2019, 8, 370. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, S.; Chen, X.; Stauffer, S.; Yu, F.; Lele, S.M.; Fu, K.; Datta, K.; Palermo, N.; Chen, Y.; et al. The hippo pathway effector YAP regulates motility, invasion, and castration-resistant growth of prostate cancer cells. Mol. Cell Biol. 2015, 35, 1350–1362. [Google Scholar] [CrossRef]
- Yim, H.; Sung, C.K.; You, J.; Tian, Y.; Benjamin, T. Nek1 and TAZ interact to maintain normal levels of polycystin 2. J. Am. Soc. Nephrol. 2011, 22, 832–837. [Google Scholar] [CrossRef]
- Yang, C.E.; Lee, W.Y.; Cheng, H.W.; Chung, C.H.; Mi, F.L.; Lin, C.W. The antipsychotic chlorpromazine suppresses YAP signaling, stemness properties, and drug resistance in breast cancer cells. Chem. Biol. Interact. 2019, 302, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Peres de Oliveira, A.; Kazuo Issayama, L.; Betim Pavan, I.C.; Riback Silva, F.; Diniz Melo-Hanchuk, T.; Moreira Simabuco, F.; Kobarg, J. Checking NEKs: Overcoming a Bottleneck in Human Diseases. Molecules 2020, 25, 1778. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Juan, L.; Xia, L.; Wang, Y.; Bao, Y.; Sun, G. Thioridazine Sensitizes Esophageal Carcinoma Cell Lines to Radiotherapy-Induced Apoptosis In Vitro and In Vivo. Med. Sci. Monit. 2016, 22, 2624–2634. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Strano, S.; Monti, O.; Pediconi, N.; Baccarini, A.; Fontemaggi, G.; Lapi, E.; Mantovani, F.; Damalas, A.; Citro, G.; Sacchi, A.; et al. The transcriptional coactivator Yes-associated protein drives p73 gene-target specificity in response to DNA Damage. Mol. Cell 2005, 18, 447–459. [Google Scholar] [CrossRef]
- Lu, M.; Li, J.; Luo, Z.; Zhang, S.; Xue, S.; Wang, K.; Shi, Y.; Zhang, C.; Chen, H.; Li, Z. Roles of dopamine receptors and their antagonist thioridazine in hepatoma metastasis. Oncol. Targets Ther. 2015, 8, 1543–1552. [Google Scholar]
- Shoshan-Barmatz, V.; Golan, M. Mitochondrial VDAC1, function in cell life and death and a target for cancer therapy. Curr. Med. Chem. 2012, 19, 714–735. [Google Scholar] [CrossRef]
- Khalil, M.I.; Singh, V.; King, J.; De Benedetti, A. TLK1-mediated MK5-S354 phosphorylation drives prostate cancer cell motility and may signify distinct pathologies. Mol. Oncol. 2022, 16, 2537–2557. [Google Scholar] [CrossRef]
- Wu, C.S.; Tsai, Y.T.; Tsai, H.J. Antipsychotic drugs and the risk of ventricular arrhythmia and/or sudden cardiac death: A nation-wide case-crossover study. J. Am. Heart Assoc. 2015, 4, e001568. [Google Scholar] [CrossRef]
- Singh, V.; Bhoir, S.; Chikhale, R.V.; Hussain, J.; Dwyer, D.; Bryce, R.A.; Kirubakaran, S.; De Benedetti, A. Generation of Phenothiazine with Potent Anti-TLK1 Activity for Prostate Cancer Therapy. iScience 2020, 23, 101474. [Google Scholar] [CrossRef]
- Kirubakaran, S.; Johnson, D.; Hussain, J.; Bhoir, S.; Chandrasekaran, V.; Sahrawat, P.; Hans, T.; Khalil, I.; De Benedetti, A.; Thiruvenkatam, V. Synthesis, kinetics and cellular studies of new phenothiazine analogs as potent human-TLK inhibitors. Org. Biomol. Chem. 2023, 21, 1980–1991. [Google Scholar]
- Lee, J.K.; Nam, D.H.; Lee, J. Repurposing antipsychotics as glioblastoma therapeutics: Potentials and challenges. Oncol. Lett. 2016, 11, 1281–1286. [Google Scholar] [CrossRef] [PubMed]
- Yong, M.; Yu, T.; Tian, S.; Liu, S.; Xu, J.; Hu, J.; Hu, L. DR2 blocker thioridazine: A promising drug for ovarian cancer therapy. Oncol. Lett. 2017, 14, 8171–8177. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Roth, B.L.; Tandra, S.; Burgess, L.H.; Sibley, D.R.; Meltzer, H.Y. D4 dopamine receptor binding affinity does not distinguish between typical and atypical antipsychotic drugs. Psychopharmacology 1995, 120, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Dutta, N.K.; Pinn, M.L.; Karakousis, P.C. Reduced emergence of isoniazid resistance with concurrent use of thioridazine against acute murine tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 4048–4053. [Google Scholar] [CrossRef]
- Ronald, S.; Sunavala-Dossabhoy, G.; Adams, L.; Williams, B.; De Benedetti, A. The expression of Tousled kinases in CaP cell lines and its relation to radiation response and DSB repair. Prostate 2011, 71, 1367–1373. [Google Scholar] [CrossRef]
- Karanika, S.; Karantanos, T.; Li, L.; Wang, J.; Park, S.; Yang, G.; Zuo, X.; Song, J.H.; Maity, S.N.; Manyam, G.C.; et al. Targeting DNA Damage Response in Prostate Cancer by Inhibiting Androgen Receptor-CDC6-ATR-Chk1 Signaling. Cell Rep. 2017, 18, 1970–1981. [Google Scholar] [CrossRef]
- Donohoe, D.R.; Aamodt, E.J.; Osborn, E.; Dwyer, D.S. Antipsychotic drugs disrupt normal development in Caenorhabditis elegans via additional mechanisms besides dopamine and serotonin receptors. Pharmacol. Res. 2006, 54, 361–372. [Google Scholar] [CrossRef]
- Donohoe, D.R.; Jarvis, R.A.; Weeks, K.; Aamodt, E.J.; Dwyer, D.S. Behavioral adaptation in C. elegans produced by antipsychotic drugs requires serotonin and is associated with calcium signaling and calcineurin inhibition. Neurosci. Res. 2009, 64, 280–289. [Google Scholar] [CrossRef]
- Thompson, T.C.; Li, L.; Broom, B.M. Combining enzalutamide with PARP inhibitors: Pharmaceutically induced BRCAness. Oncotarget 2017, 8, 93315–93316. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Riefler, G.M.; Saam, J.R.; Mango, S.E.; Schumacher, J.M. The C. elegans Tousled-like kinase contributes to chromosome segregation as a substrate and regulator of the Aurora B kinase. Curr. Biol. 2005, 15, 894–904. [Google Scholar] [CrossRef]
- Han, Z.; Saam, J.R.; Adams, H.P.; Mango, S.E.; Schumacher, J.M. The C. elegans Tousled-like kinase (TLK-1) has an essential role in transcription. Curr. Biol. 2003, 13, 1921–1929. [Google Scholar] [CrossRef] [PubMed]
- Carrera, P.; Moshkin, Y.M.; Grönke, S.; Silljé, H.H.; Nigg, E.A.; Jäckle, H.; Karch, F. Tousled-like kinase functions with the chromatin assembly pathway regulating nuclear divisions. Genes Dev. 2003, 17, 2578–2590. [Google Scholar] [CrossRef] [PubMed]
- Segura-Bayona, S.; A Knobel, P.; González-Burón, H.; A Youssef, S.; Peña-Blanco, A.; Coyaud; López-Rovira, T.; Rein, K.; Palenzuela, L.; Colombelli, J.; et al. Differential requirements for Tousled-like kinases 1 and 2 in mammalian development. Cell Death Differ. 2017, 24, 1872–1885. [Google Scholar] [CrossRef] [PubMed]
- Pavinato, L.; Villamor-Payà, M.; Sanchiz-Calvo, M.; Andreoli, C.; Gay, M.; Vilaseca, M.; Arauz-Garofalo, G.; Ciolfi, A.; Bruselles, A.; Pippucci, T.; et al. Functional analysis of TLK2 variants and their proximal interactomes implicates impaired kinase activity and chromatin maintenance defects in their pathogenesis. J. Med. Genet. 2020, 52, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Lelieveld, S.H.; Reijnders, M.R.F.; Pfundt, R.; Yntema, H.G.; Kamsteeg, E.-J.; de Vries, P.; A de Vries, B.B.; Willemsen, M.H.; Kleefstra, T.; Löhner, K.; et al. Meta-analysis of 2104 trios provides support for 10 new genes for intellectual disability. Nat. Neurosci. 2016, 19, 1194–1196. [Google Scholar] [CrossRef]
- Kim, J.-A.; Tan, Y.; Wang, X.; Cao, X.; Veeraraghavan, J.; Liang, Y.; Edwards, D.P.; Huang, S.; Pan, X.; Li, K.; et al. Comprehensive functional analysis of the tousled-like kinase 2 frequently amplified in aggressive luminal breast cancers. Nat. Commun. 2016, 7, 12991. [Google Scholar] [CrossRef]
- Lee, S.-B.; Segura-Bayona, S.; Villamor-Payà, M.; Saredi, G.; Todd, M.A.M.; Attolini, C.S.-O.; Chang, T.-Y.; Stracker, T.H.; Groth, A. Tousled-like kinases stabilize replication forks and show synthetic lethality with checkpoint and PARP inhibitors. Sci. Adv. 2018, 4, eaat4985. [Google Scholar] [CrossRef]
- Huang, T.H.; Fowler, F.; Chen, C.C.; Shen, Z.J.; Sleckman, B.; Tyler, J.K. The Histone Chaperones ASF1 and CAF-1 Promote MMS22L-TONSL-Mediated Rad51 Loading onto ssDNA during Homologous Recombination in Human Cells. Mol. Cell 2018, 69, 879–892.e875. [Google Scholar] [CrossRef]
- Simon, B.; Lou, H.J.; Huet-Calderwood, C.; Shi, G.; Boggon, T.J.; Turk, B.E.; Calderwood, D.A. Tousled-like kinase 2 targets ASF1 histone chaperones through client mimicry. Nat. Commun. 2022, 13, 749. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Davies, S.P.; Augustin, M.; Woodward, A.; Patel, U.A.; Kovelman, R.; Harvey, K.J. A broad activity screen in support of a chemogenomic map for kinase signalling research and drug discovery. Biochem. J. 2013, 451, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Hoessel, R.; Leclerc, S.; Endicott, J.; Nobel, M.E.M.; Lawrie, A.; Tunnah, P.; Leost, M.; Damiens, E.; Marie, D.; Marko, D.; et al. Indirubin, the active constituent of a Chinese antileukaemia medicine, inhibits cyclin-dependent kinases. Nat. Cell Biol. 1999, 1, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.L.; Tan, X.; Chen, M.; Kusi, M.; Hung, C.N.; Chou, C.W.; Hsu, Y.T.; Wang, C.M.; Kirma, N.; Chen, C.L.; et al. ERalpha-related chromothripsis enhances concordant gene transcription on chromosome 17q11.1-q24.1 in luminal breast cancer. BMC Med. Genom. 2020, 13, 69. [Google Scholar] [CrossRef]
- Kang, S.; Dong, S.M.; Kim, B.-R.; Park, M.S.; Trink, B.; Byun, H.-J.; Rho, S.B. Thioridazine induces apoptosis by targeting the PI3K/Akt/mTOR pathway in cervical and endometrial cancer cells. Apoptosis 2012, 17, 989–997. [Google Scholar] [CrossRef]
- Park, M.S.; Dong, S.M.; Kim, B.R.; Seo, S.H.; Kang, S.; Lee, E.J.; Lee, S.H.; Rho, S.B. Thioridazine inhibits angiogenesis and tumor growth by targeting the VEGFR-2/PI3K/mTOR pathway in ovarian cancer xenografts. Oncotarget 2014, 5, 4929–4934. [Google Scholar] [CrossRef]
- Allen-Petersen, B.L.; Risom, T.; Feng, Z.; Wang, Z.; Jenny, Z.P.; Thoma, M.C.; Pelz, K.R.; Morton, J.P.; Sansom, O.J.; Lopez, C.D.; et al. Activation of PP2A and Inhibition of mTOR Synergistically Reduce MYC Signaling and Decrease Tumor Growth in Pancreatic Ductal Adenocarcinoma. Cancer Res. 2019, 79, 209–219. [Google Scholar] [CrossRef]
- Kaistha, B.P.; Honstein, T.; Müller, V.; Bielak, S.; Sauer, M.; Kreider, R.; Fassan, M.; Scarpa, A.; Schmees, C.; Volkmer, H.; et al. Key role of dual specificity kinase TTK in proliferation and survival of pancreatic cancer cells. Br. J. Cancer 2014, 111, 1780–1787. [Google Scholar] [CrossRef]
- Bisson, W.H.; Cheltsov, A.V.; Bruey-Sedano, N.; Lin, B.; Chen, J.; Goldberger, N.; May, L.T.; Christopoulos, A.; Dalton, J.T.; Sexton, P.M.; et al. Discovery of antiandrogen activity of nonsteroidal scaffolds of marketed drugs. Proc. Natl. Acad. Sci. USA 2007, 104, 11927–11932. [Google Scholar] [CrossRef]
- Chen, Y.; Clegg, N.J.; Scher, H.I. Anti-androgens and androgen-depleting therapies in prostate cancer: New agents for an established target. Lancet Oncol. 2009, 10, 981–991. [Google Scholar] [CrossRef]
- Chism, D.D.; De Silva, D.; Whang, Y.E. Mechanisms of acquired resistance to androgen receptor targeting drugs in castration-resistant prostate cancer. Expert Rev. Anticancer Ther. 2014, 14, 1369–1378. [Google Scholar] [CrossRef] [PubMed]
- Malaquin, N.; Vancayseele, A.; Gilbert, S.; Antenor-Habazac, L.; Olivier, M.A.; Ait Ali Brahem, Z.; Saad, F.; Delouya, G.; Rodier, F. DNA Damage- But Not Enzalutamide-Induced Senescence in Prostate Cancer Promotes Senolytic Bcl-xL Inhibitor Sensitivity. Cells 2020, 9, 1593. [Google Scholar] [CrossRef] [PubMed]
- Litvinov, I.V.; Vander Griend, D.J.; Antony, L.; Dalrymple, S.; De Marzo, A.M.; Drake, C.G.; Isaacs, J.T. Androgen receptor as a licensing factor for DNA replication in androgen-sensitive prostate cancer cells. Proc. Natl. Acad. Sci. USA 2006, 103, 15085–15090. [Google Scholar] [CrossRef] [PubMed]
- Vaishampayan, U.N.; Heilbrun, L.K.; Monk, P.; Tejwani, S.; Sonpavde, G.; Hwang, C.; Smith, D.; Jasti, P.; Dobson, K.; Dickow, B.; et al. Clinical Efficacy of Enzalutamide vs Bicalutamide Combined with Androgen Deprivation Therapy in Men With Metastatic Hormone-Sensitive Prostate Cancer: A Randomized Clinical Trial. JAMA Netw. Open 2021, 4, e2034633. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, C.J.; Martin, A.J.; Stockler, M.R.; Begbie, S.; Cheung, L.; Chi, K.N.; Chowdhury, S.; Frydenberg, M.; Horvath, L.G.; Joshua, A.M.; et al. Testosterone suppression plus enzalutamide versus testosterone suppression plus standard antiandrogen therapy for metastatic hormone-sensitive prostate cancer (ENZAMET): An international, open-label, randomised, phase 3 trial. Lancet Oncol. 2023, 24, 323–334. [Google Scholar] [CrossRef]
- Teyssonneau, D.; Margot, H.; Cabart, M.; Anonnay, M.; Sargos, P.; Vuong, N.-S.; Soubeyran, I.; Sevenet, N.; Roubaud, G. Prostate cancer and PARP inhibitors: Progress and challenges. J. Hematol. Oncol. 2021, 14, 51. [Google Scholar] [CrossRef]
- Tsujino, T.; Takai, T.; Hinohara, K.; Gui, F.; Tsutsumi, T.; Bai, X.; Miao, C.; Feng, C.; Bin Gui, B.; Sztupinszki, Z.; et al. CRISPR screens reveal genetic determinants of PARP inhibitor sensitivity and resistance in prostate cancer. Nat. Commun. 2023, 14, 252. [Google Scholar] [CrossRef]
- Abida, W.; Campbell, D.; Patnaik, A.; Shapiro, J.D.; Sautois, B.; Vogelzang, N.J.; Voog, E.G.; Bryce, A.H.; McDermott, R.; Ricci, F.; et al. Non-BRCA DNA Damage Repair Gene Alterations and Response to the PARP Inhibitor Rucaparib in Metastatic Castration-Resistant Prostate Cancer: Analysis From the Phase II TRITON2 Study. Clin. Cancer Res. 2020, 26, 2487–2496. [Google Scholar] [CrossRef]
- Stopsack, K.H. Efficacy of PARP Inhibition in Metastatic Castration-resistant Prostate Cancer is Very Different with Non-BRCA DNA Repair Alterations: Reconstructing Prespecified Endpoints for Cohort B from the Phase 3 PROfound Trial of Olaparib. Eur. Urol. 2021, 79, 442–445. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhoir, S.; De Benedetti, A. Targeting Prostate Cancer, the ‘Tousled Way’. Int. J. Mol. Sci. 2023, 24, 11100. https://doi.org/10.3390/ijms241311100
Bhoir S, De Benedetti A. Targeting Prostate Cancer, the ‘Tousled Way’. International Journal of Molecular Sciences. 2023; 24(13):11100. https://doi.org/10.3390/ijms241311100
Chicago/Turabian StyleBhoir, Siddhant, and Arrigo De Benedetti. 2023. "Targeting Prostate Cancer, the ‘Tousled Way’" International Journal of Molecular Sciences 24, no. 13: 11100. https://doi.org/10.3390/ijms241311100
APA StyleBhoir, S., & De Benedetti, A. (2023). Targeting Prostate Cancer, the ‘Tousled Way’. International Journal of Molecular Sciences, 24(13), 11100. https://doi.org/10.3390/ijms241311100