
From the Bush to the Brain: Preclinical Stages of Ethnobotanical Anti-Inflammatory and Neuroprotective Drug Discovery—An Australian Example

 , , and
, , and
Abstract
1. Chronic Neuroinflammation and Its Role in Alzheimer’s Disease
- (a)
- Various inflammatory triggers can cause the initial activation of microglia. These triggers can be peripheral (e.g., systemic infections or peripheral chronic inflammation) or central (e.g., trauma, degenerating/dying neurons, or amyloid deposits) [33]. In our GFAP-IL6 mouse model, the trigger is the brain-specific production of the cytokine IL6 [34,35].
- (b)
- Activated microglia release neurotoxic factors, including cytotoxic cytokines (such as TNF-α) and reactive oxygen and nitrogen species, which cause damage to neighboring neurons.
- (c)
- These damaged neurons release various microglia activators, such as damage-associated molecular pattern molecules (DAMPs) [36], which results in further microglial activation. Consequently, targeting chronic neuroinflammation has been suggested as a disease-modifying treatment for many neurodegenerative diseases, including AD.
2. Cytokine Suppressive Anti-Inflammatory Drugs—A Better Alternative to Conventional Anti-Inflammatory Drugs?
2.1. Nonsteroidal Anti-Inflammatory Drugs (NSAIDs)
2.2. Steroidal Anti-Inflammatory Drugs (Corticosteroids)
2.3. Cytokine Suppressive Anti-Inflammatory Drugs
3. Australian Native Plants as a Source of Novel Anti-Inflammatory Drugs
3.1. Aboriginal Knowledge as a Source of Information about the Medical Use of Australian Plants
3.2. Ecology of the Australian Rainforest as a Further Source of Information about the Potential Medical Use of Australian Plants
4. Cell-Based Screening Assays for Novel Anti-Inflammatory Drugs
4.1. Macrophages as Screening Tools for Anti-Inflammatory Drugs
4.1.1. The RAW 264.7 Murine Cell Line
4.1.2. The J774 Murine Cell Line
4.2. Microglial Cells as Screening Tools for Anti-Inflammatory Drugs
4.2.1. The BV-2 Microglial Cell Line
4.2.2. The N11 Microglial Cell Line
4.3. Pro-Inflammatory Activation Pathways of Macrophages and Microglia
4.3.1. Activation via the IFN-γ Pathway
4.3.2. Activation of Cells via the LPS Pathway
4.4. Pro-Inflammatory Readouts
4.4.1. Nitric Oxide
4.4.2. Tumor Necrosis Factor-α
4.5. Cytotoxicity Assays
4.5.1. The Alamar Blue (Resazurin) Assay
4.5.2. The MTT Assay
4.5.3. The Trypan Blue Assay
4.5.4. Other Cytotoxicity Assays
5. The Triculture Model for Initial Drug Screening Targeting Neuroinflammation
6. Animal Models of Neuroinflammation
6.1. Rodent Models of Acute Neuroinflammation
6.2. Rodent Models of Chronic Neuroinflammation
6.2.1. Immune Challenged-Based Models
6.2.2. Toxin-Induced Inflammation Models
6.2.3. Transgenic Inflammatory Mouse Models
6.2.4. Validation of the Anti-Inflammatory Effect of Curcumin in the GFAP-Il6 Mouse, a Transgenic Inflammatory Mouse Model
7. Conclusions and Future Studies
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shehjar, F.; Maktabi, B.; Rahman, Z.A.; Bahader, G.A.; James, A.W.; Naqvi, A.; Mahajan, R.; Shah, Z.A. Stroke: Molecular mechanisms and therapies: Update on recent developments. Neurochem. Int. 2023, 162, 105458. [Google Scholar] [CrossRef] [PubMed]
- Ji, B.; Ono, M.; Yamasaki, T.; Fujinaga, M.; Zhang, M.-R.; Seki, C.; Aoki, I.; Kito, S.; Sawada, M.; Suhara, T.; et al. Detection of Alzheimer’s disease-related neuroinflammation by a PET ligand selective for glial versus vascular translocator protein. J. Cereb. Blood Flow Metab. 2021, 41, 2076–2089. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Singh, R.; Ganeshpurkar, A.; Pokle, A.V.; Singh, R.B.; Singh, S.K.; Kumar, A. Cellular and molecular influencers of neuroinflammation in Alzheimer’s disease: Recent concepts & roles. Neurochem. Int. 2021, 151, 105212. [Google Scholar] [PubMed]
- Dai, L.; Shen, Y. Insights into T-cell dysfunction in Alzheimer’s disease. Aging Cell 2021, 20, e13511. [Google Scholar] [CrossRef]
- Muzio, L.; Viotti, A.; Martino, G. Microglia in Neuroinflammation and Neurodegeneration: From Understanding to Therapy. Front. Neurosci. 2021, 15, 742065. [Google Scholar] [CrossRef]
- Mey, G.M.; Mahajan, K.R.; DeSilva, T.M. Neurodegeneration in multiple sclerosis. WIREs Mech. Dis. 2023, 15, e1583. [Google Scholar] [CrossRef]
- Naegele, M.; Martin, R. The good and the bad of neuroinflammation in multiple sclerosis. Handb. Clin. Neurol. 2014, 122, 59–87. [Google Scholar]
- Liu, Y.; Si, Z.-Z.; Zou, C.-J.; Mei, X.; Li, X.-F.; Luo, H.; Shen, Y.; Hu, J.; Li, X.-X.; Wu, L. Targeting neuroinflammation in Alzheimer’s disease: From mechanisms to clinical applications. Neural Regen. Res. 2023, 18, 708. [Google Scholar] [CrossRef]
- Ray, B.; Lahiri, D.K. Neuroinflammation in Alzheimer’s disease: Different molecular targets and potential therapeutic agents including curcumin. Curr. Opin. Pharmacol. 2009, 9, 434–444. [Google Scholar] [CrossRef]
- Wong, C.; Gregory, J.M.; Liao, J.; Egan, K.; Vesterinen, H.M.; Khan, A.A.; Anwar, M.; Beagan, C.; Brown, F.S.; Cafferkey, J.; et al. Systematic, comprehensive, evidence-based approach to identify neuroprotective interventions for motor neuron disease: Using systematic reviews to inform expert consensus. BMJ Open 2023, 13, e064169. [Google Scholar] [CrossRef]
- Town, T. Inflammation, immunity, and Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2010, 9, 129–131. [Google Scholar] [CrossRef] [PubMed]
- Münch, G.; Thome, J.; Foley, P.; Schinzel, R.; Riederer, P. Advanced glycation endproducts in ageing and Alzheimer’s disease. Brain Res. Brain Res. Rev. 1997, 23, 134–143. [Google Scholar] [CrossRef]
- Thameem Dheen, S.; Kaur, C.; Ling, E.-A. Microglial Activation and its Implications in the Brain Diseases. Curr. Med. Chem. 2007, 14, 1189–1197. [Google Scholar] [CrossRef]
- Venegas, C.; Heneka, M.T. Danger-associated molecular patterns in Alzheimer’s disease. J. Leukoc. Biol. 2017, 101, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Zhang, Y.; Huang, Y.; Deng, H. Pathophysiology of RAGE in inflammatory diseases. Front. Immunol. 2022, 13, 931473. [Google Scholar] [CrossRef]
- Münch, G.; Kuhla, B.; Luth, H.J.; Arendt, T.; Robinson, S.R. Anti-AGEing defences against Alzheimer’s disease. Biochem. Soc. Trans. 2003, 31, 1397–1399. [Google Scholar] [CrossRef] [PubMed]
- Thome, J.; Münch, G.; Muller, R.; Schinzel, R.; Kornhuber, J.; Blum-Degen, D.; Sitzmann, L.; Rosler, M.; Heidland, A.; Riederer, P. Advanced glycation endproducts-associated parameters in the peripheral blood of patients with Alzheimer’s disease. Life Sci. 1996, 59, 679–685. [Google Scholar] [CrossRef]
- Thome, J.; Kornhuber, J.; Münch, G.; Schinzel, R.; Taneli, Y.; Zielke, B.; Rosler, M.; Riederer, P. New hypothesis on etiopathogenesis of Alzheimer syndrome. Advanced glycation end products (AGEs). Der. Nervenarzt. 1996, 67, 924–929. [Google Scholar] [CrossRef]
- Yan, S.D.; Chen, X.; Schmidt, A.M.; Brett, J.; Godman, G.; Zou, Y.S.; Scott, C.W.; Caputo, C.; Frappier, T.; Smith, M.A. Glycated tau protein in Alzheimer disease: A mechanism for induction of oxidant stress. Proc. Natl. Acad. Sci. USA 1994, 91, 7787–7791. [Google Scholar] [CrossRef]
- Kimura, T.; Takamatsu, J.; Araki, N.; Goto, M.; Kondo, A.; Miyakawa, T.; Horiuchi, S. Are advanced glycation end-products associated with amyloidosis in Alzheimer’s disease? Neuroreport 1995, 6, 866–868. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Griffin, S.; Münch, G.; Pasinetti, G.M. Amyloid beta-peptide and amyloid pathology are central to the oxidative stress and inflammatory cascades under which Alzheimer’s disease brain exists. J. Alzheimer’s Dis. 2002, 4, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Stuchbury, G.; Münch, G. Alzheimer’s associated inflammation, potential drug targets and future therapies. J. Neural Transm. 2005, 112, 429–453. [Google Scholar] [CrossRef] [PubMed]
- Mrak, R.E.; Griffin, W.S.T. Glia and their cytokines in progression of neurodegeneration. Neurobiol. Aging 2005, 26, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Andronie-Cioara, F.L.; Ardelean, A.I.; Nistor-Cseppento, C.D.; Jurcau, A.; Jurcau, M.C.; Pascalau, N.; Marcu, F. Molecular Mechanisms of Neuroinflammation in Aging and Alzheimer’s Disease Progression. Int. J. Mol. Sci. 2023, 24, 1869. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.; Strauss, S.; Volk, B.; Berger, M. IL-6-mediated events in Alzheimer’s disease pathology. Immunol. Today 1991, 12, 422. [Google Scholar] [CrossRef] [PubMed]
- Singh-Manoux, A.; Dugravot, A.; Brunner, E.; Kumari, M.; Shipley, M.; Elbaz, A.; Kivimaki, M. Interleukin-6 and C-reactive protein as predictors of cognitive decline in late midlife. Neurology 2014, 83, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Wichmann, M.A.; Cruickshanks, K.J.; Carlsson, C.M.; Chappell, R.; Fischer, M.E.; Klein, B.E.K.; Klein, R.; Tsai, M.Y.; Schubert, C.R. Long-Term Systemic Inflammation and Cognitive Impairment in a Population-Based Cohort. J. Am. Geriatr. Soc. 2014, 62, 1683–1691. [Google Scholar] [CrossRef]
- Patel, A.; Rees, S.D.; Kelly, M.A.; Bain, S.C.; Barnett, A.H.; Prasher, A.; Arshad, H.; Prasher, V.P. Genetic variants conferring susceptibility to Alzheimer’s disease in the general population; do they also predispose to dementia in Down’s syndrome. BMC Res. Notes 2014, 7, 42. [Google Scholar] [CrossRef]
- Rodríguez-Giraldo, M.; González-Reyes, R.E.; Ramírez-Guerrero, S.; Bonilla-Trilleras, C.E.; Guardo-Maya, S.; Nava-Mesa, M.O. Astrocytes as a Therapeutic Target in Alzheimer’s Disease–Comprehensive Review and Recent Developments. Int. J. Mol. Sci. 2022, 23, 13630. [Google Scholar] [CrossRef]
- Fuller, S.; Steele, M.; Munch, G. Activated astroglia during chronic inflammation in Alzheimer’s disease—Do they neglect their neurosupportive roles? Mutat. Res. 2010, 690, 40–49. [Google Scholar] [CrossRef]
- Block, M.L.; Zecca, L.; Hong, J.-S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef]
- Münch, G.; Deuther-Conrad, W.; Gasic-Milenkovic, J. Glycoxidative stress creates a vicious cycle of neurodegeneration in Alzheimer’s disease—A target for neuroprotective treatment strategies? J. Neural Transm. Suppl. 2002, 62, 303–307. [Google Scholar]
- Gasic-Milenkovic, J.; Dukic-Stefanovic, S.; Deuther-Conrad, W.; Gartner, U.; Münch, G. beta-Amyloid peptide potentiates inflammatory responses induced by lipopolysaccharide, interferon-gamma and ‘advanced glycation endproducts’ in a murine microglia cell line. Eur. J. Neurosci. 2003, 17, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Childs, R.; Gamage, R.; Münch, G.; Gyengesi, E. The effect of aging and chronic microglia activation on the morphology and numbers of the cerebellar Purkinje cells. Neurosci. Lett. 2021, 751, 135807. [Google Scholar] [CrossRef] [PubMed]
- Chesworth, R.; Gamage, R.; Ullah, F.; Sonego, S.; Millington, C.; Fernandez, A.; Liang, H.; Karl, T.; Münch, G.; Niedermayer, G.; et al. Spatial Memory and Microglia Activation in a Mouse Model of Chronic Neuroinflammation and the Anti-inflammatory Effects of Apigenin. Front. Neurosci. 2021, 15, 699329. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, H.M.; Carl, S.M.; Weber, S.G.; Ramanujan, S.A.; Festoff, B.W.; Linseman, D.A.; Swerdlow, R.H. Mitochondrial lysates induce inflammation and Alzheimer’s disease-relevant changes in microglial and neuronal cells. J. Alzheimer’s Dis. 2015, 45, 305–318. [Google Scholar] [CrossRef]
- Ahmadi, M.; Bekeschus, S.; Weltmann, K.-D.; von Woedtke, T.; Wende, K. Non-steroidal anti-inflammatory drugs: Recent advances in the use of synthetic COX-2 inhibitors. RSC Med. Chem. 2022, 13, 471–496. [Google Scholar] [CrossRef]
- Stewart, W.F.; Kawas, C.; Corrada, M.; Metter, E.J. Risk of Alzheimer’s disease and duration of NSAID use. Neurology 1997, 48, 626–632. [Google Scholar] [CrossRef]
- Wang, J.; Tan, L.; Wang, H.F.; Tan, C.C.; Meng, X.F.; Wang, C.; Tang, S.W.; Yu, J.T. Anti-inflammatory drugs and risk of Alzheimer’s disease: An updated systematic review and meta-analysis. J. Alzheimer’s Dis. 2015, 44, 385–396. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Weggen, S.; Bulic, B.; Multhaup, G.; Munter, L.; Hull, M.; Pflanzner, T.; Pietrzik, C.U. Molecular mechanisms and therapeutic application of NSAIDs and derived compounds in Alzheimer’s disease. Curr. Alzheimer Res. 2011, 8, 115–131. [Google Scholar] [CrossRef]
- Bacchi, S.; Palumbo, P.; Sponta, A.; Coppolino, M. Clinical Pharmacology of Non-Steroidal Anti-Inflammatory Drugs: A Review. Anti-Inflamm. Anti-Allergy Agents Med. Chem. 2012, 11, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Breitner, J.C.; Gau, B.A.; Welsh, K.A.; Plassman, B.L.; McDonald, W.M.; Helms, M.J.; Anthony, J.C. Inverse association of anti-inflammatory treatments and Alzheimer’s disease: Initial results of a co-twin control study. Neurology 1994, 44, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Nerius, M.; Haenisch, B.; Gomm, W.; Doblhammer, G.; Schneider, A. Glucocorticoid Therapy is Associated with a Lower Risk of Dementia. J. Alzheimer’s Dis. 2020, 73, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Beeri, M.S.; Schmeidler, J.; Lesser, G.T.; Maroukian, M.; West, R.; Leung, S.; Wysocki, M.; Perl, D.P.; Purohit, D.P.; Haroutunian, V. Corticosteroids, but not NSAIDs, are associated with less Alzheimer neuropathology. Neurobiol. Aging 2012, 33, 1258–1264. [Google Scholar] [CrossRef]
- Aisen, P.S.; Davis, K.L.; Berg, J.D.; Schafer, K.; Campbell, K.; Thomas, R.G.; Weiner, M.F.; Farlow, M.R.; Sano, M.; Grundman, M.; et al. A randomized controlled trial of prednisone in Alzheimer’s disease. Alzheimer’s Disease Cooperative Study. Neurology 2000, 54, 588–593. [Google Scholar] [CrossRef]
- Villarejo-Galende, A.; Gonzalez-Sanchez, M.; Blanco-Palmero, V.A.; Llamas-Velasco, S.; Benito-Leon, J. Non-steroidal Anti-inflammatory Drugs as Candidates for the Prevention or Treatment of Alzheimer’s Disease: Do they Still Have a Role? Curr. Alzheimer Res. 2020, 17, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Steele, M.; Stuchbury, G.; Münch, G. The molecular basis of the prevention of Alzheimer’s disease through healthy nutrition. Exp. Gerontol. 2007, 42, 28–36. [Google Scholar] [CrossRef]
- Gunawardena, D.; Karunaweera, N.; Lee, S.; van Der Kooy, F.; Harman, D.G.; Raju, R.; Bennett, L.; Gyengesi, E.; Sucher, N.J.; Münch, G. Anti-inflammatory activity of cinnamon (C. zeylanicum and C. cassia) extracts—Identification of E-cinnamaldehyde and o-methoxy cinnamaldehyde as the most potent bioactive compounds. Food Funct. 2015, 6, 910–919. [Google Scholar] [CrossRef]
- Dinarello, C.A. Reduction of inflammation by decreasing production of interleukin-1 or by specific receptor antagonism. Int. J. Tissue React. 1992, 14, 65–75. [Google Scholar]
- Ataseven, A.; Temiz, S.A.; Eren, G.; Özer, I.; Dursun, R. Comparison of anti-TNF and IL-inhibitors treatments in patients with psoriasis in terms of response to routine laboratory parameter dynamics. J. Dermatol. Treat. 2022, 33, 1091–1096. [Google Scholar] [CrossRef]
- McDermott, M. Rilonacept in the treatment of chronic inflammatory disorders. Drugs Today 2009, 45, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Harada, C.; Namekata, K.; Matsuzawa, A.; Camps, M.; Ji, H.; Swinnen, D.; Jorand-Lebrun, C.; Muzerelle, M.; Vitte, P.; et al. Regulation of the severity of neuroinflammation and demyelination by TLR-ASK1-p38 pathway. EMBO Mol. Med. 2010, 2, 504–515. [Google Scholar] [CrossRef]
- Strassburger, M.; Braun, H.; Reymann, K.G. Anti-inflammatory treatment with the p38 mitogen-activated protein kinase inhibitor SB239063 is neuroprotective, decreases the number of activated microglia and facilitates neurogenesis in oxygen-glucose-deprived hippocampal slice cultures. Eur. J. Pharmacol. 2008, 592, 55–61. [Google Scholar] [CrossRef]
- Sun, Y.X.; Dai, D.K.; Liu, R.; Wang, T.; Luo, C.L.; Bao, H.J.; Yang, R.; Feng, X.Y.; Qin, Z.H.; Chen, X.P.; et al. Therapeutic effect of SN50, an inhibitor of nuclear factor-kappaB, in treatment of TBI in mice. Neurol. Sci. 2013, 34, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.S.; Konczak, I.; Zhao, J. Identification and quantification of phenolics in Australian native mint (Mentha australis R. Br.). Food Chem. 2016, 192, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Seididamyeh, M.; Phan, A.D.T.; Sivakumar, D.; Netzel, M.E.; Mereddy, R.; Sultanbawa, Y. Valorisation of Three Underutilised Native Australian Plants: Phenolic and Organic Acid Profiles and In Vitro Antimicrobial Activity. Foods 2023, 12, 623. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.; Dendale, P.; Beelen, M.; Jonkers, R.A.M.; Mullens, A.; Corluy, L.; Meeusen, R.; van Loon, L.J.C. Plasma adipokine and inflammatory marker concentrations are altered in obese, as opposed to non-obese, type 2 diabetes patients. Eur. J. Appl. Physiol. 2010, 109, 397–404. [Google Scholar] [CrossRef]
- Popović, M.; Caballero-Bleda, M.; Benavente-García, O.; Castillo, J. The flavonoid apigenin delays forgetting of passive avoidance conditioning in rats. J. Psychopharmacol. 2014, 28, 498–501. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, J.L.; Liu, R.; Li, X.X.; Li, J.F.; Zhang, L. Neuroprotective, anti-amyloidogenic and neurotrophic effects of apigenin in an Alzheimer’s disease mouse model. Molecules 2013, 18, 9949–9965. [Google Scholar] [CrossRef]
- DeRango-Adem, E.F.; Blay, J. Does Oral Apigenin Have Real Potential for a Therapeutic Effect in the Context of Human Gastrointestinal and Other Cancers? Front. Pharmacol. 2021, 12, 681477. [Google Scholar] [CrossRef]
- Anusha, C.; Sumathi, T.; Joseph, L.D. Protective role of apigenin on rotenone induced rat model of Parkinson’s disease: Suppression of neuroinflammation and oxidative stress mediated apoptosis. Chem. Biol. Interact. 2017, 269, 67–79. [Google Scholar] [CrossRef]
- Bharti, A.C.; Donato, N.; Aggarwal, B.B. Curcumin (Diferuloylmethane) Inhibits Constitutive and IL-6-Inducible STAT3 Phosphorylation in Human Multiple Myeloma Cells. J. Immunol. 2003, 171, 3863–3871. [Google Scholar] [CrossRef] [PubMed]
- Begum, A.N.; Jones, M.R.; Lim, G.P.; Morihara, T.; Kim, P.; Heath, D.D.; Rock, C.L.; Pruitt, M.A.; Yang, F.; Hudspeth, B.; et al. Curcumin structure-function, bioavailability, and efficacy in models of neuroinflammation and Alzheimer’s disease. J. Pharmacol. Exp. Ther. 2008, 326, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Liu, J.; Yu, H.; Wang, Q.; Chen, Y.; Xiang, L. Curcumin promotes nerve regeneration and functional recovery in rat model of nerve crush injury. Neurosci. Lett. 2013, 547, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Quispe, C.; Cruz-Martins, N.; Manca, M.L.; Manconi, M.; Sytar, O.; Hudz, N.; Shanaida, M.; Kumar, M.; Taheri, Y.; Martorell, M.; et al. Nano-Derived Therapeutic Formulations with Curcumin in Inflammation-Related Diseases. Oxidative Med. Cell. Longev. 2021, 2021, 3149223. [Google Scholar] [CrossRef] [PubMed]
- Pennacchio, M.; Kemp, A.S.; Taylor, R.P.; Wickens, K.M.; Kienow, L. Interesting biological activities from plants traditionally used by Native Australians. J. Ethnopharmacol. 2005, 96, 597–601. [Google Scholar] [CrossRef]
- Ngaanyatjarra Pitjantjatjara Yankunytjatjara Women’s Council Aboriginal Corporation. Ngangkari; Magabala Books: Broome, Australia, 2013; p. 272. [Google Scholar]
- McConvell, P.; Thieberger, N.A. Keeping track of Indigenous language endangerment in Australia. In Language Diversity in the Pacific: Endangerment and Survival; Multilingual Matters Ltd.: Bristol, UK, 2006. [Google Scholar]
- Yeshi, K.; Turpin, G.; Jamtsho, T.; Wangchuk, P. Indigenous Uses, Phytochemical Analysis, and Anti-Inflammatory Properties of Australian Tropical Medicinal Plants. Molecules 2022, 27, 3849. [Google Scholar] [CrossRef]
- Palombo, E.A.; Semple, S.J. Antibacterial activity of traditional Australian medicinal plants. J. Ethnopharmacol. 2001, 77, 151–157. [Google Scholar] [CrossRef]
- Guo, Y.; Sakulnarmrat, K.; Konczak, I. Anti-inflammatory potential of native Australian herbs polyphenols. Toxicol. Rep. 2014, 1, 385–390. [Google Scholar] [CrossRef]
- Ali, A.; Bashmil, Y.M.; Cottrell, J.J.; Suleria, H.A.R.; Dunshea, F.R. LC-MS/MS-QTOF Screening and Identification of Phenolic Compounds from Australian Grown Herbs and Their Antioxidant Potential. Antioxidants 2021, 10, 1770. [Google Scholar] [CrossRef]
- Bashmil, Y.M.; Ali, A.; Bk, A.; Dunshea, F.R.; Suleria, H.A.R. Screening and Characterization of Phenolic Compounds from Australian Grown Bananas and Their Antioxidant Capacity. Antioxidants 2021, 10, 1521. [Google Scholar] [CrossRef]
- Akhtar, M.A.; Raju, R.; Beattie, K.D.; Bodkin, F.; Münch, G. Medicinal Plants of the Australian Aboriginal Dharawal People Exhibiting Anti-Inflammatory Activity. Evid.-Based Complement. Altern. Med. 2016, 2016, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Raju, R.; Münch, G. Potential anti-neuroinflammatory compounds from Australian plants—A review. Neurochem. Int. 2021, 142, 104897. [Google Scholar] [CrossRef] [PubMed]
- Carson, C.F.; Hammer, K.A.; Riley, T.V. Melaleuca alternifolia (Tea Tree) Oil: A Review of Antimicrobial and Other Medicinal Properties. Clin. Microbiol. Rev. 2006, 19, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Pazyar, N.; Yaghoobi, R.; Bagherani, N.; Kazerouni, A. A review of applications of tea tree oil in dermatology. Int. J. Dermatol. 2013, 52, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Simpson, B.; Claudie, D.; Smith, N.; Wang, J.; McKinnon, R.; Semple, S. Evaluation of the anti-inflammatory properties of Dodonaea polyandra, a Kaanju traditional medicine. J. Ethnopharmacol. 2010, 132, 340–343. [Google Scholar] [CrossRef]
- Simpson, B.S.; Luo, X.; Wang, J.; Song, Y.; Claudie, D.; Garg, S.; Smith, N.; McKinnon, R.; Semple, S. Development and Evaluation of a Topical Anti-Inflammatory Preparation Containing Dodonaea polyandra Extract. J. Pharm. Pharm. Sci. 2015, 18, 578–599. [Google Scholar] [CrossRef]
- Bodkin, F. Dharawal Pharmacopeia Collection; Western Sydney University: Penrith, Australia, 2021. [Google Scholar]
- Dhakad, A.K.; Pandey, V.V.; Beg, S.; Rawat, J.M.; Singh, A. Biological, medicinal and toxicological significance of Eucalyptus leaf essential oil: A review. J. Sci. Food Agric. 2018, 98, 833–848. [Google Scholar] [CrossRef]
- Akthar, M.A.; Münch, G.; Bodkin, F.; Raju, R. A New Anti-inflammatory Chromone from the leaves of Eucalyptus viminalis. Nat. Prod. Commun. 2018, 13, 1297–1300. [Google Scholar]
- Raju, R.; Singh, A.; Bodkin, F.; Münch, G. Costatamins A–C, new 4-phenylcoumarins with anti-inflammatory activity from the Australian woodland tree Angophora costata (Myrtaceae). Fitoterapia 2019, 133, 171–174. [Google Scholar] [CrossRef]
- Barbosa, L.C.; Filomeno, C.A.; Teixeira, R.R. Chemical Variability and Biological Activities of Eucalyptus spp. Essential Oils. Molecules 2016, 21, 1671. [Google Scholar] [CrossRef] [PubMed]
- Aljaafari, M.N.; AlAli, A.O.; Baqais, L.; Alqubaisy, M.; AlAli, M.; Molouki, A.; Ong-Abdullah, J.; Abushelaibi, A.; Lai, K.-S.; Lim, S.-H.E. An Overview of the Potential Therapeutic Applications of Essential Oils. Molecules 2021, 26, 628. [Google Scholar] [CrossRef] [PubMed]
- Cock, I.E.; Baghtchedjian, L.; Cordon, M.-E.; Dumont, E.J.M. Phytochemistry, Medicinal Properties, Bioactive Compounds, and Therapeutic Potential of the Genus Eremophila (Scrophulariaceae). Molecules 2022, 27, 7734. [Google Scholar] [CrossRef]
- Williams, C.J. Phytochemistry of Australia’s Tropical Rainforest: Medicinal Potential of Ancient Plants; Csiro Publishing: Clayton, Australia, 2021. [Google Scholar]
- Cock, I.E.J.E. Medicinal and Aromatic Plants-Australia; Encyclopedia of Life Support Systems: Brisbane, Australia, 2011; pp. 16–31. [Google Scholar]
- Endt, D.V.; Kijne, J.W.; Memelink, J. Transcription factors controlling plant secondary metabolism: What regulates the regulators? Phytochemistry 2002, 61, 107–114. [Google Scholar] [CrossRef] [PubMed]
- An, M.; Pratley, J. In Searching native Australian plants for natural herbicides-a case study. In Proceedings of the World Congress on Allelopathy, New South Wales, Australia, 21–26 August 2005. [Google Scholar]
- Mani, J.S.; Johnson, J.B.; Hosking, H.; Ashwath, N.; Walsh, K.B.; Neilsen, P.M.; Broszczak, D.A.; Naiker, M. Antioxidative and therapeutic potential of selected Australian plants: A review. J. Ethnopharmacol. 2020, 268, 113580. [Google Scholar] [CrossRef]
- Shanks, G.D. Historical Review: Problematic Malaria Prophylaxis with Quinine. Am. J. Trop. Med. Hyg. 2016, 95, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-B.; Yang, X.-Y.; Du, G.-H. Tubocurarine. In Natural Small Molecule Drugs from Plants; Springer: Berlin/Heidelberg, Germany, 2018; pp. 337–341. [Google Scholar]
- Grinspoon, L.; Bakalar, J.B. Coca and cocaine as medicines: An historical review. J. Ethnopharmacol. 1981, 3, 149–159. [Google Scholar] [CrossRef]
- Boyle, G.M.; D’Souza, M.M.A.; Pierce, C.J.; Adams, R.A.; Cantor, A.S.; Johns, J.P.; Maslovskaya, L.; Gordon, V.A.; Reddell, P.; Parsons, P. Intra-Lesional Injection of the Novel PKC Activator EBC-46 Rapidly Ablates Tumors in Mouse Models. PLoS ONE 2014, 9, e108887. [Google Scholar] [CrossRef]
- Barnett, C.M.E.; Broit, N.; Yap, P.-Y.; Cullen, J.K.; Parsons, P.G.; Panizza, B.J.; Boyle, G.M. Optimising intratumoral treatment of head and neck squamous cell carcinoma models with the diterpene ester Tigilanol tiglate. Investig. New Drugs 2019, 37, 1–8. [Google Scholar] [CrossRef]
- Raju, R.; Mathew, S.; Reddell, P.; Münch, G. Ternstroenol F: A new pentacyclic triterpenoid saponin isolated from the Australian rainforest plant Ternstroemia cherryi. Nat. Prod. Res. 2023, 37, 2421–2426. [Google Scholar] [CrossRef]
- Mathew, S.; Zhang, K.; Zhou, X.; Münch, G.; Bodkin, F.; Li, F.; Raju, R. Myrtinols A–F: New Anti-Inflammatory Peltogynoid Flavonoid Derivatives from the Leaves of Australian Indigenous Plant Backhousia myrtifolia. Molecules 2023, 28, 2160. [Google Scholar] [CrossRef] [PubMed]
- Mathew, S.; Raju, R.; Zhou, X.; Bodkin, F.; Govindaraghavan, S.; Münch, G. A Method and Formula for the Quantitative Analysis of the Total Bioactivity of Natural Products. Int. J. Mol. Sci. 2023, 24, 6850. [Google Scholar] [CrossRef]
- Raju, R.; Mathew, S.; Singh, A.; Reddell, P.; Münch, G. Acronyols A and B, new anti-inflammatory prenylated phloroglucinols from the fruits of Acronychia crassipetala. Nat. Prod. Res. 2022, 36, 4364–4370. [Google Scholar] [CrossRef] [PubMed]
- Mathew, S.; Zhou, X.; Münch, G.; Bodkin, F.; Wallis, M.; Li, F.; Raju, R. Tristaenone A: A New Anti-Inflammatory Compound Isolated from the Australian Indigenous Plant Tristaniopsis laurina. Molecules 2022, 27, 6592. [Google Scholar] [CrossRef]
- del Palacio, J.P.; Díaz, C.; de la Cruz, M.; Annang, F.; Martín, J.; Pérez-Victoria, I.; González-Menéndez, V.; de Pedro, N.; Tormo, J.R.; Algieri, F.; et al. High-throughput screening platform for the discovery of new immunomodulator molecules from natural product extract libraries. J. Biomol. Screen. 2016, 21, 567–578. [Google Scholar] [CrossRef]
- Raju, R.; Gunawardena, D.; Ahktar, M.A.; Low, M.; Reddell, P.; Münch, G. Anti-Inflammatory Chemical Profiling of the Australian Rainforest Tree Alphitonia petriei (Rhamnaceae). Molecules 2016, 21, 1521. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Münch, G.; Reddell, P.; Radzieta, M.; Jensen, S.O.; Raju, R. A New Anti-inflammatory Phenolic Monosaccharide from the Australian Native Rainforest Plant Elaeocarpus Eumundi. Nat. Prod. Commun. 2018, 13, 1934578X1801300619. [Google Scholar] [CrossRef]
- Raju, R.; Cullen, J.K.; Bruce, Z.C.; Reddell, P.; Münch, G.J.F. Eupomatenes A–E: Neolignans isolated from the leaves of Australian rainforest plant Eupomatia laurina. Fitoterapia 2021, 153, 104972. [Google Scholar] [CrossRef]
- Singh, A.; Cullen, J.K.; Bruce, Z.C.; Reddell, P.; Münch, G.; Raju, R.J.P. Ternstroenols A–E: Undescribed pentacyclic triterpenoids from the Australian rainforest plant Ternstroemia cherryi. Phytochemistry 2020, 176, 112426. [Google Scholar] [CrossRef]
- Raju, R.; Gunawardena, D.; Reddell, P.; Münch, G.J.F. Cryptocaryoic acids A–C: New phenyl alkyl acids isolated from the leaves of Australian rainforest plant Cryptocarya mackinnoniana. Fitoterapia 2022, 162, 105266. [Google Scholar] [CrossRef]
- Venigalla, M.; Roberts, T.L.; Raju, R.; Mrad, M.; Bodkin, F.; Kopp, K.; Doyle, K.; Münch, G. Identification of tetragocarbone C and sideroxylin as the most potent anti-inflammatory components of Syncarpia glomulifera. Fitoterapia 2021, 150, 104843. [Google Scholar] [CrossRef] [PubMed]
- Raju, R.; Singh, A.; Gunawardena, D.; Reddell, P.; Münch, G. Mulgravanols A and B, rare oxidized xanthenes and a new phloroglucinol isolated from the Australian rainforest plant Waterhousea mulgraveana (Myrtaceae). Fitoterapia 2020, 143, 104595. [Google Scholar] [CrossRef] [PubMed]
- Raju, R.; Singh, A.; Gunawardena, D.; Reddell, P.; Münch, G. Diarylheptanoids with anti-inflammatory activity from the rhizomes of Pleuranthodium racemigerum (Zingiberaceae). Phytochem. Lett. 2019, 30, 10–13. [Google Scholar] [CrossRef]
- Raju, R.; Singh, A.; Reddell, P.; Münch, G. Anti-inflammatory activity of prenyl and geranyloxy furanocoumarins from Citrus garrawayi (Rutaceae). Phytochem. Lett. 2018, 27, 197–202. [Google Scholar] [CrossRef]
- Elhelu, M.A. The role of macrophages in immunology. J. Natl. Med. Assoc. 1983, 75, 314–317. [Google Scholar]
- Gordon, S.; Martinez-Pomares, L. Physiological roles of macrophages. Pflugers. Arch. 2017, 469, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Johnston, R.B., Jr. Current concepts: Immunology. Monocytes and macrophages. N. Engl. J. Med. 1988, 318, 747–752. [Google Scholar] [CrossRef]
- Cavaillon, J.J.B. Pharmacotherapy, Cytokines and macrophages. Biomed. Pharmacother. 1994, 48, 445–453. [Google Scholar] [CrossRef]
- Mosser, D.M.; Zhang, X. Activation of murine macrophages. Curr. Protoc. Immunol. 2008, 83, 14.2.1–14.2.8. [Google Scholar] [CrossRef]
- Classen, A.; Lloberas, J.; Celada, A. Macrophage activation: Classical vs. alternative. In Macrophages and Dendritic Cells; Springer: Berlin/Heidelberg, Germany, 2009; pp. 29–43. [Google Scholar]
- Eapen, M.S.; Myers, S.; Walters, E.H.; Sohal, S.S. Airway inflammation in chronic obstructive pulmonary disease (COPD): A true paradox. Expert Rev. Respir. Med. 2017, 11, 827–839. [Google Scholar] [CrossRef]
- Gauley, J.; Pisetsky, D.S. The release of microparticles by RAW 264.7 macrophage cells stimulated with TLR ligands. J. Leukoc. Biol. 2010, 87, 1115–1123. [Google Scholar] [CrossRef] [PubMed]
- Pinho, B.R.; Sousa, C.; Valentão, P.; Andrade, P.B. Is Nitric Oxide Decrease Observed with Naphthoquinones in LPS Stimulated RAW 264.7 Macrophages a Beneficial Property? PLoS ONE 2011, 6, e24098. [Google Scholar] [CrossRef] [PubMed]
- Migliorini, P.; Corradin, G.; Corradin, S.B. Macrophage NO2− production as a sensitive and rapid assay for the quantitation of murine IFN-γ. J. Immunol. Methods 1991, 139, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Larrick, J.W.; Fischer, D.G.; Anderson, S.J.; Koren, H.S. Characterization of a human macrophage-like cell line stimulated in vitro: A model of macrophage functions. J. Immunol. 1980, 125, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.; Herant, M.; Dembo, M.; Heinrich, V. Baseline Mechanical Characterization of J774 Macrophages. Biophys. J. 2009, 96, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Amemura, A.; Cabrera-Crespo, J.; Maeda, M.; Futai, M. Changes in morphology of J774.1 macrophage-like cells and release of prostaglandin E2 induced by bacterial acidic polysaccharides. Agric. Biol. Chem. 1987, 51, 2649–2656. [Google Scholar] [CrossRef]
- Fyfe, D.; Abbey, M. Effects of n-3 fatty acids on growth and survival of J774 macrophages. Prostaglandins Leukot. Essent. Fat. Acids 2000, 62, 201–207. [Google Scholar] [CrossRef]
- Tezuka, Y.; Irikawa, S.; Kaneko, T.; Banskota, A.H.; Nagaoka, T.; Xiong, Q.; Hase, K.; Kadota, S. Screening of Chinese herbal drug extracts for inhibitory activity on nitric oxide production and identification of an active compound of Zanthoxylum bungeanum. J. Ethnopharmacol. 2001, 77, 209–217. [Google Scholar] [CrossRef]
- Pacheco-García, U.; Legorreta-HerrEra, M.; Hernández-Rodríguez, C.; Sánchez-García, F.J. Multiple Mycobacterium microti Derived Lipids Stimulate iNOS Gene Expression in the J774 Murine Macrophage Cell Line. Scand. J. Immunol. 2002, 56, 52–58. [Google Scholar] [CrossRef]
- Kreisl, W.C. Microglial Activation and Neuroinflammation. In Hybrid PET/MR Neuroimaging; Springer: Berlin/Heidelberg, Germany, 2022; pp. 191–196. [Google Scholar]
- Polazzi, E.; Monti, B. Microglia and neuroprotection: From in vitro studies to therapeutic applications. Prog. Neurobiol. 2010, 92, 293–315. [Google Scholar] [CrossRef]
- Chen, Z.; Trapp, B.D. Microglia and neuroprotection. J. Neurochem. 2016, 136, 10–17. [Google Scholar] [CrossRef]
- Kierdorf, K.; Prinz, M. Factors regulating microglia activation. Front. Cell. Neurosci. 2013, 7, 44. [Google Scholar] [CrossRef]
- Lue, L.-F.; Kuo, Y.-M.; Beach, T.; Walker, D.G. Microglia Activation and Anti-inflammatory Regulation in Alzheimer’s Disease. Mol. Neurobiol. 2010, 41, 115–128. [Google Scholar] [CrossRef]
- Nakajima, K.; Kohsaka, S. Microglia: Activation and Their Significance in the Central Nervous System. J. Biochem. 2001, 130, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C.; Vilalta, A. How microglia kill neurons. Brain Res. 2015, 1628, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Lull, M.E.; Block, M.L. Microglial activation and chronic neurodegeneration. Neurotherapeutics 2010, 7, 354–365. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Taylor, N.; Yao, X.; Bhattacharya, A. Mouse primary microglia respond differently to LPS and poly(I:C) in vitro. Sci. Rep. 2021, 11, 10447. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Ding, D.-H.; Li, Q.-Q.; Wang, X.-Y.; Sun, Y.-Y.; Li, L.-J. Lipoxin A4 Regulates Lipopolysaccharide-Induced BV2 Microglial Activation and Differentiation via the Notch Signaling Pathway. Front. Cell. Neurosci. 2019, 13, 19. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.-J.; Li, N.; Yu, L.; Chen, Z.-Y.; Hua, R.; Qin, X.; Zhang, Y.-M. Activation of BV2 microglia by lipopolysaccharide triggers an inflammatory reaction in PC12 cell apoptosis through a toll-like receptor 4-dependent pathway. Cell Stress Chaperon. 2015, 20, 321–331. [Google Scholar] [CrossRef]
- Das, A.; Kim, S.H.; Arifuzzaman, S.; Yoon, T.; Chai, J.C.; Lee, Y.S.; Park, K.S.; Jung, K.H.; Chai, Y.G. Transcriptome sequencing reveals that LPS-triggered transcriptional responses in established microglia BV2 cell lines are poorly representative of primary microglia. J. Neuroinflamm. 2016, 13, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, Y.-S.; Choi, D.-H.; Bang, M.S.; Han, T.R.; Joh, T.H.; Kim, S.-Y. Transglutaminase 2 induces nuclear factor-κB activation via a novel pathway in BV-2 microglia. J. Biol. Chem. 2004, 279, 53725–53735. [Google Scholar] [CrossRef] [PubMed]
- Henn, A.; Lund, S.; Hedtjärn, M.; Schrattenholz, A.; Pörzgen, P.; Leist, M. The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. Altex 2009, 26, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.H.; Park, H.Y. Anti-inflammatory effects of spermidine in lipopolysaccharide-stimulated BV2 microglial cells. J. Biomed. Sci. 2012, 19, 31. [Google Scholar] [CrossRef] [PubMed]
- Papageorgiou, I.E.; Lewen, A.; Galow, L.V.; Cesetti, T.; Scheffel, J.; Regen, T.; Hanisch, U.-K.; Kann, O. TLR4-activated microglia require IFN-γ to induce severe neuronal dysfunction and death in situ. Proc. Natl. Acad. Sci. USA 2016, 113, 212–217. [Google Scholar] [CrossRef]
- He, B.P.; Wen, W.; Strong, M.J. Activated microglia (BV-2) facilitation of TNF-α-mediated motor neuron death in vitro. J. Neuroimmunol. 2002, 128, 31–38. [Google Scholar] [CrossRef]
- Li, L.; Lu, J.; Tay, S.S.W.; Moochhala, S.M.; He, B.P. The function of microglia, either neuroprotection or neurotoxicity, is determined by the equilibrium among factors released from activated microglia in vitro. Brain Res. 2007, 1159, 8–17. [Google Scholar] [CrossRef]
- Latta, C.H.; Sudduth, T.L.; Weekman, E.M.; Brothers, H.M.; Abner, E.L.; Popa, G.J.; Mendenhall, M.D.; Gonzalez-Oregon, F.; Braun, K.; Wilcock, D.M. Determining the role of IL-4 induced neuroinflammation in microglial activity and amyloid-β using BV2 microglial cells and APP/PS1 transgenic mice. J. Neuroinflamm. 2015, 12, 41. [Google Scholar] [CrossRef]
- Li, L.; Wu, Y.; Wang, Y.; Wu, J.; Song, L.; Xian, W.; Yuan, S.; Pei, L.; Shang, Y. Resolvin D1 promotes the interleukin-4-induced alternative activation in BV-2 microglial cells. J. Neuroinflamm. 2014, 11, 72. [Google Scholar] [CrossRef]
- Chrysanthou, M.; Estruch, I.M.; Rietjens, I.M.C.M.; Wichers, H.J.; Hoppenbrouwers, T. In Vitro Methodologies to Study the Role of Advanced Glycation End Products (AGEs) in Neurodegeneration. Nutrients 2022, 14, 363. [Google Scholar] [CrossRef]
- Sassano, M.; Granucci, F.; Seveso, M.; Marconi, G.; Foti, M.; Ricciardi-Castagnoli, P. Molecular cloning of a recombinant retrovirus carrying a mutated envAKR-mycMH2 fusion gene immortalizing cells of the monocytic-macrophage lineage. Oncogene 1994, 9, 1473–1477. [Google Scholar]
- Wong, A.; Dukic-Stefanovic, S.; Gasic-Milenkovic, J.; Schinzel, R.; Wiesinger, H.; Riederer, P.; Münch, G. Anti-inflammatory antioxidants attenuate the expression of inducible nitric oxide synthase mediated by advanced glycation endproducts in murine microglia. Eur. J. Neurosci. 2001, 14, 1961–1967. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Zhou, X.; Chang, D.; Bhuyan, D.J.; Zhang, J.P.; Yu, W.; Jiang, X.; Seto, S.W.; Yeon, S.Y.; Li, J.; et al. A novel tri-culture model for neuroinflammation. J. Neurochem. 2021, 156, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Münch, G.; Gasic-Milenkovic, J.; Dukic-Stefanovic, S.; Kuhla, B.; Heinrich, K.; Riederer, P.; Huttunen, H.; Founds, H.; Sajithlal, G. Microglial activation induces cell death, inhibits neurite outgrowth and causes neurite retraction of differentiated neuroblastoma cells. Exp. Brain Res. 2003, 150, 1–8. [Google Scholar] [CrossRef]
- Münch, G.; Apelt, J.; Rosemarie-Kientsch-Engel; Stahl, P.; Lüth, H.J.; Schliebs, R. Advanced glycation endproducts and pro-inflammatory cytokines in transgenic Tg2576 mice with amyloid plaque pathology. J. Neurochem. 2003, 86, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Allendorf, D.H.; Franssen, E.H.; Brown, G.C. Lipopolysaccharide activates microglia via neuraminidase 1 desialylation of Toll-like Receptor 4. J. Neurochem. 2020, 155, 403–416. [Google Scholar] [CrossRef]
- Berbaum, K.; Shanmugam, K.; Stuchbury, G.; Wiede, F.; Körner, H.; Münch, G. Induction of novel cytokines and chemokines by advanced glycation endproducts determined with a cytometric bead array. Cytokine 2008, 41, 198–203. [Google Scholar] [CrossRef]
- Pestka, S.; Krause, C.D.; Walter, M.R. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 2004, 202, 8–32. [Google Scholar] [CrossRef]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef]
- Rauch, I.; Müller, M.; Decker, T. The regulation of inflammation by interferons and their STATs. Jak-Stat 2013, 2, e23820. [Google Scholar] [CrossRef]
- Teixeira, L.K.; Fonseca, B.P.; Barboza, B.A.; Viola, J.P. The role of interferon-gamma on immune and allergic responses. Memórias Inst. Oswaldo Cruz 2005, 100, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Mühl, H.; Pfeilschifter, J. Anti-inflammatory properties of pro-inflammatory interferon-γ. Int. Immunopharmacol. 2003, 3, 1247–1255. [Google Scholar] [CrossRef]
- Jorgovanovic, D.; Song, M.; Wang, L.; Zhang, Y. Roles of IFN-γ in tumor progression and regression: A review. Biomark. Res. 2020, 8, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Ivashkiv, L.B. Cross-regulation of Signaling Pathways by Interferon-γ: Implications for Immune Responses and Autoimmune Diseases. Immunity 2009, 31, 539–550. [Google Scholar] [CrossRef]
- Kak, G.; Raza, M.; Tiwari, B.K. Interferon-gamma (IFN-γ): Exploring its implications in infectious diseases. Biomol. Concepts 2018, 9, 64–79. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-C.; Yeh, W.-C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef]
- Zong, Y.; Sun, L.; Liu, B.; Deng, Y.-S.; Zhan, D.; Chen, Y.-L.; He, Y.; Liu, J.; Zhang, Z.-J.; Sun, J.; et al. Resveratrol Inhibits LPS-Induced MAPKs Activation via Activation of the Phosphatidylinositol 3-Kinase Pathway in Murine RAW 264.7 Macrophage Cells. PLoS ONE 2012, 7, e44107. [Google Scholar] [CrossRef]
- Peri, F.; Piazza, M.; Calabrese, V.; Damore, G.; Cighetti, R. Exploring the LPS/TLR4 signal pathway with small molecules. Biochem. Soc. Trans. 2010, 38, 1390–1395. [Google Scholar] [CrossRef]
- Diogenes, A.; Ferraz, C.; Akopian, A.; Henry, M.; Hargreaves, K. LPS Sensitizes TRPV1 via Activation of TLR4 in Trigeminal Sensory Neurons. J. Dent. Res. 2011, 90, 759–764. [Google Scholar] [CrossRef]
- Guha, M.; Mackman, N. LPS induction of gene expression in human monocytes. Cell. Signal. 2001, 13, 85–94. [Google Scholar] [CrossRef]
- Shi, Y.; Tu, Z.; Tang, D.; Zhang, H.; Liu, M.; Wang, K.; Calderwood, S.K.; Xiao, X. The inhibition of LPS-induced production of inflammatory cytokines by HSP70 involves inactivation of the NF-κB pathway but not the MAPK pathways. Shock 2006, 26, 277–284. [Google Scholar] [CrossRef]
- Senftleben, U.; Karin, M. The Ikk/nf-κb pathway. Crit. Care Med. 2002, 30, S18–S26. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, P.; Tripathi, P.; Kashyap, L.; Singh, V.J.F.I.; Microbiology, M. The role of nitric oxide in inflammatory reactions. FEMS Immunol. Med. Microbiol. 2007, 51, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Nathan, S.B.; Bryan, N.S.; Bian, K.; Murad, F. Discovery of the nitric oxide signaling pathway and targets for drug development. Front. Biosci. 2009, 14, 1–18. [Google Scholar] [CrossRef]
- Moilanen, E.; Vapaatalo, H. Nitric Oxide in Inflammation and Immune Response. Ann. Med. 1995, 27, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Sharma, J.N.; Al-Omran, A.; Parvathy, S.S. Role of nitric oxide in inflammatory diseases. Inflammopharmacology 2007, 15, 252–259. [Google Scholar] [CrossRef]
- Laskin, J.D.; Heck, D.E.; Laskin, D.L. Multifunctional role of nitric oxide in inflammation. Trends Endocrinol. Metab. 1994, 5, 377–382. [Google Scholar] [CrossRef]
- Wiart, C. Ethnopharmacology of Medicinal Plants: Asia and the Pacific; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2007. [Google Scholar]
- Kopincová, J.; Púzserová, A.; Bernátová, I. Biochemical aspects of nitric oxide synthase feedback regulation by nitric oxide. Interdiscip. Toxicol. 2011, 4, 63–68. [Google Scholar] [CrossRef]
- Schmölz, L.; Wallert, M.; Lorkowski, S. Optimized incubation regime for nitric oxide measurements in murine macrophages using the Griess assay. J. Immunol. Methods 2017, 449, 68–70. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, X.; Broderick, M.; Fein, H. Measurement of Nitric Oxide Production in Biological Systems by Using Griess Reaction Assay. Sensors 2003, 3, 276–284. [Google Scholar] [CrossRef]
- Baud, V.; Karin, M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 2001, 11, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Zelová, H.; Hošek, J. TNF-α signalling and inflammation: Interactions between old acquaintances. Inflamm. Res. 2013, 62, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Feuerstein, G.Z.; Liu, T.; Barone, F.C. Cytokines, inflammation, and brain injury: Role of tumor necrosis factor-alpha. Cerebrovasc. Brain Metab. Rev. 1994, 6, 341–360. [Google Scholar]
- Esposito, E.; Cuzzocrea, S. TNF-alpha as a therapeutic target in inflammatory diseases, ischemia-reperfusion injury and trauma. Curr. Med. Chem. 2009, 16, 3152–3167. [Google Scholar] [CrossRef] [PubMed]
- Corti, A.; Poiesi, C.; Merli, S.; Cassani, G. Tumor Necrosis Factor (TNF) α quantification by ELISA and bioassay: Effects of TNFα-soluble TNF receptor (p55) complex dissociation during assay incubations. J. Immunol. Methods 1994, 177, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Hamid, R.; Rotshteyn, Y.; Rabadi, L.; Parikh, R.; Bullock, P. Comparison of alamar blue and MTT assays for high through-put screening. Toxicol. Vitr. 2004, 18, 703–710. [Google Scholar] [CrossRef]
- Mikus, J.; Steverding, D. A simple colorimetric method to screen drug cytotoxicity against Leishmania using the dye Alamar Blue®. Parasitol. Int. 1999, 48, 265–269. [Google Scholar] [CrossRef]
- Nociari, M.M.; Shalev, A.; Benias, P.; Russo, C. A novel one-step, highly sensitive fluorometric assay to evaluate cell-mediated cytotoxicity. J. Immunol. Methods 1998, 213, 157–167. [Google Scholar] [CrossRef]
- Tolosa, L.; Donato, M.T.; Gómez-Lechón, M.J. General cytotoxicity assessment by means of the MTT assay. In Protocols in In Vitro Hepatocyte Research; Springer: Berlin/Heidelberg, Germany, 2015; pp. 333–348. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- van de Loosdrecht, A.; Beelen, R.; Ossenkoppele, G.; Broekhoven, M.; Langenhuijsen, M. A tetrazolium-based colorimetric MTT assay to quantitate human monocyte mediated cytotoxicity against leukemic cells from cell lines and patients with acute myeloid leukemia. J. Immunol. Methods 1994, 174, 311–320. [Google Scholar] [CrossRef]
- Larramendy, M.; Soloneski, S. Genotoxicity: A Predictable Risk to Our Actual World; BoD–Books on Demand: London, UK, 2018. [Google Scholar]
- Avelar-Freitas, B.; Almeida, V.; Pinto, M.C.X.; Mourão, F.; Massensini, A.; Martins-Filho, O.A.; Rocha-Vieira, E.; Brito-Melo, G. Trypan blue exclusion assay by flow cytometry. Braz. J. Med. Biol. Res. 2014, 47, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Strober, W. Trypan Blue Exclusion Test of Cell Viability. Curr. Protoc. Immunol. 1997, 21, A.3B.1–A.3B.2. [Google Scholar] [CrossRef]
- Tennant, J.R. Evaluation of the trypan blue technique for determination of cell viability. Transplantation 1964, 2, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Bigl, K.; Schmitt, A.; Meiners, I.; Münch, G.; Arendt, T. Comparison of results of the CellTiter Blue, the tetrazolium (3-[4,5-dimethylthioazol-2-yl]-2,5-diphenyl tetrazolium bromide), and the lactate dehydrogenase assay applied in brain cells after exposure to advanced glycation endproducts. Toxicol. Vitr. 2007, 21, 962–971. [Google Scholar] [CrossRef]
- Chao, F.C. Determination of platelet viability. The physiological and biochemical parameters. Vox Sang. 1971, 20, 428–432. [Google Scholar] [CrossRef]
- Zhang, S.-Z.; Lipsky, M.; Trump, B.; Hsu, I.-C. Neutral red (NR) assay for cell viability and xenobiotic-induced cytotoxicity in primary cultures of human and rat hepatocytes. Cell Biol. Toxicol. 1990, 6, 219–234. [Google Scholar] [CrossRef]
- Hansen, E.; Krautwald, M.; Maczurek, A.E.; Stuchbury, G.; Fromm, P.; Steele, M.; Schulz, O.; Garcia, O.B.; Castillo, J.; Körner, H.; et al. A Versatile High Throughput Screening System for the Simultaneous Identification of Anti-Inflammatory and Neuroprotective Compounds. J. Alzheimer’s Dis. 2010, 19, 451–464. [Google Scholar] [CrossRef]
- Banks, W.A.; Gray, A.M.; Erickson, M.A.; Salameh, T.S.; Damodarasamy, M.; Sheibani, N.; Meabon, J.S.; Wing, E.E.; Morofuji, Y.; Cook, D.G.; et al. Lipopolysaccharide-induced blood-brain barrier disruption: Roles of cyclooxygenase, oxidative stress, neuroinflammation, and elements of the neurovascular unit. J. Neuroinflamm. 2015, 12, 223. [Google Scholar] [CrossRef]
- Banks, W.A.; Kastin, A.J.; Broadwell, R.D. Passage of Cytokines across the Blood-Brain Barrier. Neuroimmunomodulation 1995, 2, 241–248. [Google Scholar] [CrossRef]
- Nazem, A.; Sankowski, R.; Bacher, M.; Al-Abed, Y. Rodent models of neuroinflammation for Alzheimer’s disease. J. Neuroinflamm. 2015, 12, 1–15. [Google Scholar] [CrossRef]
- Peng, G.-S.; Li, G.; Tzeng, N.-S.; Chen, P.-S.; Chuang, D.-M.; Hsu, Y.-D.; Yang, S.; Hong, J.-S. Valproate pretreatment protects dopaminergic neurons from LPS-induced neurotoxicity in rat primary midbrain cultures: Role of microglia. Mol. Brain Res. 2005, 134, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Liu, Z.; Ren, W.; Jiang, W. Acute lipopolysaccharide exposure facilitates epileptiform activity via enhanced excitatory synaptic transmission and neuronal excitability in vitro. Neuropsychiatr. Dis. Treat. 2014, 10, 1489–1495. [Google Scholar] [CrossRef] [PubMed]
- Gullo, F.; Amadeo, A.; Donvito, G.; Lecchi, M.; Costa, B.; Constanti, A.; Wanke, E. Atypical “seizure-like” activity in cortical reverberating networks in vitro can be caused by LPS-induced inflammation: A multi-electrode array study from a hundred neurons. Front. Cell. Neurosci. 2014, 8, 361. [Google Scholar] [CrossRef]
- Godbout, J.P.; Chen, J.; Abraham, J.; Richwine, A.F.; Berg, B.M.; Kelley, K.W.; Johnson, R.W. Exaggerated neuroinflammation and sickness behavior in aged mice after activation of the peripheral innate immune system. FASEB J. 2005, 19, 1329–1331. [Google Scholar] [CrossRef] [PubMed]
- Godbout, J.P.; Moreau, M.; Lestage, J.; Chen, J.; Sparkman, N.L.; Connor, J.O.; Castanon, N.; Kelley, K.W.; Dantzer, R.; Johnson, R.W. Aging Exacerbates Depressive-like Behavior in Mice in Response to Activation of the Peripheral Innate Immune System. Neuropsychopharmacology 2008, 33, 2341–2351. [Google Scholar] [CrossRef]
- Chen, J.; Buchanan, J.B.; Sparkman, N.L.; Godbout, J.P.; Freund, G.G.; Johnson, R.W. Neuroinflammation and disruption in working memory in aged mice after acute stimulation of the peripheral innate immune system. Brain Behav. Immun. 2008, 22, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Henry, C.J.; Huang, Y.; Wynne, A.; Hanke, M.; Himler, J.; Bailey, M.T.; Sheridan, J.F.; Godbout, J.P. Minocycline attenuates lipopolysaccharide (LPS)-induced neuroinflammation, sickness behavior, and anhedonia. J. Neuroinflamm. 2008, 5, 15. [Google Scholar] [CrossRef]
- Henry, C.J.; Huang, Y.; Wynne, A.M.; Godbout, J.P. Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1β and anti-inflammatory IL-10 cytokines. Brain Behav. Immun. 2009, 23, 309–317. [Google Scholar] [CrossRef]
- Chugh, D.; Nilsson, P.; Afjei, S.-A.; Bakochi, A.; Ekdahl, C.T. Brain inflammation induces post-synaptic changes during early synapse formation in adult-born hippocampal neurons. Exp. Neurol. 2013, 250, 176–188. [Google Scholar] [CrossRef]
- Noh, H.; Jeon, J.; Seo, H. Systemic injection of LPS induces region-specific neuroinflammation and mitochondrial dysfunction in normal mouse brain. Neurochem. Int. 2014, 69, 35–40. [Google Scholar] [CrossRef]
- Qiao, X.; Cummins, D.J.; Paul, S.M. Neuroinflammation-induced acceleration of amyloid deposition in the APPV717F transgenic mouse. Eur. J. Neurosci. 2001, 14, 474–482. [Google Scholar] [CrossRef]
- Qin, L.; Wu, X.; Block, M.L.; Liu, Y.; Breese, G.R.; Hong, J.-S.; Knapp, D.J.; Crews, F.T. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 2007, 55, 453–462. [Google Scholar] [CrossRef]
- Lee, J.W.; Lee, Y.K.; Yuk, D.Y.; Choi, D.Y.; Ban, S.B.; Oh, K.W.; Hong, J.T. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J. Neuroinflamm. 2008, 5, 37. [Google Scholar] [CrossRef]
- Kékesi, O.; Liang, H.; Münch, G.; Morley, J.W.; Gyengesi, E.; Buskila, Y. The differential impact of acute microglia activation on the excitability of cholinergic neurons in the mouse medial septum. Anat. Embryol. 2019, 224, 2297–2309. [Google Scholar] [CrossRef]
- Millington, C.; Sonego, S.; Karunaweera, N.; Rangel, A.; Aldrich-Wright, J.; Campbell, I.L.; Gyengesi, E.; Münch, G. Chronic Neuroinflammation in Alzheimer’s Disease: New Perspectives on Animal Models and Promising Candidate Drugs. BioMed Res. Int. 2014, 2014, 1–10. [Google Scholar] [CrossRef]
- Lee, B.; Shim, I.; Lee, H. Gypenosides Attenuate Lipopolysaccharide-Induced Neuroinflammation and Memory Impairment in Rats. Evid.-Based Complement. Altern. Med. 2018, 2018, 1–10. [Google Scholar] [CrossRef]
- Zhao, J.; Bi, W.; Xiao, S.; Lan, X.; Cheng, X.; Zhang, J.; Lu, D.; Wei, W.; Wang, Y.; Li, H.; et al. Neuroinflammation induced by lipopolysaccharide causes cognitive impairment in mice. Sci. Rep. 2019, 9, 5790. [Google Scholar] [CrossRef]
- González-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sánchez, K.; Ariza-Salamanca, D.; Mora-Muñoz, L. Involvement of astrocytes in Alzheimer’s disease from a neuroinflammatory and oxidative stress perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef]
- De Miranda, J.; Yaddanapudi, K.; Hornig, M.; Villar, G.; Serge, R.; Lipkin, W.I. Induction of Toll-Like Receptor 3-Mediated Immunity during Gestation Inhibits Cortical Neurogenesis and Causes Behavioral Disturbances. mBio 2010, 1, e00176-10. [Google Scholar] [CrossRef]
- Krstic, D.; Madhusudan, A.; Doehner, J.; Vogel, P.; Notter, T.; Imhof, C.; Manalastas, A.; Hilfiker, M.; Pfister, S.; Schwerdel, C.; et al. Systemic immune challenges trigger and drive Alzheimer-like neuropathology in mice. J. Neuroinflamm. 2012, 9, 151. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Sawikr, Y.; Yarla, N.S.; Peluso, I.; Kamal, M.A.; Aliev, G.; Bishayee, A. Neuroinflammation in Alzheimer’s disease: The preventive and therapeutic potential of polyphenolic nutraceuticals. Adv. Protein Chem. Struct. Biol. 2017, 108, 33–57. [Google Scholar] [PubMed]
- Liddelow, S.A.; Sofroniew, M.V. Astrocytes usurp neurons as a disease focus. Nat. Neurosci. 2019, 22, 512–513. [Google Scholar] [CrossRef] [PubMed]
- Town, T.; Jeng, D.; Alexopoulou, L.; Tan, J.; Flavell, R.A. Microglia Recognize Double-Stranded RNA via TLR3. J. Immunol. 2006, 176, 3804–3812. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-Q.; Yin, J.; Song, Y.-F.; Zhang, L.; Ren, Y.-X.; Wang, D.-G.; Gao, L.-P.; Jing, Y.-H. Brain Aging and AD-Like Pathology in Streptozotocin-Induced Diabetic Rats. J. Diabetes Res. 2014, 2014, 1–12. [Google Scholar] [CrossRef]
- Kraska, A.; Santin, M.D.; Dorieux, O.; Joseph-Mathurin, N.; Bourrin, E.; Petit, F.; Jan, C.; Chaigneau, M.; Hantraye, P.; Lestage, P.; et al. In Vivo Cross-sectional Characterization of Cerebral Alterations Induced by Intracerebroventricular Administration of Streptozotocin. PLoS ONE 2012, 7, e46196. [Google Scholar] [CrossRef]
- Sil, S.; Ghosh, R.; Sanyal, M.; Guha, D.; Ghosh, T. A comparison of neurodegeneration linked with neuroinflammation in different brain areas of rats after intracerebroventricular colchicine injection. J. Immunotoxicol. 2016, 13, 181–190. [Google Scholar] [CrossRef]
- Shaftel, S.S.; Kyrkanides, S.; Olschowka, J.A.; Miller, J.-N.H.; Johnson, R.E.; O’banion, M.K. Sustained hippocampal IL-1β overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J. Clin. Investig. 2007, 117, 1595–1604. [Google Scholar] [CrossRef]
- Matousek, S.B.; Ghosh, S.; Shaftel, S.S.; Kyrkanides, S.; Olschowka, J.A.; O’banion, M.K. Chronic IL-1β-Mediated Neuroinflammation Mitigates Amyloid Pathology in a Mouse Model of Alzheimer’s Disease without Inducing Overt Neurodegeneration. J. Neuroimmune Pharmacol. 2011, 7, 156–164. [Google Scholar] [CrossRef]
- Geula, C.; Mesulam, M.-M. Cortical cholinergic fibers in aging and Alzheimer’s disease: A morphometric study. Neuroscience 1989, 33, 469–481. [Google Scholar] [CrossRef]
- Tago, H.; McGeer, P.; McGeer, E. Acetylcholinesterase fibers and the development of senile plaques. Brain Res. 1987, 406, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, J.R.; Chan, E.S.; Poore, C.P.; Pareek, T.K.; Cheong, W.F.; Shui, G.; Tang, N.; Low, C.-M.; Wenk, M.R.; Kesavapany, S. Cdk5/p25-induced cytosolic PLA2-mediated lysophosphatidylcholine production regulates neuroinflammation and triggers neurodegeneration. J. Neurosci. 2012, 32, 1020–1034. [Google Scholar] [CrossRef] [PubMed]
- Patrick, G.N.; Zukerberg, L.; Nikolic, M.; De La Monte, S.; Dikkes, P.; Tsai, L.-H. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 1999, 402, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Giese, K.P. Generation of the Cdk5 activator p25 is a memory mechanism that is affected in early Alzheimer’s disease. Front. Mol. Neurosci. 2014, 7, 36. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Sananbenesi, F.; Pang, P.T.; Lu, B.; Tsai, L.-H. Opposing Roles of Transient and Prolonged Expression of p25 in Synaptic Plasticity and Hippocampus-Dependent Memory. Neuron 2005, 48, 825–838. [Google Scholar] [CrossRef] [PubMed]
- Rothaug, M.; Becker-Pauly, C.; Rose-John, S. The role of interleukin-6 signaling in nervous tissue. Biochim. Biophys. Acta BBA -Mol. Cell Res. 2016, 1863, 1218–1227. [Google Scholar] [CrossRef]
- Campbell, I.L.; Abraham, C.R.; Masliah, E.; Kemper, P.; Inglis, J.D.; Oldstone, M.B.; Mucke, L. Neurologic disease induced in transgenic mice by cerebral overexpression of interleukin 6. Proc. Natl. Acad. Sci. USA 1993, 90, 10061–10065. [Google Scholar] [CrossRef]
- Vallières, L.; Campbell, I.L.; Gage, F.H.; Sawchenko, P.E. Reduced Hippocampal Neurogenesis in Adult Transgenic Mice with Chronic Astrocytic Production of Interleukin-6. J. Neurosci. 2002, 22, 486–492. [Google Scholar] [CrossRef]
- Quintana, A.; Muüller, M.; Frausto, R.F.; Ramos, R.; Getts, D.R.; Sanz, E.; Hofer, M.J.; Krauthausen, M.; King, N.J.C.; Hidalgo, J.; et al. Site-Specific Production of IL-6 in the Central Nervous System Retargets and Enhances the Inflammatory Response in Experimental Autoimmune Encephalomyelitis. J. Immunol. 2009, 183, 2079–2088. [Google Scholar] [CrossRef]
- Gyengesi, E.; Rangel, A.; Ullah, F.; Liang, H.; Niedermayer, G.; Asgarov, R.; Venigalla, M.; Gunawardena, D.; Karl, T.; Münch, G. Chronic Microglial Activation in the GFAP-IL6 Mouse Contributes to Age-Dependent Cerebellar Volume Loss and Impairment in Motor Function. Front. Neurosci. 2019, 13, 303. [Google Scholar] [CrossRef]
- Campbell, I.L.; Hofer, M.J.; Pagenstecher, A. Transgenic models for cytokine-induced neurological disease. Biochim. Biophys. Acta BBA -Mol. Basis Dis. 2010, 1802, 903–917. [Google Scholar] [CrossRef] [PubMed]
- Heyser, C.J.; Masliah, E.; Samimi, A.; Campbell, I.L.; Gold, L.H. Progressive decline in avoidance learning paralleled by inflammatory neurodegeneration in transgenic mice expressing interleukin 6 in the brain. Proc. Natl. Acad. Sci. USA 1997, 94, 1500–1505. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.L.; Erta, M.; Lim, S.L.; Frausto, R.; May, U.; Rose-John, S.; Scheller, J.; Hidalgo, J. Trans-Signaling Is a Dominant Mechanism for the Pathogenic Actions of Interleukin-6 in the Brain. J. Neurosci. 2014, 34, 2503–2513. [Google Scholar] [CrossRef] [PubMed]
- Goel, A.; Jhurani, S.; Aggarwal, B.B. Multi-targeted therapy by curcumin: How spicy is it? Mol. Nutr. Food Res. 2008, 52, 1010–1030. [Google Scholar] [CrossRef] [PubMed]
- Goel, A.; Aggarwal, B.B. Curcumin, the Golden Spice from Indian Saffron, Is a Chemosensitizer and Radiosensitizer for Tumors and Chemoprotector and Radioprotector for Normal Organs. Nutr. Cancer 2010, 62, 919–930. [Google Scholar] [CrossRef]
- Storka, A.; Vcelar, B.; Klickovic, U.; Gouya, G.; Weisshaar, S.; Aschauer, S.; Bolger, G.; Helson, L.; Woltz, M. Safety, tolerability and pharmacokinetics of liposomal curcumin (Lipocurc™) in healthy humans. Int. J. Clin. Pharmacol. Ther. 2015, 53, 54–65. [Google Scholar] [CrossRef]
- Panahi, Y.; Kianpour, P.; Mohtashami, R.; Jafari, R.; Simental-Mendía, L.E.; Sahebkar, A. Efficacy and Safety of Phytosomal Curcumin in Non-Alcoholic Fatty Liver Disease: A Randomized Controlled Trial. Drug Res. 2017, 67, 244–251. [Google Scholar] [CrossRef]
- Qin, S.; Huang, L.; Gong, J.; Shen, S.; Huang, J.; Ren, H.; Hu, H. Efficacy and safety of turmeric and curcumin in lowering blood lipid levels in patients with cardiovascular risk factors: A meta-analysis of randomized controlled trials. Nutr. J. 2017, 16, 1–10. [Google Scholar] [CrossRef]
- Tsai, Y.-M.; Chien, C.-F.; Lin, L.-C.; Tsai, T.-H. Curcumin and its nano-formulation: The kinetics of tissue distribution and blood–brain barrier penetration. Int. J. Pharm. 2011, 416, 331–338. [Google Scholar] [CrossRef]
- Ma, Q.-L.; Zuo, X.; Yang, F.; Ubeda, O.J.; Gant, D.J.; Alaverdyan, M.; Teng, E.; Hu, S.; Chen, P.-P.; Maiti, P.; et al. Curcumin Suppresses Soluble Tau Dimers and Corrects Molecular Chaperone, Synaptic, and Behavioral Deficits in Aged Human Tau Transgenic Mice. J. Biol. Chem. 2013, 288, 4056–4065. [Google Scholar] [CrossRef]
- Ullah, F.; Asgarov, R.; Venigalla, M.; Liang, H.; Niedermayer, G.; Münch, G.; Gyengesi, E. Effects of a solid lipid curcumin particle formulation on chronic activation of microglia and astroglia in the GFAP-IL6 mouse model. Sci. Rep. 2020, 10, 2365. [Google Scholar] [CrossRef] [PubMed]
- Ullah, F.; Liang, H.; Niedermayer, G.; Münch, G.; Gyengesi, E. Evaluation of Phytosomal Curcumin as an Anti-inflammatory Agent for Chronic Glial Activation in the GFAP-IL6 Mouse Model. Front. Neurosci. 2020, 14, 170. [Google Scholar] [CrossRef] [PubMed]



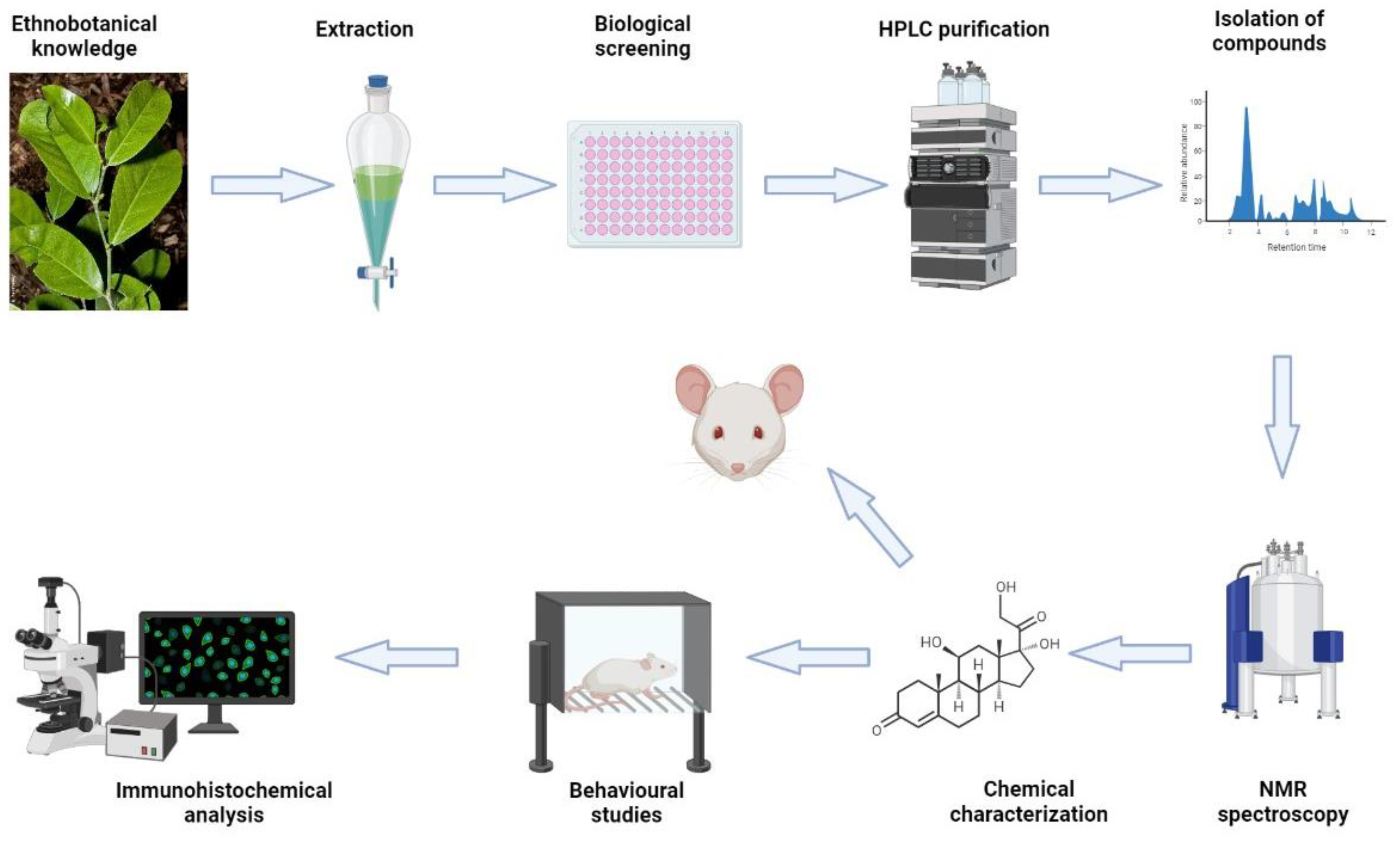{kind=link}
{kind=link}
| Plant Species | Constituent(s) Identified | IC50 in μM (NO Inhibition) | IC50 in μM (TNF-α Inhibition) | Cytotoxicity/LC50 in μM | Therapeutic Index (TI) (Compared to NO) | Reference |
|---|---|---|---|---|---|---|
| Alphitonia petriei (Rhamnaceae) | trans- coumaroyl ester of alphitolic acid | 1.7 | 10.9 | 4.8 | 2.8 | [103] |
| cis-coumaroyl esters of alphitolic acid | 3.5 | 5.6 | 8.0 | 2.3 | ||
| Elaeocarpus Eumundi (Elaeocarpaceae) | New phenolic monosaccharide related to pieceid-2-Ogallate | 22.6 | 39.7 | >165.4 | >7.3 | [104] |
| dihydropieceid | 73.4 | 25.6 | >230.2 | >3.1 | ||
| Eupomatia laurina (Eupomatiaceae) | eupomatenoid-2 | 28.2 | 57.4 | 21.9 | 0.8 | [105] |
| Eupomatene 4 | 32.7 | 44.1 | 38.8 | 1.2 | ||
| Ternstroemia cherryi (Theaceae) | Ternstroenol B | 0.7 | 2.1 | 2.3 | 3.2 | [106] |
| Ternstroenol D | 0.9 | 3.8 | 3.2 | 3.5 | ||
| Cryptocarya mackinnoniana (Lauraceae) | Cryptocaryoic acid A | 18.4 | >100 | >100 | >5.4 | [107] |
| Cryptocaryoic acid B | 27.6 | >100 | >100 | >3.6 | ||
| Tristaniopsis laurina (Myrtaceae) | Tristaenone A | 37.6 | 80.6 | >250 | >6.7 | [101] |
| 8-desmethyleucalyptin | 16.2 | 46.6 | >250 | >15.1 | ||
| Backhousia mytifolia (Myrtaceae) | Myrtinol A | 11.5 | 24.5 | 18.8 | 1.6 | [98] |
| Myrtinol E | 8.5 | 17.2 | 15.5 | 1.8 | ||
| Syncarpia glomulifera (Myrtaceae) | Sideroxylin | 2.3 | 20.8 | 19.4 | 8.4 | [108] |
| Tetragocarbone C | 3.9 | 16.9 | 18.4 | 4.7 | ||
| Waterhousia Mulgraveana (Myrtaceae) | Mulgravanol B | 42.0 | 140.8 | 202.0 | 4.8 | [109] |
| Mulgravanol C | 35.2 | 128.0 | 135.0 | 3.8 | ||
| Pleuranthodium racemigerum (Zingiberaceae) | 1-(4″-Methoxyphenyl)-7-(3′,4′-di-hydroxyphenyl)-(E)-hept-2-ene | 25.1 | 15.9 | 78.8 | 3.1 | [110] |
| 1-(4″-Methoxyphenyl)-7-(3′,4′-di-methoxyphenyl)-(E)-hept-2-ene | 28.3 | 5.3 | 56.2 | 2.0 | ||
| Acronychia crassipetala (Rutaceae) | Acronyol A | 31.4 | 45.3 | 108.0 | 3.4 | [100] |
| Acronyol B | 55.4 | 82.6 | 114.0 | 2.1 | ||
| Angophora costata (Myrtaceae) | Costatamin A | 89.5 | 91.1 | >115.0 | >1.3 | [83] |
| Costatamin B | 67.7 | 115.0 | >115.0 | >1.7 | ||
| Citrus garrawayi (Rutaceae) | Garracoumarin C | 44.0 | 15.4 | <90 | <2.1 | [111] |
| Garracoumarin D | 53.9 | <90 | <90 | <1.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, P.; Mathew, S.; Gamage, R.; Bodkin, F.; Doyle, K.; Rossetti, I.; Wagnon, I.; Zhou, X.; Raju, R.; Gyengesi, E.; et al. From the Bush to the Brain: Preclinical Stages of Ethnobotanical Anti-Inflammatory and Neuroprotective Drug Discovery—An Australian Example. Int. J. Mol. Sci. 2023, 24, 11086. https://doi.org/10.3390/ijms241311086
Kumar P, Mathew S, Gamage R, Bodkin F, Doyle K, Rossetti I, Wagnon I, Zhou X, Raju R, Gyengesi E, et al. From the Bush to the Brain: Preclinical Stages of Ethnobotanical Anti-Inflammatory and Neuroprotective Drug Discovery—An Australian Example. International Journal of Molecular Sciences. 2023; 24(13):11086. https://doi.org/10.3390/ijms241311086
Chicago/Turabian StyleKumar, Payaal, Shintu Mathew, Rashmi Gamage, Frances Bodkin, Kerrie Doyle, Ilaria Rossetti, Ingrid Wagnon, Xian Zhou, Ritesh Raju, Erika Gyengesi, and et al. 2023. "From the Bush to the Brain: Preclinical Stages of Ethnobotanical Anti-Inflammatory and Neuroprotective Drug Discovery—An Australian Example" International Journal of Molecular Sciences 24, no. 13: 11086. https://doi.org/10.3390/ijms241311086
APA StyleKumar, P., Mathew, S., Gamage, R., Bodkin, F., Doyle, K., Rossetti, I., Wagnon, I., Zhou, X., Raju, R., Gyengesi, E., & Münch, G. (2023). From the Bush to the Brain: Preclinical Stages of Ethnobotanical Anti-Inflammatory and Neuroprotective Drug Discovery—An Australian Example. International Journal of Molecular Sciences, 24(13), 11086. https://doi.org/10.3390/ijms241311086