
Dysfunction of an Anaphase-Promoting Complex Subunit 8 Homolog Leads to Super-Short Petioles and Enlarged Petiole Angles in Soybean


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
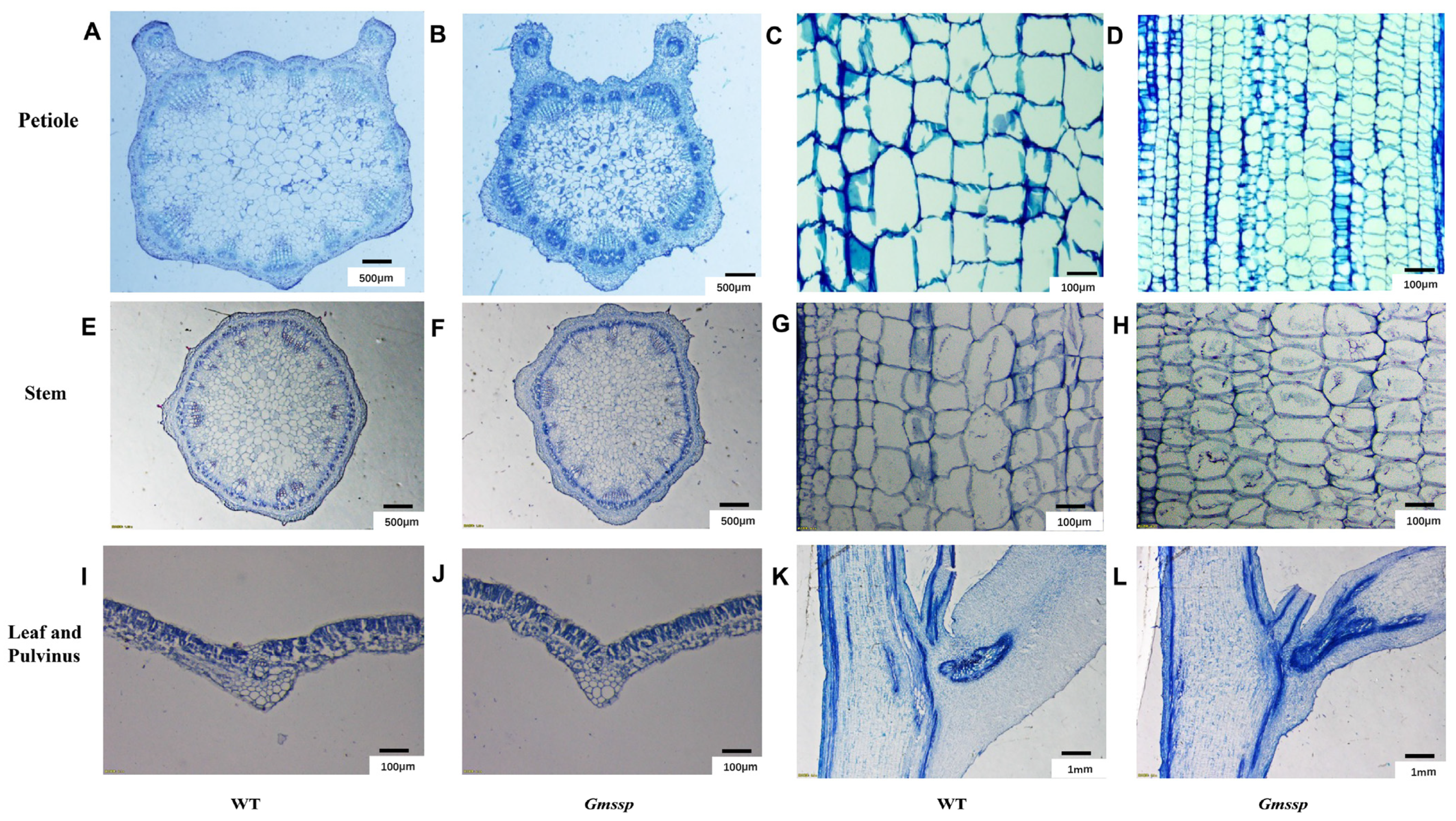
2.1. The Gmssp Mutant Has a Dwarf Phenotype with Super-Short Petioles and Enlarged Petiole Angles
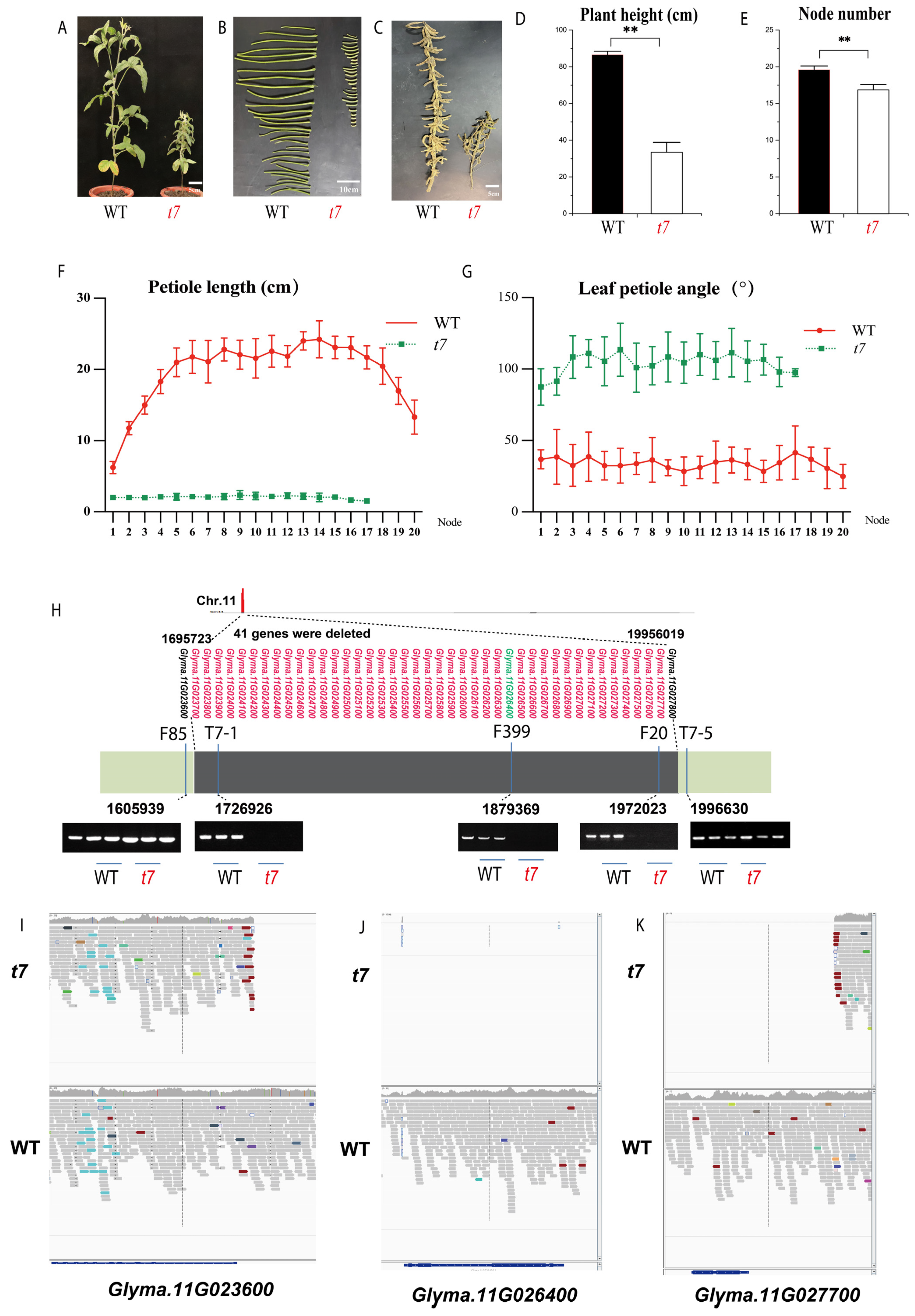
2.2. The BVF-IGV Pipeline Identified Glyma.11G026400 as the Candidate Gene for the Gmssp Phenotype
2.3. QTL Mapping Validates Glyma.11G026400 as the Candidate Gene for Gmssp
2.4. Characterization of the t7 Mutant Supports Glyma.11G026400 as the Causal Gene for the Gmssp Phenotype
2.5. Glyma.11G026400 Encodes a Truncated Protein in Gmssp
2.6. The Mutation in Gmapc8 in the Gmssp Mutant Does Not Alter Its Subcellular Localization
2.7. DEGs Are Enriched in Ubiquitin-Mediated Proteolysis and Hormone Signal Transduction
2.8. Gibberellin Treatment Partially Rescues the Phenotype of Gmssp
2.9. WGCNA Reveals Modules Associated with Traits Affecting Both Tissues and Growth Stages of Gmssp
3. Discussion
3.1. Short Petioles and Enlarged Leaf Petiole Angles Are Important Traits Determining Plant Architecture in Soybean
3.2. The BVF-IGV Pipeline Identifies the Causal Mutation for the Gmssp Phenotype
3.3. Glyma.11G026400 Affects Plant Architecture via Ubiquitin-Mediated Proteolysis (APC) and Plant Hormone Signal Transduction
4. Materials and Methods
4.1. Mutant Library Development, Phenotyping, and Analysis of Anatomical Structure
4.2. Resequencing and Identification of the Causal Gene by MutMap and BVF-IGV
4.3. Generation of the Mapping Population and Markers
4.4. RNA Sequencing and Transcriptome Analysis
4.5. Application of Exogenous GA3
4.6. Subcellular Localization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, Y.F.; Zhang, D.J.; Ping, J.Q.; Li, S.; Chen, Z.X.; Ma, J.X. Innovation of a regulatory mechanism modulating semi-determinate stem growth through artificial selection in soybean. PLoS Genet. 2016, 12, e1005818. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Watanabe, S.; Yamada, T.; Tsubokura, Y.; Nakashima, H.; Zhai, H.; Anai, T.; Sato, S.; Yamazaki, T.; Lu, S.X. Positional cloning and characterization reveal the molecular basis for soybean maturity locus E1 that regulates photoperiodic flowering. Proc. Natl. Acad. Sci. USA 2012, 109, E2155–E2164. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Zhai, H.; Zhang, Y.; Wang, Y.; Wang, L.; Xu, K.; Wu, H.; Zhu, J.; Jiao, S.; Wan, Z.; et al. QNE1 is a key flowering regulator determining the length of the vegetative period in soybean cultivars. Sci. China Life Sci. 2022, 65, 2472–2490. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.; Ross, V.; Koppel, A. Estimation of leaf area and its vertical distribution during growth period. Agric. For. Meteorol. 2000, 101, 237–246. [Google Scholar] [CrossRef]
- Zhu, J.T.; Li, X.Y.; Zhang, X.M.; Lin, L.S.; Yang, S.G. Nitrogen allocation and partitioning within a leguminous and two non-leguminous plant species growing at the southern fringe of China’s Taklamakan Desert. J. Plant Ecol. 2010, 34, 1025–1032. [Google Scholar]
- Egli, D.B. Plant density and soybean yield. Crop Sci. 1988, 28, 977–981. [Google Scholar] [CrossRef]
- Jun, T.H.; Kang, S.T. Genetic map of lps3: A new short petiole gene in soybeans. Genome 2012, 55, 140–146. [Google Scholar] [CrossRef]
- Liu, S.L.; Zhang, M.; Feng, F.; Tian, Z.X. Toward a “Green Revolution” for Soybean. Mol. Plant 2020, 13, 688–697. [Google Scholar] [CrossRef]
- Gao, H.W.; Sun, R.J.; Yang, M.Y.; Yan, L.; Hu, X.Z.; Fu, G.H.; Hong, H.L.; Guo, B.F.; Zhang, X.; Liu, L.K.; et al. Characterization of the petiole length in soybean compact architecture mutant M657 and the breeding of new lines. J. Integr. Agric. 2022, 21, 2508–2520. [Google Scholar] [CrossRef]
- Kilen, T.C. Inheritance of a short petiole trait in soybean. Crop Sci. 1983, 23, 1208–1210. [Google Scholar] [CrossRef]
- You, M.G.; Zhao, T.J.; Gai, J.Y.; Yen, Y. Genetic analysis of short petiole and abnormal pulvinus in soybean. Euphytica 1998, 102, 329–333. [Google Scholar] [CrossRef]
- Liu, M.; Wang, Y.; Gai, J.; Bhat, J.; Li, Y.; Kong, J.; Zhao, T. Genetic analysis and gene mapping for a short-petiole mutant in soybean (Glycine max (L.) Merr.). Agronomy 2019, 9, 709. [Google Scholar] [CrossRef]
- Clark, C.B.; Wang, W.D.; Wang, Y.; Fear, G.J.; Wen, Z.X.; Wang, D.C.; Ren, B.; Ma, J.K. Identification and molecular mapping of a major quantitative trait locus underlying branch angle in soybean. Theor. Appl. Genet. 2022, 135, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Q.; Gao, L.; Ke, L.; Gao, Z.; Tu, T.; Huang, L.; Chen, J.; Guan, Y.; Huang, X.; Chen, X. GmPIN1-mediated auxin asymmetry regulates leaf petiole angle and plant architecture in soybean. J. Integr. Plant Biol. 2022, 64, 1325–1338. [Google Scholar] [CrossRef] [PubMed]
- Pesin, J.A.; Orr-Weaver, T.L. Regulation of APC/C activators in mitosis and meiosis. Annu. Rev. Cell Dev. Biol. 2008, 24, 475–499. [Google Scholar] [CrossRef]
- Xu, C.; Wang, Y.; Yu, Y.; Duan, J.; Liao, Z.; Xiong, G.; Meng, X.; Liu, G.; Qian, Q.; Li, J. Degradation of monoculm 1 by apc/ctad1 regulates rice tillering. Nat. Commun. 2012, 3, 750. [Google Scholar] [CrossRef]
- Gao, J.; Yang, S.; Cheng, W.; Fu, Y.; Leng, J.; Yuan, X.; Jiang, N.; Ma, J.X.; Feng, X.Z. GmILPA1, encoding an anaphase-promoting complex-like protein, affects leaf petiole angle. Plant Physiol. 2017, 174, 1167–1176. [Google Scholar] [CrossRef]
- Jin, Y.; Li, J.; Zhu, Q.; Du, X.; Liu, F.; Li, Y.; Ahmar, S.; Zhang, X.; Sun, J.; Xue, F. GhAPC8 regulates leaf blade angle by modulating multiple hormones in cotton (Gossypium hirsutum L.). Int. J. Biol. Macromol. 2022, 195, 217–228. [Google Scholar] [CrossRef]
- Abe, A.; Kosugi, S.; Yoshida, K.; Natsume, S.; Takagi, H.; Kanzaki, H.; Matsumura, H.; Yoshida, K.; Mitsuoka, C.; Tamiru, M.; et al. Genome sequencing reveals agronomically important loci in rice using MutMap. Nat. Biotechnol. 2012, 30, 174–178. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows—Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Chiang, C.; Layer, R.M.; Faust, G.G.; Lindberg, M.R.; Rose, D.B.; Garrison, E.P.; Marth, G.; Quinlan, A.; Hall, I.M. SpeedSeq: Ultra-fast personal genome analysis and interpretation. Nat. Methods 2015, 12, 966–968. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Zhu, J.; Zhai, H.; Wu, H.; Gao, Y.; Li, Y.; Zhu, X.; Xia, Z. A critical role of PvFtsH2 in the degradation of photodamaged D1 protein in common bean. Hortic. Res. 2021, 8, 126. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.Y.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Volkov, A.G.; Foster, J.C.; Baker, K.D.; Markin, V.S. Mechanical and electrical anisotropy in Mimosa pudica pulvini. Plant Signal. Behav. 2010, 5, 1211–1221. [Google Scholar] [CrossRef] [PubMed]
- Ashton, G.D.; Blythe, M.; Sang, F.; Zadik, D.; Holmes, N.; Malla, S.; Camps, S.M.T.; Wright, V.; Melchers, W.J.G.E.; Verweij, P.S. Use of bulk segregant analysis for determining the genetic basis of azole resistance in the opportunistic pathogen aspergillus fumigatus. Front. Cell. Infect. Microbiol. 2022, 12, 841138. [Google Scholar] [CrossRef]
- Tran, Q.H.; Bui, N.H.; Kappel, C.; Dau, N.T.N.; Nguyen, L.T.; Tran, T.T.; Khanh, D.K. Mapping-by-sequencing via MutMap identifies a mutation in ZmCLE7 underlying fasciation in a newly developed EMS mutant population in an elite tropical maize inbred. Genes 2020, 11, 281. [Google Scholar] [CrossRef]
- Zhou, W.; Soghigian, J.; Xiang, Q.Y. A new pipeline for removing paralogs in target enrichment data. Syst. Biol. 2022, 71, 410–425. [Google Scholar] [CrossRef]
- Zhou, H.; Tang, K.; Li, G.; Liu, W.; Yu, H.; Yuan, X.; Yang, S.; Bhattacharyya, M.; Feng, X. A robust and rapid candidate gene mapping pipeline based on M2 populations. Front. Plant Sci. 2021, 12, 681816. [Google Scholar] [CrossRef]
- Hunter, C.T.; Suzuki, M.; Saunders, J.; Wu, S.; Tasi, A.; McCarty, D.R.; Koch, K.E. Phenotype to genotype using forward-genetic Mu-seq for identification and functional classification of maize mutants. Front. Plant Sci. 2014, 4, 545. [Google Scholar] [CrossRef]
- Wang, L.; Lu, Z.; Regulski, M.; Jiao, Y.; Chen, J.; Ware, D.; Xin, Z. BSAseq: An interactive and integrated web-based workflow for identification of causal mutations in bulked F2 populations. Bioinformatics 2021, 37, 382–387. [Google Scholar] [CrossRef]
- Hill, J.T.; Demarest, B.L.; Bisgrove, B.W.; Gorsi, B.; Su, Y.C.; Yost, H.J. MMAPPR: Mutation mapping analysis pipeline for pooled RNA-seq. Genome Res. 2013, 23, 687–697. [Google Scholar] [CrossRef]
- Willems, A.; De Veylder, L. The plant anaphase-promoting complex/cyclosome. Annu. Rev. Cell Dev. Biol. 2022, 38, 25–48. [Google Scholar] [CrossRef]
- Schwickart, M.; Havlis, J.; Habermann, B.; Bogdanova, A.; Camasses, A.; Oelschlaegel, T.; Shevchenko, A.; Zachariae, W. Swm1/Apc13 is an evolutionarily conserved subunit of the anaphase-promoting complex stabilizing the association of Cdc16 and Cdc27. Mol. Cell. Biol. 2004, 24, 3562–3576. [Google Scholar] [CrossRef]
- Zhou, Y.; He, Q.; Chen, J.; Liu, Y.; Mao, Z.; Lyu, Z.; Ni, D.; Long, Y.; Ju, P.; Liu, J.; et al. The expression patterns of Tetratricopeptide repeat domain 36 (Ttc36). Gene Expr. Patterns 2016, 22, 37–45. [Google Scholar] [CrossRef]
- Wang, Q.; Moyret-Lalle, C.; Couzon, F.; Surbiguet-Clippe, C.; Saurin, J.C.; Lorca, T.; Navarro, C.; Puisieux, A. Alterations of anaphase-promoting complex genes in human colon cancer cells. Oncogene 2003, 22, 1486–1490. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.Y.; Xu, J.; Wang, L.; Niu, B.; Copenhaver, G.P.; Ma, H.; Zheng, B.; Wang, Y. The Arabidopsis anaphase-promoting complex/cyclosome subunit 8 is required for male meiosis. New Phytol. 2019, 224, 229–241. [Google Scholar] [CrossRef]
- Zheng, B.; Chen, X.; Mc Cormick, S. The anaphase-promoting complex is a dual integrator that regulates both MicroRNA-mediated transcriptional regulation of cyclin B1 and degradation of Cyclin B1 during arabidopsis male gametophyte development. Plant Cell 2011, 23, 1033–1046. [Google Scholar] [CrossRef] [PubMed]
- Eloy, N.B.; Coppens, F.; Beemster, G.T.; Hemerly, A.S.; Ferreira, P.C.G. The Arabidopsis anaphase promoting complex (APC): Regulation through subunit availability in plant tissues. Cell Cycle 2006, 5, 1957–1965. [Google Scholar] [CrossRef] [PubMed]
- Moran, N. Osmoregulation of leaf motor cells. FEBS Lett. 2007, 581, 2337–2347. [Google Scholar] [CrossRef]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, 1–7. [Google Scholar] [CrossRef]
- Hu, B.; Zhu, J.; Wu, H.; Xu, K.; Zhai, H.; Guo, N.; Gao, Y.; Yang, J.; Zhu, D.; Xia, Z. Enhanced Chlorophyll Degradation Triggers the Pod Degreening of “Golden Hook”, a Special Ecotype in Common Bean (Phaseolus vulgaris L.). Front. Genet. 2020, 11, 570816. [Google Scholar] [CrossRef]
- Li, J.J.; Hong, H.L.; Wan, M.Y.; Chu, L.; Zhao, J.H.; Wang, M.H. Construction and application of detection model for the chemical composition content of soybean stem based on near infrared spectroscopy. Sci. Agric. Sin. 2021, 54, 887–900. [Google Scholar] [CrossRef]
- Patel, R.K.; Jain, M. NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE 2012, 7, 2. [Google Scholar] [CrossRef]
- Jo, H. Multi-threading the generation of burrows-wheeler alignment. Genet. Mol. Res. 2016, 15, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant review with the integrative genomics viewer. Cancer Res. 2017, 77, e31–e34. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.J.; Tsubokura, Y.; Hoshi, M.; Hanawa, M.; Yano, C.; Okamura, K.; Ahmed, T.A.; Anai, T.; Watanabe, S.; Hayashi, M.; et al. An integrated high-density linkage map of soybean with RFLP, SSR, STS, and AFLP markers using a single F2 population. DNA Res. 2007, 14, 257–269. [Google Scholar] [CrossRef]
- Meng, L.; Li, H.H.; Zhang, L.Y.; Wang, J.K. QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J. 2015, 3, 269–283. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pages, F.; Trajanoski, A.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef]
- Lyu, X.; Li, M.; Li, X.; Li, S.; Yan, C.; Ma, C.; Gong, Z. Assessing the systematic effects of the concentration of nitrogen supplied to dual-root systems of soybean plants on nodulation and nitrogen fixation. Agronomy 2020, 10, 763. [Google Scholar] [CrossRef]
- Shan, F.; Zhang, R.; Zhang, J.; Wang, C.; Lyu, X.; Xin, T.; Yan, C.; Dong, S.; Ma, C.; Gong, Z. Study on the regulatory effects of GA3 on soybean internode elongation. Plants 2021, 10, 1737. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, Y.; Zhu, J.; Zhai, H.; Xu, K.; Zhu, X.; Wu, H.; Zhang, W.; Wu, S.; Chen, X.; Xia, Z. Dysfunction of an Anaphase-Promoting Complex Subunit 8 Homolog Leads to Super-Short Petioles and Enlarged Petiole Angles in Soybean. Int. J. Mol. Sci. 2023, 24, 11024. https://doi.org/10.3390/ijms241311024
Gao Y, Zhu J, Zhai H, Xu K, Zhu X, Wu H, Zhang W, Wu S, Chen X, Xia Z. Dysfunction of an Anaphase-Promoting Complex Subunit 8 Homolog Leads to Super-Short Petioles and Enlarged Petiole Angles in Soybean. International Journal of Molecular Sciences. 2023; 24(13):11024. https://doi.org/10.3390/ijms241311024
Chicago/Turabian StyleGao, Yi, Jinlong Zhu, Hong Zhai, Kun Xu, Xiaobin Zhu, Hongyan Wu, Wenjing Zhang, Shihao Wu, Xin Chen, and Zhengjun Xia. 2023. "Dysfunction of an Anaphase-Promoting Complex Subunit 8 Homolog Leads to Super-Short Petioles and Enlarged Petiole Angles in Soybean" International Journal of Molecular Sciences 24, no. 13: 11024. https://doi.org/10.3390/ijms241311024
APA StyleGao, Y., Zhu, J., Zhai, H., Xu, K., Zhu, X., Wu, H., Zhang, W., Wu, S., Chen, X., & Xia, Z. (2023). Dysfunction of an Anaphase-Promoting Complex Subunit 8 Homolog Leads to Super-Short Petioles and Enlarged Petiole Angles in Soybean. International Journal of Molecular Sciences, 24(13), 11024. https://doi.org/10.3390/ijms241311024