
Cyclophilin A Isomerisation of Septin 2 Mediates Abscission during Cytokinesis

 , , ,
, , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
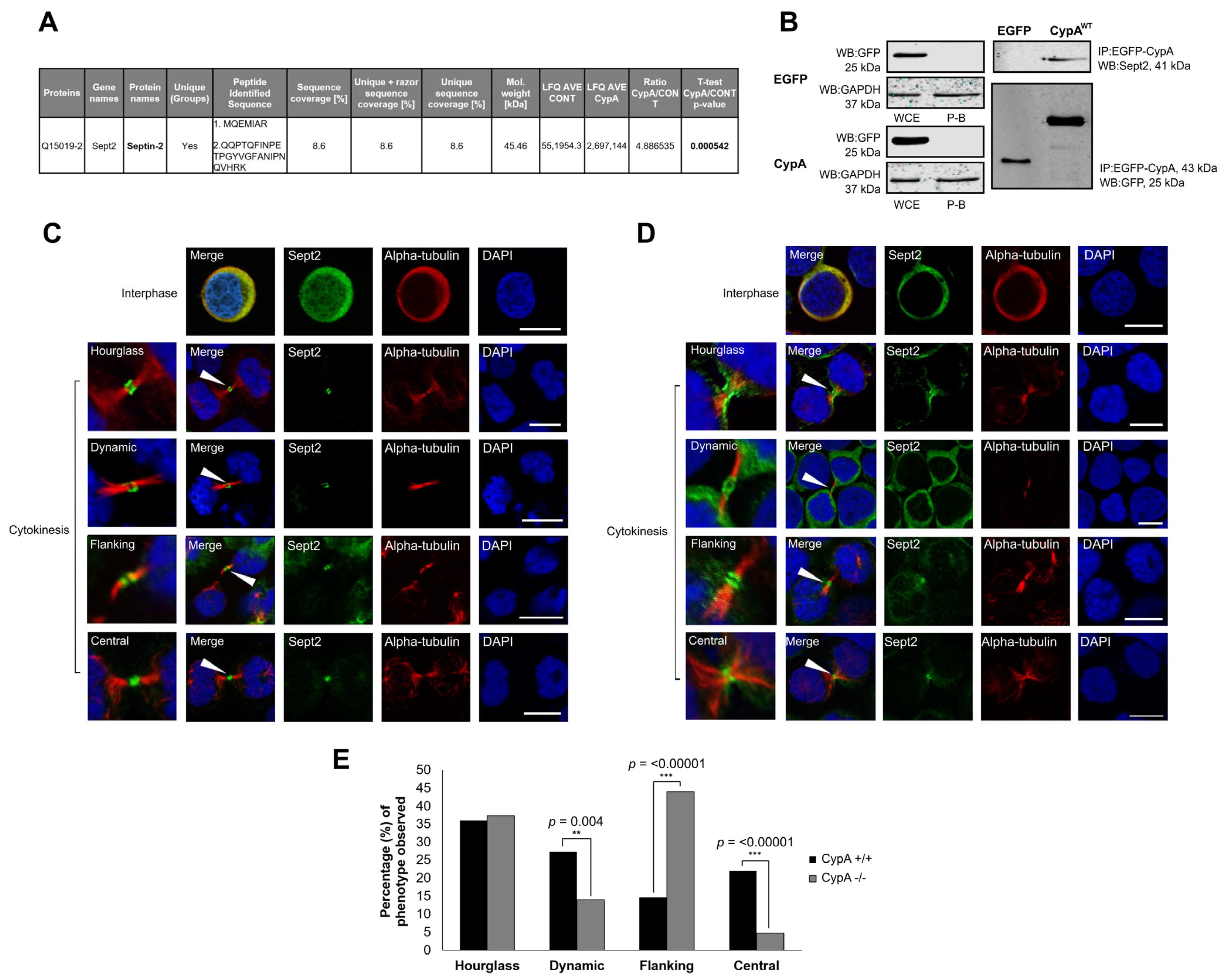
2.1. Sept2 Is a Novel Interacting Partner of CypA
2.2. Localisation of Sept2 at the Midbody in Human Cancer Cells
2.3. Depletion of CypA Disrupts Sept2 Localisation at the Midbody
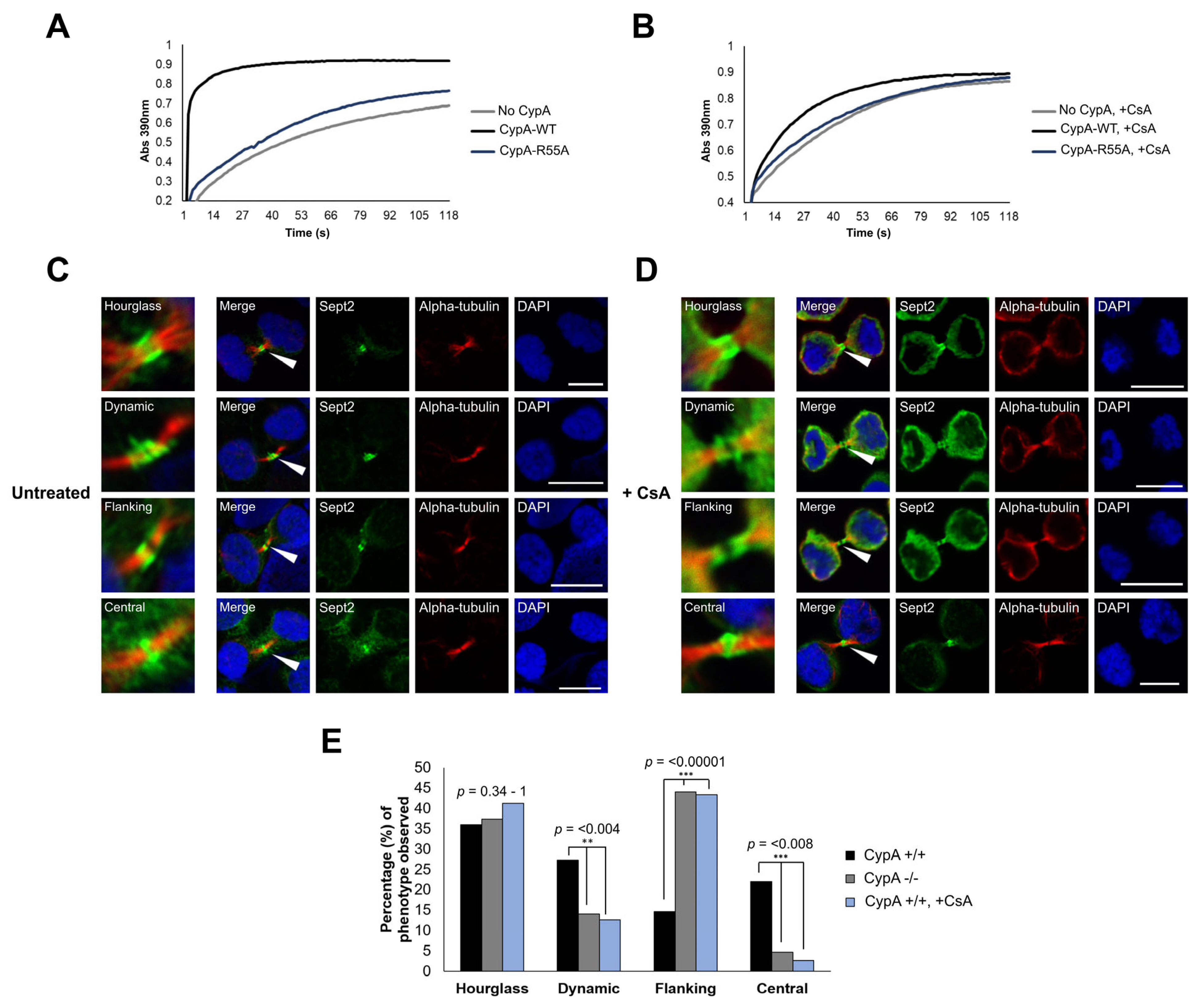
2.4. Inhibition of CypA Isomerase Activity Alters Sept2 Dynamics at the Midbody
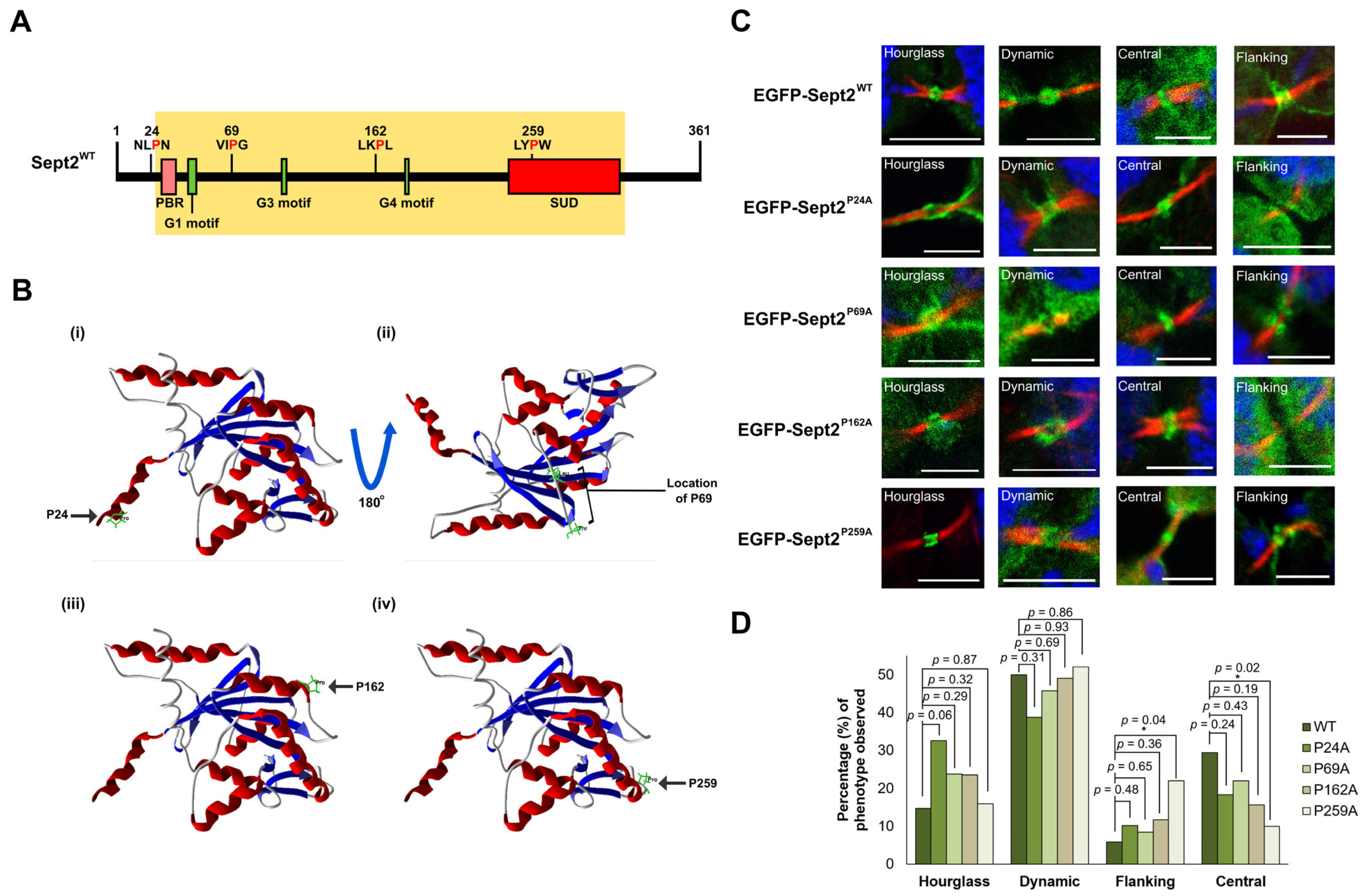
2.5. Isomerisation of Pro259 Is Required for Central Midbody Localisation of Sept2

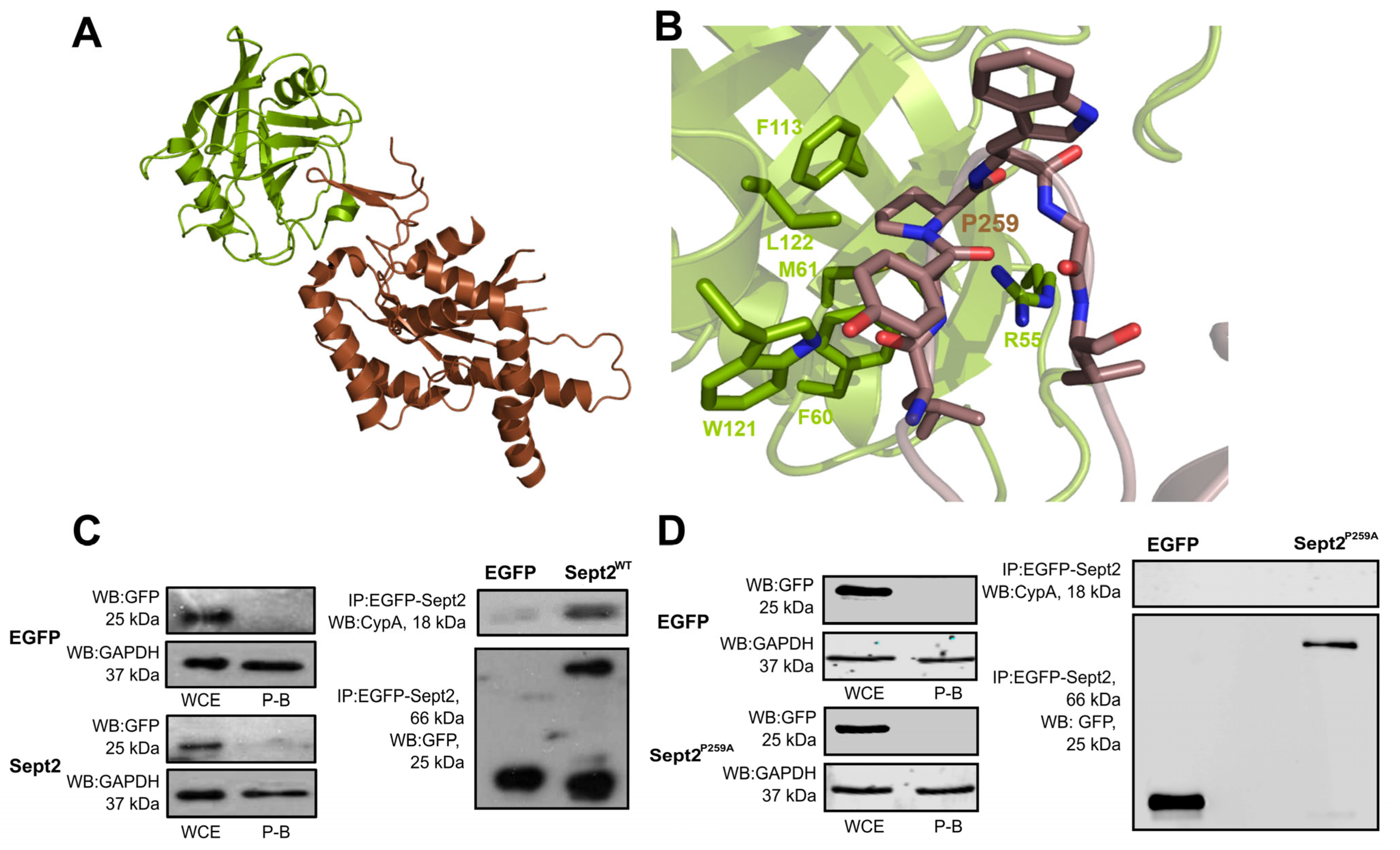
2.6. Structural and Molecular Analysis of CypA-Sept2P259A Interaction
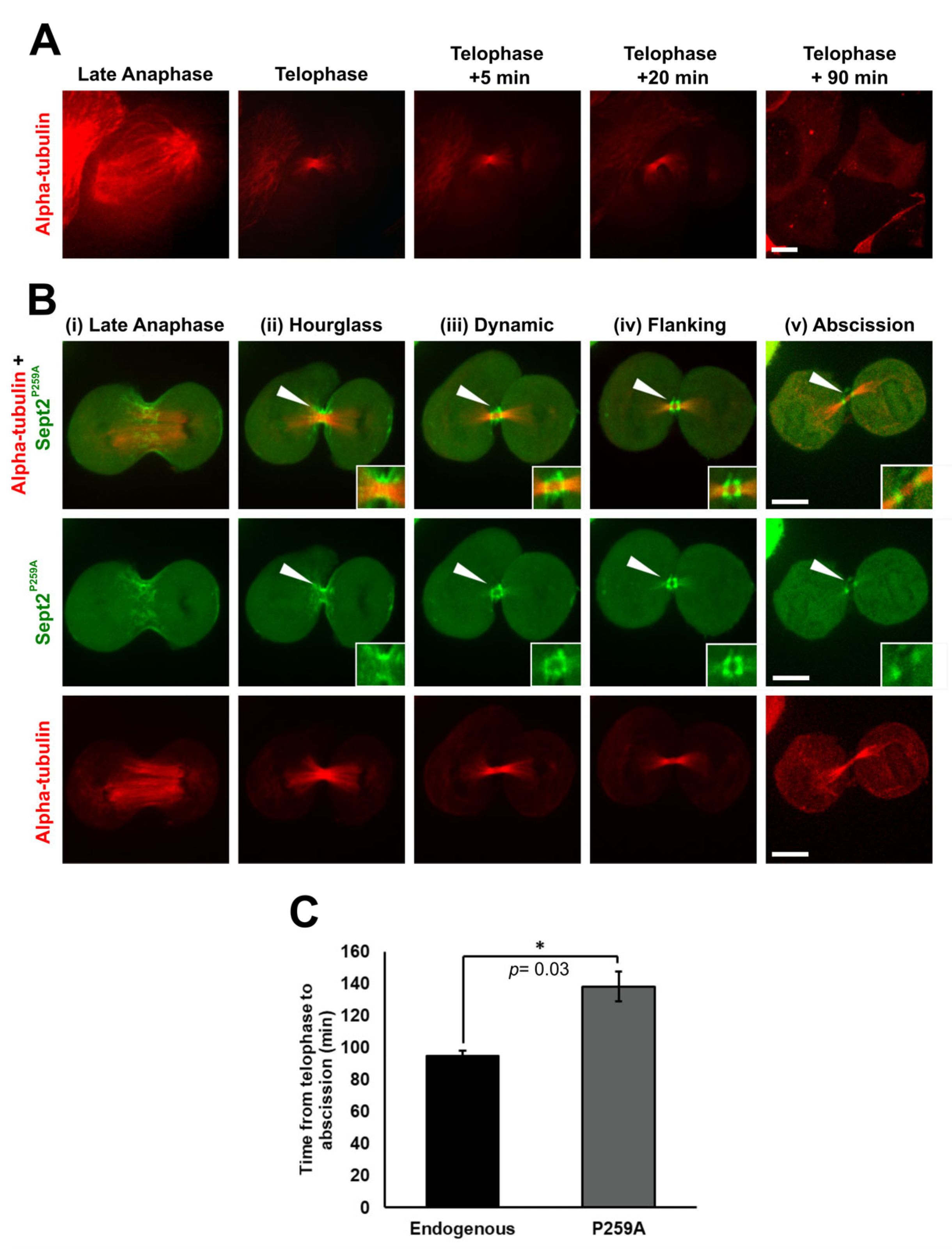
2.7. Isomerisation of Sept2 Pro259 Is Required for the Completion of Cell Division
3. Discussion
4. Materials and Methods
4.1. Cell Culture, Synchronisation and Transfection
4.2. Proteomics Sample Preparation, Analysis and Identification of Sept2 as a CypA Interactor
4.3. Co-Immunoprecipitation
4.4. Immunoblotting
4.5. Immunofluorescence
4.6. Data and Image Analysis
4.7. Isomerase Assay
4.8. Plasmids and Site-Directed Mutagenesis (SDM)
4.9. Modelling of the Interaction between CypA and Sept2
4.10. Time-Lapse Spinning Disk Microscopy
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Renshaw, M.J.; Liu, J.; Lavoie, B.D.; Wilde, A. Anillin-dependent organization of septin filaments promotes intercellular bridge elongation and Chmp4B targeting to the abscission site. Open Biol. 2014, 4, 130190. [Google Scholar] [CrossRef] [PubMed]
- Green, R.A.; Paluch, E.; Oegema, K. Cytokinesis in Animal Cells. Annu. Rev. Cell Dev. Biol. 2012, 28, 29–58. [Google Scholar] [CrossRef] [PubMed]
- Cheffings, T.H.; Burroughs, N.J.; Balasubramanian, M.K. Actomyosin Ring Formation and Tension Generation in Eukaryotic Cytokinesis. Curr. Biol. 2016, 26, R719–R737. [Google Scholar] [CrossRef] [PubMed]
- Antanavičiūtė, I.; Gibieža, P.; Prekeris, R.; Skeberdis, V. Midbody: From the Regulator of Cytokinesis to Postmitotic Signaling Organelle. Medicina 2018, 54, 53. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; Elia, N.; Ghirlando, R.; Lippincott-Schwartz, J.; Hurley, J.H. Midbody Targeting of the ESCRT Machinery by a Noncanonical Coiled Coil in CEP55. Science 2008, 322, 576–580. [Google Scholar] [CrossRef]
- Zheng, Y.; Guo, J.; Li, X.; Xie, Y.; Hou, M.; Fu, X.; Dai, S.; Diao, R.; Miao, Y.; Ren, J. An integrated overview of spatiotemporal organization and regulation in mitosis in terms of the proteins in the functional supercomplexes. Front. Microbiol. 2014, 5, 573. [Google Scholar] [CrossRef]
- Leipe, D.D.; Wolf, Y.I.; Koonin, E.V.; Aravind, L. Classification and evolution of P-loop GTPases and related ATPases. J. Mol. Biol. 2002, 317, 41–72. [Google Scholar] [CrossRef]
- Mostowy, S.; Cossart, P. Septins: The fourth component of the cytoskeleton. Nat. Rev. Mol. Cell Biol. 2012, 13, 183–194. [Google Scholar] [CrossRef]
- Macara, I.G.; Baldarelli, R.; Field, C.M.; Glotzer, M.; Hayashi, Y.; Hsu, S.C.; Kennedy, M.B.; Kinoshita, M.; Longtine, M.; Low, C.; et al. Mammalian septins nomenclature. Mol. Biol. Cell 2002, 13, 4111–4113. [Google Scholar] [CrossRef]
- Kinoshita, M. The septins. Genome Biol. 2003, 4, 236. [Google Scholar] [CrossRef]
- Kang, H.; Lew, D.J. How do cells know what shape they are? Curr. Genet. 2017, 63, 75–77. [Google Scholar] [CrossRef]
- Spiliotis, E.T. A Mitotic Septin Scaffold Required for Mammalian Chromosome Congression and Segregation. Science 2005, 307, 1781–1785. [Google Scholar] [CrossRef]
- Nakos, K.; Radler, M.R.; Spiliotis, E.T. Septin 2/6/7 complexes tune microtubule plus-end growth and EB1 binding in a concentration- and filament-dependent manner. Mol. Biol. Cell 2019, 30, 2913–2928. [Google Scholar] [CrossRef]
- Kinoshita, M.; Noda, M. Roles of septins in the mammalian cytokinesis machinery. Cell Struct. Funct. 2001, 26, 667–670. [Google Scholar] [CrossRef]
- Joo, E.; Surka, M.C.; Trimble, W.S. Mammalian SEPT2 Is Required for Scaffolding Nonmuscle Myosin II and Its Kinases. Dev. Cell 2007, 13, 677–690. [Google Scholar] [CrossRef]
- Karasmanis, E.P.; Hwang, D.; Nakos, K.; Bowen, J.R.; Angelis, D.; Spiliotis, E.T. A Septin Double Ring Controls the Spatiotemporal Organization of the ESCRT Machinery in Cytokinetic Abscission. Curr. Biol. 2019, 29, 2174–2182.e7. [Google Scholar] [CrossRef]
- Shaw, P.E. Peptidyl-prolyl isomerases: A new twist to transcription. EMBO Rep. 2002, 3, 521–526. [Google Scholar] [CrossRef]
- Kim, H.; Oh, Y.; Kim, K.; Jeong, S.; Chon, S.; Kim, D.; Jung, M.H.; Pak, Y.K.; Ha, J.; Kang, I.; et al. Cyclophilin A regulates JNK/p38-MAPK signaling through its physical interaction with ASK1. Biochem. Biophys. Res. Commun. 2015, 464, 112–117. [Google Scholar] [CrossRef]
- Tong, M.; Jiang, Y. FK506-Binding Proteins and Their Diverse Functions. Curr. Mol. Pharmacol. 2015, 9, 48–65. [Google Scholar] [CrossRef]
- Matena, A.; Rehic, E.; Hönig, D.; Kamba, B.; Bayer, P. Structure and function of the human parvulins Pin1 and Par14/17. Biol. Chem. 2018, 399, 101–125. [Google Scholar] [CrossRef]
- Scholz, C.; Rahfeld, J.; Fischer, G.; Schmid, F.X. Catalysis of protein folding by Parvulin. J. Mol. Biol. 1997, 273, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Schonbrunner, E.R.; Mayer, S.; Tropschug, M.; Fischer, G.; Takahashi, N.; Schmid, F.X. Catalysis of protein folding by cyclophilins from different species. J. Biol. Chem. 1991, 266, 3630–3635. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Lee, J.A.; Park, S.G.; Lee, D.H.; Kim, S.J.; Kim, H.-J.; Uchida, C.; Uchida, T.; Park, B.C.; Cho, S. A critical step for JNK activation: Isomerization by the prolyl isomerase Pin1. Cell Death Differ. 2012, 19, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Lavin, P.T.M.; Mc Gee, M.M. Cyclophilin function in Cancer; lessons from virus replication. Curr. Mol. Pharmacol. 2015, 9, 148–164. [Google Scholar] [CrossRef]
- Gorry, R.; Brennan, K.; Lavin, P.T.; Sheridan, R.; Mc Gee, M.M. Phosphorylation of the prolyl isomerase Cyclophilin A regulates its localisation and release from the centrosome during mitosis. Cell Cycle 2023, 22, 951–966. [Google Scholar] [CrossRef] [PubMed]
- Bannon, J.H.; O’Donovan, D.S.; Kennelly, S.M.E.; Mc Gee, M.M. The peptidyl prolyl isomerase cyclophilin A localizes at the centrosome and the midbody and is required for cytokinesis. Cell Cycle 2012, 11, 1340–1353. [Google Scholar] [CrossRef]
- Van Der Horst, A.; Khanna, K.K. The peptidyl-prolyl isomerase Pin1 regulates cytokinesis through Cep55. Cancer Res. 2009, 69, 6651–6659. [Google Scholar] [CrossRef]
- Takahashi, N.; Hayano, T.; Suzuki, M. Peptidyl-prolyl cis-trans isomerase is the cyclosporin A-binding protein cyclophilin. Nature 1989, 337, 473–475. [Google Scholar] [CrossRef]
- Davis, T.L.; Walker, J.R.; Campagna-Slater, V.; Finerty, P.J.; Finerty, P.J.; Paramanathan, R.; Bernstein, G.; Mackenzie, F.; Tempel, W.; Ouyang, H.; et al. Structural and biochemical characterization of the human cyclophilin family of peptidyl-prolyl isomerases. PLoS Biol. 2010, 8, e1000439. [Google Scholar] [CrossRef]
- Yang, H.; Chen, J.; Yang, J.; Qiao, S.; Zhao, S.; Yu, L. Cyclophilin A is upregulated in small cell lung cancer and activates ERK1/2 signal. Biochem. Biophys. Res. Commun. 2007, 361, 763–767. [Google Scholar] [CrossRef]
- Li, Z.; Zhao, X.; Bai, S.; Wang, Z.; Chen, L.; Wei, Y.; Huang, C. Proteomics identification of cyclophilin A as a potential prognostic factor and therapeutic target in endometrial carcinoma. Mol. Cell. Proteomics 2008, 7, 1810–1823. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.J.; Piao, Y.J.; Lim, M.J.; Kim, J.H.; Ha, J.; Choe, W.; Kim, S.S. Overexpressed cyclophilin A in cancer cells renders resistance to hypoxia- and cisplatin-induced cell death. Cancer Res. 2007, 67, 3654–3662. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.P.; Finn, G.; Lee, T.H.; Nicholson, L.K. Prolyl cis-trans isomerization as a molecular timer. Nat. Chem. Biol. 2007, 3, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.P.; Zhou, X.Z. The prolyl isomerase PIN1: A pivotal new twist in phosphorylation signalling and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 904–916. [Google Scholar] [CrossRef] [PubMed]
- Estey, M.P.; Di Ciano-Oliveira, C.; Froese, C.D.; Fung, K.Y.Y.; Steels, J.D.; Litchfield, D.W.; Trimble, W.S. Mitotic Regulation of SEPT9 Protein by Cyclin-dependent Kinase 1 (Cdk1) and Pin1 Protein Is Important for the Completion of Cytokinesis. J. Biol. Chem. 2013, 288, 30075–30086. [Google Scholar] [CrossRef]
- Kinoshita, M.; Kumar, S.; Mizoguchi, A.; Ide, C.; Kinoshita, A.; Haraguchi, T.; Hiraoka, Y.; Noda, M. Nedd5, a mammalian septin, is a novel cytoskeletal component interacting with actin-based structures. Genes Dev. 1997, 11, 1535–1547. [Google Scholar] [CrossRef]
- Fung, K.Y.Y.; Dai, L.; Trimble, W.S. Cell and Molecular Biology of Septins. Int. Rev. Cell Mol. Biol. 2014, 310, 289–339. [Google Scholar]
- Menon, M.B.; Gaestel, M. Sep(t)arate or not—How some cells take septin-independent routes through cytokinesis. J. Cell Sci. 2015, 128, 1877–1886. [Google Scholar] [CrossRef]
- Glomb, O.; Gronemeyer, T. Septin Organization and Functions in Budding Yeast. Front. Cell Dev. Biol. 2016, 4, 123. [Google Scholar] [CrossRef]
- Ong, K.; Wloka, C.; Okada, S.; Svitkina, T.; Bi, E. Architecture and dynamic remodelling of the septin cytoskeleton during the cell cycle. Nat. Commun. 2014, 5, 5698. [Google Scholar] [CrossRef]
- Estey, M.P.; Di Ciano-Oliveira, C.; Froese, C.D.; Bejide, M.T.; Trimble, W.S. Distinct roles of septins in cytokinesis: SEPT9 mediates midbody abscission. J. Cell Biol. 2010, 191, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Farmer, J.D.; Lane, W.S.; Friedman, J.; Weissman, I.; Schreiber, S.L. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 1991, 66, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Kofron, J.L.; Kuzmic, P.; Kishore, V.; Colon-Bonilla, E.; Rich, D.H. Determination of kinetic constants for peptidyl prolyl cis-trans isomerases by an improved spectrophotometric assay. Biochemistry 1991, 30, 6127–6134. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, L.; Ehrlich, L.S.; LaGrassa, T.J.; Ebbets-Reed, D.; Carter, C. Structural Consequences of Cyclophilin A Binding on Maturational Refolding in Human Immunodeficiency Virus Type 1 Capsid Protein. J. Virol. 2001, 75, 4721–4733. [Google Scholar] [CrossRef] [PubMed]
- Yurchenko, V.; Pushkarsky, T.; Li, J.-H.; Dai, W.W.; Sherry, B.; Bukrinsky, M. Regulation of CD147 Cell Surface Expression. J. Biol. Chem. 2005, 280, 17013–17019. [Google Scholar] [CrossRef] [PubMed]
- Piotukh, K.; Gu, W.; Kofler, M.; Labudde, D.; Helms, V.; Freund, C. Cyclophilin A binds to linear peptide motifs containing a consensus that is present in many human proteins. J. Biol. Chem. 2005, 280, 23668–23674. [Google Scholar] [CrossRef]
- Schlegel, J.; Redzic, J.S.; Porter, C.C.; Yurchenko, V.; Bukrinsky, M.; Labeikovsky, W.; Armstrong, G.S.; Zhang, F.; Isern, N.G.; DeGregori, J.; et al. Solution Characterization of the Extracellular Region of CD147 and Its Interaction with Its Enzyme Ligand Cyclophilin A. J. Mol. Biol. 2009, 391, 518–535. [Google Scholar] [CrossRef]
- Valadares, N.F.; d’Muniz Pereira, H.; Ulian Araujo, A.P.; Garratt, R.C. Septin structure and filament assembly. Biophys. Rev. 2017, 9, 481–500. [Google Scholar] [CrossRef]
- Sirajuddin, M.; Farkasovsky, M.; Hauer, F.; Kühlmann, D.; Macara, I.G.; Weyand, M.; Stark, H.; Wittinghofer, A. Structural insight into filament formation by mammalian septins. Nature 2007, 449, 311–315. [Google Scholar] [CrossRef]
- Howard, B.R.; Vajdos, F.F.; Li, S.; Sundquist, W.I.; Hill, C.P. Structural insights into the catalytic mechanism of cyclophilin A. Nat. Struct. Mol. Biol. 2003, 10, 475–481. [Google Scholar] [CrossRef]
- Liu, M.; Shen, S.; Chen, F.; Yu, W.; Yu, L. Linking the septin expression with carcinogenesis. Mol. Biol. Rep. 2010, 37, 3601–3608. [Google Scholar] [CrossRef]
- Yu, J.; Zhang, W.; Tang, H.; Qian, H.; Yang, J.; Zhu, Z.; Ren, P.; Lu, B. Septin 2 accelerates the progression of biliary tract cancer and is negatively regulated by mir-140-5p. Gene 2016, 589, 20–26. [Google Scholar] [CrossRef]
- He, H.; Li, J.; Xu, M.; Kan, Z.; Gao, Y.; Yuan, C. Expression of septin 2 and association with clinicopathological parameters in colorectal cancer. Oncol. Lett. 2019, 18, 2376–2383. [Google Scholar] [CrossRef]
- Lens, S.M.A.; Medema, R.H. Cytokinesis defects and cancer. Nat. Rev. Cancer 2019, 19, 32–45. [Google Scholar] [CrossRef]
- Yang, X.; Yu, K.; Hao, Y.; Li, D.; Stewart, R.; Insogna, K.L.; Xu, T. LATS1 tumour suppressor affects cytokinesis by inhibiting LIMK1. Nat. Cell Biol. 2004, 6, 609–617. [Google Scholar] [CrossRef]
- Kops, G.J.P.L.; Foltz, D.R.; Cleveland, D.W. Lethality to human cancer cells through massive chromosome loss by inhibition of the mitotic checkpoint. Proc. Natl. Acad. Sci. USA 2004, 101, 8699–8704. [Google Scholar] [CrossRef]
- Michel, L.; Diaz-Rodriguez, E.; Narayan, G.; Hernando, E.; Murty, V.V.V.S.; Benezra, R. Complete loss of the tumor suppressor MAD2 causes premature cyclin B degradation and mitotic failure in human somatic cells. Proc. Natl. Acad. Sci. USA. 2004, 101, 4459–4464. [Google Scholar] [CrossRef]
- Hanks, S.; Coleman, K.; Reid, S.; Plaja, A.; Firth, H.; FitzPatrick, D.; Kidd, A.; Méhes, K.; Nash, R.; Robin, N.; et al. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat. Genet. 2004, 36, 1159–1161. [Google Scholar] [CrossRef]
- Suijkerbuijk, S.J.E.; Van Osch, M.H.J.; Bos, F.L.; Hanks, S.; Rahman, N.; Kops, G.J.P.L. Molecular causes for BUBR1 dysfunction in the human cancer predisposition syndrome mosaic variegated aneuploidy. Cancer Res. 2010, 70, 4891–4900. [Google Scholar] [CrossRef]
- Liu, X.; Li, Y.; Meng, L.; Liu, X.-Y.; Peng, A.; Chen, Y.; Liu, C.; Chen, H.; Sun, S.; Miao, X.; et al. Reducing protein regulator of cytokinesis 1 as a prospective therapy for hepatocellular carcinoma. Cell Death Dis. 2018, 9, 534. [Google Scholar] [CrossRef]
- Nigro, P.; Pompilio, G.; Capogrossi, M.C. Cyclophilin A: A key player for human disease. Cell Death Dis. 2013, 4, e888. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Xin, Y.; Xiao, Y.; Li, W.; Sun, D. Cyclophilin A Enhances Cell Proliferation and Xenografted Tumor Growth of Early Gastric Cancer. Dig. Dis. Sci. 2015, 60, 2700–2711. [Google Scholar] [CrossRef] [PubMed]
- Kremer, B.E.; Haystead, T.; Macara, I.G. Mammalian Septins Regulate Microtubule Stability through Interaction with the Microtubule-binding Protein MAP4. Mol. Biol. Cell 2005, 16, 4648–4659. [Google Scholar] [CrossRef] [PubMed]
- Menon, M.B.; Wu, L.-W.; Vagin, O.; Baillet, A.; Poüs, C.; Klipfel, L. Cancer-Related Functions and Subcellular Localizations of Septins. Front. Cell Dev. Biol. 2016, 1, 126. [Google Scholar] [CrossRef]
- Angelis, D.; Spiliotis, E.T. Septin Mutations in Human Cancers. Front. Cell Dev. Biol. 2016, 4, 122. [Google Scholar] [CrossRef]
- Moriya, H. Quantitative nature of overexpression experiments. Mol. Biol. Cell 2015, 26, 3932–3939. [Google Scholar] [CrossRef]
- Turriziani, B.; Garcia-Munoz, A.; Pilkington, R.; Raso, C.; Kolch, W.; von Kriegsheim, A. On-beads digestion in conjunction with data-dependent mass spectrometry: A shortcut to quantitative and dynamic interaction proteomics. Biology 2014, 3, 320–332. [Google Scholar] [CrossRef]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.H.; Vreven, T.; Weng, Z. ZDOCK server: Interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2011, 27, 343–350. [Google Scholar] [CrossRef]
- Madden, T. The BLAST sequence analysis tool. BLAST Seq. Anal. Tool. 2013.
- Adeira, F.; Park, Y.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.; Potter, S.; Finn, R.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorry, R.L.; Brennan, K.; Lavin, P.T.M.; Mazurski, T.; Mary, C.; Matallanas, D.; Guichou, J.-F.; Mc Gee, M.M. Cyclophilin A Isomerisation of Septin 2 Mediates Abscission during Cytokinesis. Int. J. Mol. Sci. 2023, 24, 11084. https://doi.org/10.3390/ijms241311084
Gorry RL, Brennan K, Lavin PTM, Mazurski T, Mary C, Matallanas D, Guichou J-F, Mc Gee MM. Cyclophilin A Isomerisation of Septin 2 Mediates Abscission during Cytokinesis. International Journal of Molecular Sciences. 2023; 24(13):11084. https://doi.org/10.3390/ijms241311084
Chicago/Turabian StyleGorry, Rebecca L., Kieran Brennan, Paul T. M. Lavin, Tayler Mazurski, Charline Mary, David Matallanas, Jean-François Guichou, and Margaret M. Mc Gee. 2023. "Cyclophilin A Isomerisation of Septin 2 Mediates Abscission during Cytokinesis" International Journal of Molecular Sciences 24, no. 13: 11084. https://doi.org/10.3390/ijms241311084
APA StyleGorry, R. L., Brennan, K., Lavin, P. T. M., Mazurski, T., Mary, C., Matallanas, D., Guichou, J.-F., & Mc Gee, M. M. (2023). Cyclophilin A Isomerisation of Septin 2 Mediates Abscission during Cytokinesis. International Journal of Molecular Sciences, 24(13), 11084. https://doi.org/10.3390/ijms241311084