

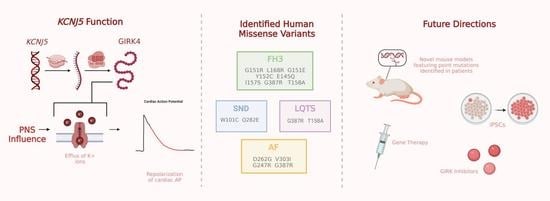
Relevance of KCNJ5 in Pathologies of Heart Disease


Abstract
{kind=link}
{kind=link}
{kind=link}
1. Introduction
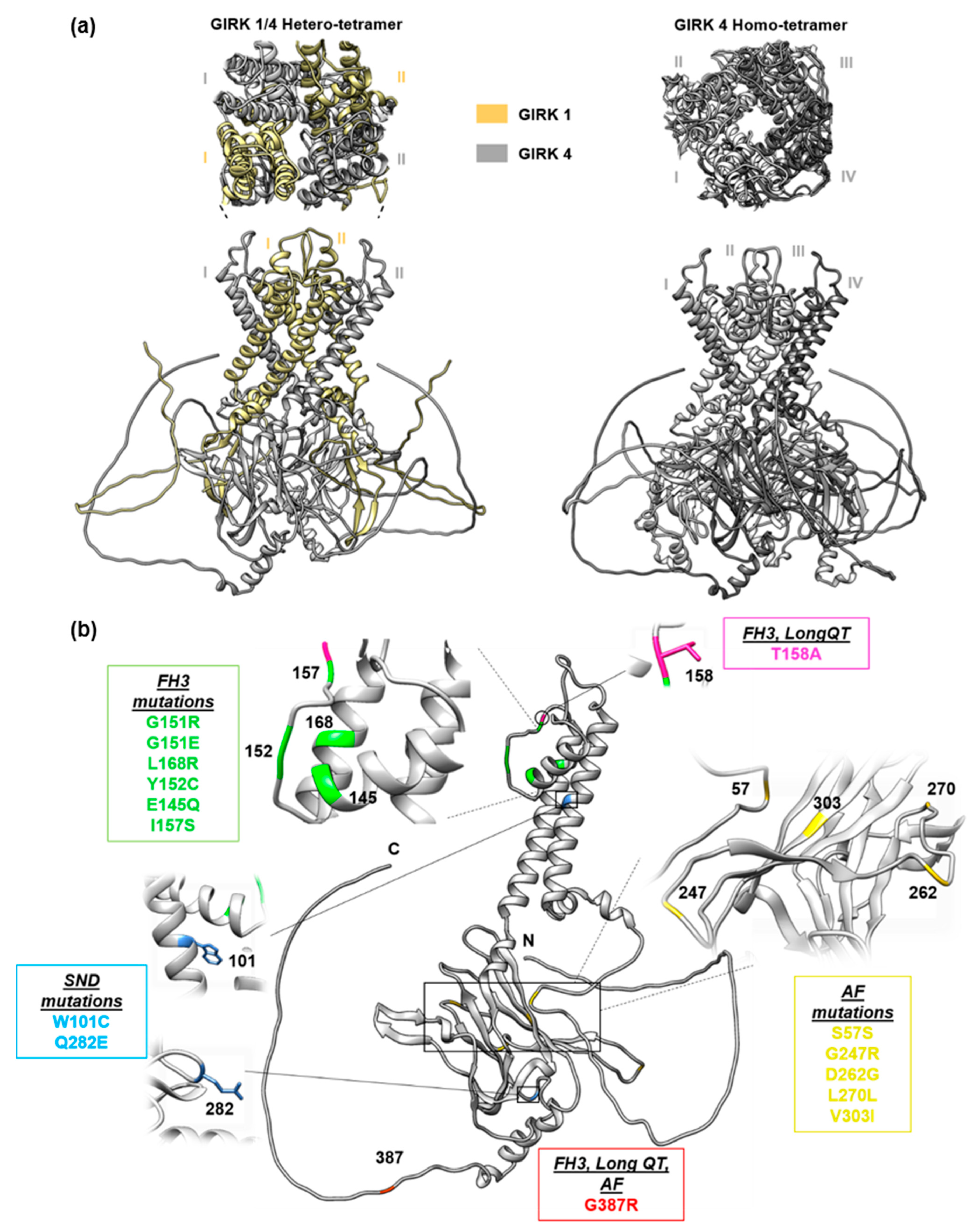
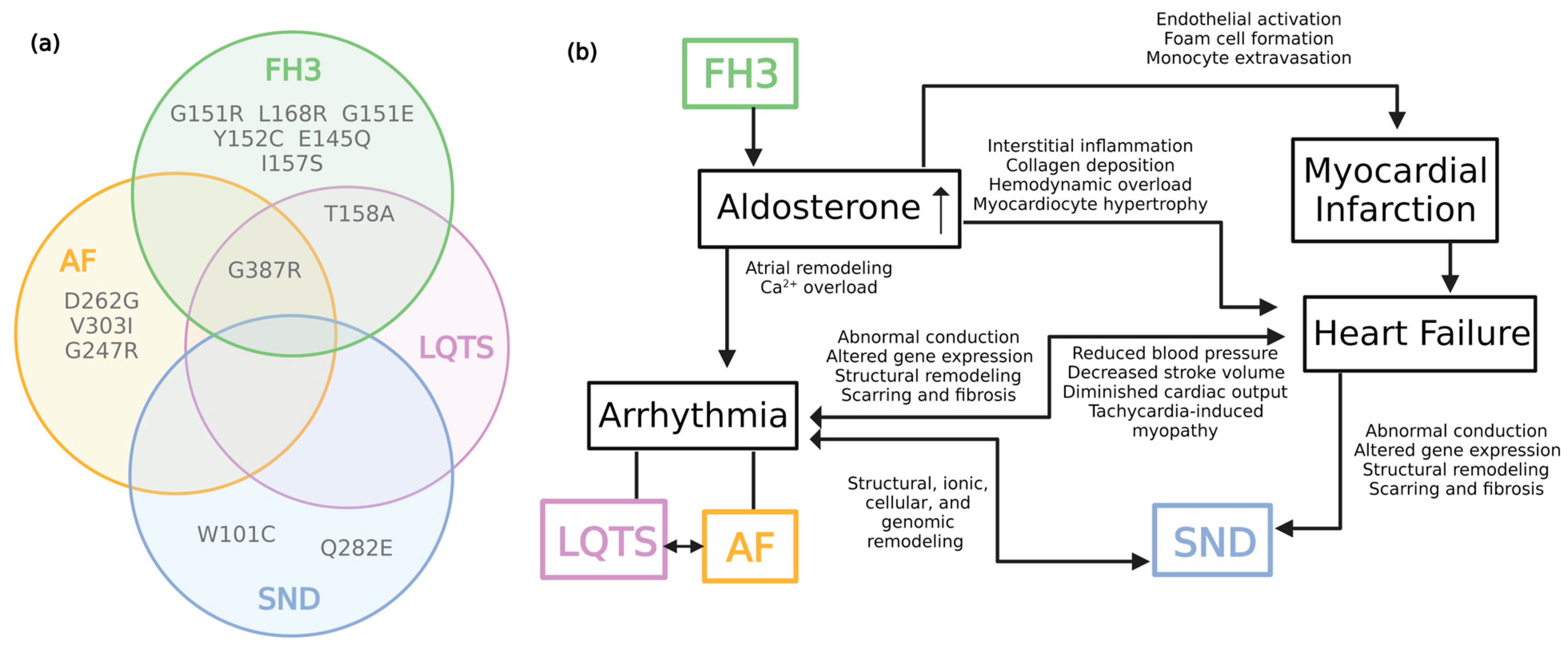
2. Familial Hyperaldosteronism Type III
3. KCNJ5 in Long QT Syndrome
4. KCNJ5 in Atrial Fibrillation
5. KCNJ5 in Sinus Node Dysfunction
6. Complexities of KCNJ5 in Cardiovascular Disease
7. Future Implications
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Krapivinsky, G.; Gordon, E.A.; Wickman, K.; Velimirović, B.; Krapivinsky, L.; Clapham, D.E. The G-protein-gated atrial K+ channel IKACh is a heteromultimer of two inwardly rectifying K+-channel proteins. Nature 1995, 374, 135–141. [Google Scholar] [CrossRef]
- Ferrer, J.; Nichols, C.G.; Makhina, E.N.; Salkoff, L.; Bernstein, J.; Gerhard, D.; Wasson, J.; Ramanadham, S.; Permutt, A. Pancreatic islet cells express a family of inwardly rectifying K+ channel subunits which interact to form G-protein-activated channels. J. Biol. Chem. 1995, 270, 26086–26091. [Google Scholar] [CrossRef]
- Choi, M.; Scholl, U.I.; Yue, P.; Björklund, P.; Zhao, B.; Nelson-Williams, C.; Ji, W.; Cho, Y.; Patel, A.; Men, C.J.; et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science 2011, 331, 768–772. [Google Scholar] [CrossRef] [PubMed]
- Wickman, K.; Nemec, J.; Gendler, S.J.; Clapham, D.E. Abnormal heart rate regulation in GIRK4 knockout mice. Neuron 1998, 20, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Mesirca, P.; Marger, L.; Toyoda, F.; Rizzetto, R.; Audoubert, M.; Dubel, S.; Torrente, A.G.; Difrancesco, M.L.; Muller, J.C.; Leoni, A.L.; et al. The G-protein-gated K+ channel, IKACh, is required for regulation of pacemaker activity and recovery of resting heart rate after sympathetic stimulation. J. Gen. Physiol. 2013, 142, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.; Kulkarni, K.; Marron Fernandez de Velasco, E.; Carlblom, N.; Xia, Z.; Nakano, A.; Martemyanov, K.A.; Tolkacheva, E.G.; Wickman, K. Expression and relevance of the G-protein-gated K+ channel in the mouse ventricle. Sci. Rep. 2018, 8, 1192. [Google Scholar] [CrossRef]
- Lee, S.W.; Anderson, A.; Guzman, P.A.; Nakano, A.; Tolkacheva, E.G.; Wickman, K. Atrial GIRK Channels Mediate the Effects of Vagus Nerve Stimulation on Heart Rate Dynamics and Arrhythmogenesis. Front. Physiol. 2018, 9, 943. [Google Scholar] [CrossRef]
- Gordan, R.; Gwathmey, J.K.; Xie, L.H. Autonomic and endocrine control of cardiovascular function. World J. Cardiol. 2015, 7, 204–214. [Google Scholar] [CrossRef]
- Touhara, K.K.; MacKinnon, R. Molecular basis of signaling specificity between GIRK channels and GPCRs. eLife 2018, 7, e42908. [Google Scholar] [CrossRef]
- Bukiya, A.N.; Osborn, C.V.; Kuntamallappanavar, G.; Toth, P.T.; Baki, L.; Kowalsky, G.; Oh, M.J.; Dopico, A.M.; Levitan, I.; Rosenhouse-Dantsker, A. Cholesterol increases the open probability of cardiac KACh currents. Biochim. Biophys. Acta 2015, 1848, 2406–2413. [Google Scholar] [CrossRef]
- Kobayashi, T.; Ikeda, K.; Kojima, H.; Niki, H.; Yano, R.; Yoshioka, T.; Kumanishi, T. Ethanol opens G-protein-activated inwardly rectifying K+ channels. Nat. Neurosci. 1999, 2, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Tao, X.; Touhara, K.K.; MacKinnon, R. Cryo-EM analysis of PIP2 regulation in mammalian GIRK channels. eLife 2020, 9, e60552. [Google Scholar] [CrossRef] [PubMed]
- Campos-Ríos, A.; Rueda-Ruzafa, L.; Lamas, J.A. The Relevance of GIRK Channels in Heart Function. Membranes 2022, 12, 1119. [Google Scholar] [CrossRef]
- Wickman, K.; Krapivinsky, G.; Corey, S.; Kennedy, M.; Nemec, J.; Medina, I.; Clapham, D.E. Structure, G-protein activation, and functional relevance of the cardiac G-protein-gated K+ channel, IKACh. Ann. N. Y. Acad. Sci. 1999, 868, 386–398. [Google Scholar] [CrossRef]
- Kennedy, M.E.; Nemec, J.; Corey, S.; Wickman, K.; Clapham, D.E. GIRK4 confers appropriate processing and cell surface localization to G-protein-gated potassium channels. J. Biol. Chem. 1999, 274, 2571–2582. [Google Scholar] [CrossRef] [PubMed]
- Hedin, K.E.; Lim, N.F.; Clapham, D.E. Cloning of a Xenopus laevis inwardly rectifying K+ channel subunit that permits GIRK1 expression of IKACh currents in oocytes. Neuron 1996, 16, 423–429. [Google Scholar] [CrossRef]
- Consortium, T.U. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2022, 51, D523–D531. [Google Scholar] [CrossRef]
- Stevens, E.B.; Woodward, R.; Ho, I.H.; Murrell-Lagnado, R. Identification of regions that regulate the expression and activity of G-protein-gated inward rectifier K+ channels in Xenopus oocytes. J. Physiol. 1997, 503, 547–562. [Google Scholar] [CrossRef]
- Woodward, R.; Stevens, E.B.; Murrell-Lagnado, R.D. Molecular Determinants for Assembly of G-protein-activated Inwardly Rectifying K+ Channels*. J. Biol. Chem. 1997, 272, 10823–10830. [Google Scholar] [CrossRef]
- Stavrakis, S.; Kulkarni, K.; Singh, J.P.; Katritsis, D.G.; Armoundas, A.A. Autonomic Modulation of Cardiac Arrhythmias: Methods to Assess Treatment and Outcomes. JACC Clin. Electrophysiol. 2020, 6, 467–483. [Google Scholar] [CrossRef]
- Harvey, R.D. Muscarinic receptor agonists and antagonists: Effects on cardiovascular function. Handb. Exp. Pharmacol. 2012, 208, 299–316. [Google Scholar]
- Monticone, S.; Tetti, M.; Burrello, J.; Buffolo, F.; De Giovanni, R.; Veglio, F.; Williams, T.A.; Mulatero, P. Familial hyperaldosteronism type III. J. Hum. Hypertens. 2017, 31, 776–781. [Google Scholar] [CrossRef] [PubMed]
- Tauber, P.; Penton, D.; Stindl, J.; Humberg, E.; Tegtmeier, I.; Sterner, C.; Beuschlein, F.; Reincke, M.; Barhanin, J.; Bandulik, S.; et al. Pharmacology and pathophysiology of mutated KCNJ5 found in adrenal aldosterone-producing adenomas. Endocrinology 2014, 155, 1353–1362. [Google Scholar] [CrossRef]
- He, B.J.; Anderson, M.E. Aldosterone and cardiovascular disease: The heart of the matter. Trends Endocrinol. Metab. 2013, 24, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Buffolo, F.; Tetti, M.; Mulatero, P.; Monticone, S. Aldosterone as a Mediator of Cardiovascular Damage. Hypertension 2022, 79, 1899–1911. [Google Scholar] [CrossRef]
- Chueh, J.S.; Peng, K.Y.; Wu, V.C.; Wang, S.M.; Chan, C.K.; Chen, Y.M.; Ke, Y.Y.; Pan, C.Y.; Liao, H.W. Characterization of a mutated KCNJ5 gene, G387R, in unilateral primary aldosteronism. J. Mol. Endocrinol. 2021, 67, 203–215. [Google Scholar] [CrossRef]
- Scholl, U.I.; Nelson-Williams, C.; Yue, P.; Grekin, R.; Wyatt, R.J.; Dillon, M.J.; Couch, R.; Hammer, L.K.; Harley, F.L.; Farhi, A. Hypertension with or without adrenal hyperplasia due to different inherited mutations in the potassium channel KCNJ5. Proc. Natl. Acad. Sci. 2012, 109, 2533–2538. [Google Scholar] [CrossRef]
- Geller, D.S.; Zhang, J.; Wisgerhof, M.V.; Shackleton, C.; Kashgarian, M.; Lifton, R.P. A novel form of human mendelian hypertension featuring nonglucocorticoid-remediable aldosteronism. J. Clin. Endocrinol. Metab. 2008, 93, 3117–3123. [Google Scholar] [CrossRef]
- Monticone, S.; Hattangady, N.G.; Penton, D.; Isales, C.M.; Edwards, M.A.; Williams, T.A.; Sterner, C.; Warth, R.; Mulatero, P.; Rainey, W.E. A novel Y152C KCNJ5 mutation responsible for familial hyperaldosteronism type III. J. Clin. Endocrinol. Metab. 2013, 98, E1861–E1865. [Google Scholar] [CrossRef]
- Monticone, S.; Bandulik, S.; Stindl, J.; Zilbermint, M.; Dedov, I.; Mulatero, P.; Allgaeuer, M.; Lee, C.-C.R.; Stratakis, C.A.; Williams, T.A. A case of severe hyperaldosteronism caused by a de novo mutation affecting a critical salt bridge Kir3. 4 residue. J. Clin. Endocrinol. Metab. 2015, 100, E114–E118. [Google Scholar] [CrossRef]
- Charmandari, E.; Sertedaki, A.; Kino, T.; Merakou, C.; Hoffman, D.A.; Hatch, M.M.; Hurt, D.E.; Lin, L.; Xekouki, P.; Stratakis, C.A. A novel point mutation in the KCNJ5 gene causing primary hyperaldosteronism and early-onset autosomal dominant hypertension. J. Clin. Endocrinol. Metab. 2012, 97, E1532–E1539. [Google Scholar] [CrossRef] [PubMed]
- Åkerström, T.; Crona, J.; Delgado Verdugo, A.; Starker, L.F.; Cupisti, K.; Willenberg, H.S.; Knoefel, W.T.; Saeger, W.; Feller, A.; Ip, J.; et al. Comprehensive re-sequencing of adrenal aldosterone producing lesions reveal three somatic mutations near the KCNJ5 potassium channel selectivity filter. PLoS ONE 2012, 7, e41926. [Google Scholar] [CrossRef] [PubMed]
- Dibb, K.M.; Rose, T.; Makary, S.Y.; Claydon, T.W.; Enkvetchakul, D.; Leach, R.; Nichols, C.G.; Boyett, M.R. Molecular basis of ion selectivity, block, and rectification of the inward rectifier Kir3.1/Kir3.4 K+ channel. J. Biol. Chem. 2003, 278, 49537–49548. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Rusin, C.G.; Tan, Z.; Guagliardo, N.A.; Barrett, P.Q. Zona glomerulosa cells of the mouse adrenal cortex are intrinsic electrical oscillators. J. Clin. Invest. 2012, 122, 2046–2053. [Google Scholar] [CrossRef] [PubMed]
- Tester, D.J.; Ackerman, M.J. Genetics of long QT syndrome. Methodist Debakey Cardiovasc. J. 2014, 10, 29–33. [Google Scholar] [CrossRef]
- Pérez-Riera, A.R.; Barbosa-Barros, R.; Samesina, N.; Pastore, C.A.; Scanavacca, M.; Daminello-Raimundo, R.; de Abreu, L.C.; Nikus, K.; Brugada, P. Andersen-Tawil Syndrome: A Comprehensive Review. Cardiol. Rev. 2021, 29, 165–177. [Google Scholar] [CrossRef]
- Kokunai, Y.; Nakata, T.; Furuta, M.; Sakata, S.; Kimura, H.; Aiba, T.; Yoshinaga, M.; Osaki, Y.; Nakamori, M.; Itoh, H.; et al. A Kir3.4 mutation causes Andersen–Tawil syndrome by an inhibitory effect on Kir2.1. Neurology 2014, 82, 1058–1064. [Google Scholar] [CrossRef]
- Wang, F.; Liu, J.; Hong, L.; Liang, B.; Graff, C.; Yang, Y.; Christiansen, M.; Olesen, S.P.; Zhang, L.; Kanters, J.K. The phenotype characteristics of type 13 long QT syndrome with mutation in KCNJ5 (Kir3.4-G387R). Heart Rhythm 2013, 10, 1500–1506. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, Y.; Liang, B.; Liu, J.; Li, J.; Grunnet, M.; Olesen, S.P.; Rasmussen, H.B.; Ellinor, P.T.; Gao, L.; et al. Identification of a Kir3.4 mutation in congenital long QT syndrome. Am. J. Hum. Genet. 2010, 86, 872–880. [Google Scholar] [CrossRef]
- Adler, A.; Novelli, V.; Amin, A.S.; Abiusi, E.; Care, M.; Nannenberg, E.A.; Feilotter, H.; Amenta, S.; Mazza, D.; Bikker, H.; et al. An International, Multicentered, Evidence-Based Reappraisal of Genes Reported to Cause Congenital Long QT Syndrome. Circulation 2020, 141, 418–428. [Google Scholar] [CrossRef]
- Kovoor, P.; Wickman, K.; Maguire, C.T.; Pu, W.; Gehrmann, J.; Berul, C.I.; Clapham, D.E. Evaluation of the role of I(KACh) in atrial fibrillation using a mouse knockout model. J. Am. Coll. Cardiol. 2001, 37, 2136–2143. [Google Scholar] [CrossRef]
- Dhamoon, A.S.; Jalife, J. The inward rectifier current (IK1) controls cardiac excitability and is involved in arrhythmogenesis. Heart Rhythm 2005, 2, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Dobrev, D.; Graf, E.; Wettwer, E.; Himmel, H.M.; Hála, O.; Doerfel, C.; Christ, T.; Schüler, S.; Ravens, U. Molecular basis of downregulation of G-protein–coupled inward rectifying K+ current (IK,ACh) in chronic human atrial fibrillation: Decrease in GIRK4 mRNA correlates with reduced IK,ACh and muscarinic receptor–mediated shortening of action potentials. Circulation 2001, 104, 2551–2557. [Google Scholar] [CrossRef] [PubMed]
- Dobrev, D.; Friedrich, A.; Voigt, N.; Jost, N.; Wettwer, E.; Christ, T.; Knaut, M.; Ravens, U. The G-protein-gated potassium current IK,ACh is constitutively active in patients with chronic atrial fibrillation. Circulation 2005, 112, 3697–3706. [Google Scholar] [CrossRef] [PubMed]
- Voigt, N.; Trausch, A.; Knaut, M.; Matschke, K.; Varró, A.; Wagoner, D.R.V.; Nattel, S.; Ravens, U.; Dobrev, D. Left-to-Right Atrial Inward Rectifier Potassium Current Gradients in Patients With Paroxysmal Versus Chronic Atrial Fibrillation. Circ. Arrhythmia Electrophysiol. 2010, 3, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Csepe, T.A.; Hansen, B.J.; Sul, L.V.; Kalyanasundaram, A.; Zakharkin, S.O.; Zhao, J.; Guha, A.; Wagoner, D.R.V.; Kilic, A.; et al. Adenosine-Induced Atrial Fibrillation. Circulation 2016, 134, 486–498. [Google Scholar] [CrossRef]
- Strickberger, S.A.; Man, K.C.; Daoud, E.G.; Goyal, R.; Brinkman, K.; Knight, B.P.; Weiss, R.; Bahu, M.; Morady, F. Adenosine-induced atrial arrhythmia: A prospective analysis. Ann. Intern. Med. 1997, 127, 417–422. [Google Scholar] [CrossRef]
- Yamada, N.; Asano, Y.; Fujita, M.; Yamazaki, S.; Inanobe, A.; Matsuura, N.; Kobayashi, H.; Ohno, S.; Ebana, Y.; Tsukamoto, O.; et al. Mutant KCNJ3 and KCNJ5 Potassium Channels as Novel Molecular Targets in Bradyarrhythmias and Atrial Fibrillation. Circulation 2019, 139, 2157–2169. [Google Scholar] [CrossRef]
- Luján, R.; Maylie, J.; Adelman, J.P. New sites of action for GIRK and SK channels. Nat. Rev. Neurosci. 2009, 10, 475–480. [Google Scholar] [CrossRef]
- Calloe, K.; Ravn, L.S.; Schmitt, N.; Sui, J.L.; Duno, M.; Haunso, S.; Grunnet, M.; Svendsen, J.H.; Olesen, S.P. Characterizations of a loss-of-function mutation in the Kir3.4 channel subunit. Biochem. Biophys. Res. Commun. 2007, 364, 889–895. [Google Scholar] [CrossRef]
- Krapivinsky, G.; Kennedy, M.E.; Nemec, J.; Medina, I.; Krapivinsky, L.; Clapham, D.E. Gβγ Binding to GIRK4 Subunit Is Critical for G-protein-gated K+ Channel Activation. J. Biol. Chem. 1998, 273, 16946–16952. [Google Scholar] [CrossRef]
- Zhang, C.; Yuan, G.H.; Cheng, Z.F.; Xu, M.W.; Hou, L.F.; Wei, F.P. The single nucleotide polymorphisms of Kir3.4 gene and their correlation with lone paroxysmal atrial fibrillation in Chinese Han population. Heart Lung Circ. 2009, 18, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Jabbari, J.; Olesen, M.S.; Holst, A.G.; Nielsen, J.B.; Haunso, S.; Svendsen, J.H. Common polymorphisms in KCNJ5 [corrected] are associated with early-onset lone atrial fibrillation in Caucasians. Cardiology 2011, 118, 116–120. [Google Scholar] [CrossRef]
- Young, L.J.; Antwi-Boasiako, S.; Ferrall, J.; Wold, L.E.; Mohler, P.J.; El Refaey, M. Genetic and non-genetic risk factors associated with atrial fibrillation. Life Sci. 2022, 299, 120529. [Google Scholar] [CrossRef] [PubMed]
- Brundel, B.; Ai, X.; Hills, M.T.; Kuipers, M.F.; Lip, G.Y.H.; de Groot, N.M.S. Atrial fibrillation. Nat. Rev. Dis. Prim. 2022, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Hawks, M.K.; Paul, M.L.B.; Malu, O.O. Sinus Node Dysfunction. Am. Fam. Physician 2021, 104, 179–185. [Google Scholar]
- Monfredi, O.; Boyett, M.R. Sick sinus syndrome and atrial fibrillation in older persons—A view from the sinoatrial nodal myocyte. J. Mol. Cell. Cardiol. 2015, 83, 88–100. [Google Scholar] [CrossRef]
- Carlisle, M.A.; Fudim, M.; DeVore, A.D.; Piccini, J.P. Heart Failure and Atrial Fibrillation, Like Fire and Fury. JACC Heart Fail. 2019, 7, 447–456. [Google Scholar] [CrossRef]
- John, R.M.; Kumar, S. Sinus Node and Atrial Arrhythmias. Circulation 2016, 133, 1892–1900. [Google Scholar] [CrossRef]
- Mesirca, P.; Bidaud, I.; Briec, F.; Evain, S.; Torrente, A.G.; Le Quang, K.; Leoni, A.L.; Baudot, M.; Marger, L.; Chung You Chong, A.; et al. G-protein-gated IKACh channels as therapeutic targets for treatment of sick sinus syndrome and heart block. Proc. Natl. Acad. Sci. USA 2016, 113, E932–E941. [Google Scholar] [CrossRef]
- Bidaud, I.; Chong, A.C.Y.; Carcouet, A.; Waard, S.; Charpentier, F.; Ronjat, M.; Waard, M.; Isbrandt, D.; Wickman, K.; Vincent, A.; et al. Inhibition of G-protein-gated K+ channels by tertiapin-Q rescues sinus node dysfunction and atrioventricular conduction in mouse models of primary bradycardia. Sci. Rep. 2020, 10, 9835. [Google Scholar] [CrossRef]
- Mesirca, P.; Alig, J.; Torrente, A.G.; Müller, J.C.; Marger, L.; Rollin, A.; Marquilly, C.; Vincent, A.; Dubel, S.; Bidaud, I.; et al. Cardiac arrhythmia induced by genetic silencing of ‘funny’ (f) channels is rescued by GIRK4 inactivation. Nat. Commun. 2014, 5, 4664. [Google Scholar] [CrossRef]
- Li, N.; Hansen, B.J.; Csepe, T.A.; Zhao, J.; Ignozzi, A.J.; Sul, L.V.; Zakharkin, S.O.; Kalyanasundaram, A.; Davis, J.P.; Biesiadecki, B.J.; et al. Redundant and diverse intranodal pacemakers and conduction pathways protect the human sinoatrial node from failure. Sci. Transl. Med. 2017, 9, eaam5607. [Google Scholar] [CrossRef]
- Bidaud, I.; D’Souza, A.; Forte, G.; Torre, E.; Greuet, D.; Thirard, S.; Anderson, C.; Chung You Chong, A.; Torrente, A.G.; Roussel, J.; et al. Genetic Ablation of G-protein-Gated Inwardly Rectifying K+ Channels Prevents Training-Induced Sinus Bradycardia. Front. Physiol. 2020, 11, 519382. [Google Scholar] [CrossRef]
- DiFrancesco, D. The role of the funny current in pacemaker activity. Circ. Res. 2010, 106, 434–446. [Google Scholar] [CrossRef]
- Hagiwara, N.; Irisawa, H.; Kameyama, M. Contribution of two types of calcium currents to the pacemaker potentials of rabbit sino-atrial node cells. J. Physiol. 1988, 395, 233–253. [Google Scholar] [CrossRef]
- Mangoni, M.E.; Traboulsie, A.; Leoni, A.L.; Couette, B.; Marger, L.; Le Quang, K.; Kupfer, E.; Cohen-Solal, A.; Vilar, J.; Shin, H.S.; et al. Bradycardia and slowing of the atrioventricular conduction in mice lacking CaV3.1/α1G T-type calcium channels. Circ. Res. 2006, 98, 1422–1430. [Google Scholar] [CrossRef]
- Torrente, A.G.; Mesirca, P.; Neco, P.; Rizzetto, R.; Dubel, S.; Barrere, C.; Sinegger-Brauns, M.; Striessnig, J.; Richard, S.; Nargeot, J.; et al. L-type Cav1.3 channels regulate ryanodine receptor-dependent Ca2+ release during sino-atrial node pacemaker activity. Cardiovasc. Res. 2016, 109, 451–461. [Google Scholar] [CrossRef]
- Verheijck, E.E.; van Ginneken, A.C.; Wilders, R.; Bouman, L.N. Contribution of L-type Ca2+ current to electrical activity in sinoatrial nodal myocytes of rabbits. Am. J. Physiol. 1999, 276, H1064–H1077. [Google Scholar] [CrossRef]
- Stallmeyer, B.; Kuß, J.; Kotthoff, S.; Zumhagen, S.; Vowinkel, K.; Rinné, S.; Matschke, L.A.; Friedrich, C.; Schulze-Bahr, E.; Rust, S.; et al. A Mutation in the G-Protein Gene GNB2 Causes Familial Sinus Node and Atrioventricular Conduction Dysfunction. Circ. Res. 2017, 120, e33–e44. [Google Scholar] [CrossRef]
- Kuß, J.; Stallmeyer, B.; Goldstein, M.; Rinné, S.; Pees, C.; Zumhagen, S.; Seebohm, G.; Decher, N.; Pott, L.; Kienitz, M.C.; et al. Familial Sinus Node Disease Caused by a Gain of GIRK (G-Protein Activated Inwardly Rectifying K+ Channel) Channel Function. Circulation. Genom. Precis. Med. 2019, 12, e002238. [Google Scholar] [CrossRef]
- Baronas, V.A.; Kurata, H.T. Inward rectifiers and their regulation by endogenous polyamines. Front. Physiol. 2014, 5, 325. [Google Scholar] [CrossRef]
- Lu, Z.; MacKinnon, R. Electrostatic tuning of Mg2+ affinity in an inward-rectifier K+ channel. Nature 1994, 371, 243–246. [Google Scholar] [CrossRef]
- Bollati, M.; Lopez, C.; Bioletto, F.; Ponzetto, F.; Ghigo, E.; Maccario, M.; Parasiliti-Caprino, M. Atrial Fibrillation and Aortic Ectasia as Complications of Primary Aldosteronism: Focus on Pathophysiological Aspects. Int. J. Mol. Sci. 2022, 23, 2111. [Google Scholar] [CrossRef]
- Holmegard, H.N.; Theilade, J.; Benn, M.; Duno, M.; Haunso, S.; Svendsen, J.H. Genetic variation in the inwardly rectifying K channel subunits KCNJ3 (GIRK1) and KCNJ5 (GIRK4) in patients with sinus node dysfunction. Cardiology 2010, 115, 176–181. [Google Scholar] [CrossRef]
- Inui, M.; Miyado, M.; Igarashi, M.; Tamano, M.; Kubo, A.; Yamashita, S.; Asahara, H.; Fukami, M.; Takada, S. Rapid generation of mouse models with defined point mutations by the CRISPR/Cas9 system. Sci. Rep. 2014, 4, 5396. [Google Scholar] [CrossRef]
- Ye, L.; Ni, X.; Zhao, Z.A.; Lei, W.; Hu, S. The Application of Induced Pluripotent Stem Cells in Cardiac Disease Modeling and Drug Testing. J. Cardiovasc. Transl. Res. 2018, 11, 366–374. [Google Scholar] [CrossRef]
- Aboul-Soud, M.A.M.; Alzahrani, A.J.; Mahmoud, A. Induced Pluripotent Stem Cells (iPSCs)-Roles in Regenerative Therapies, Disease Modelling and Drug Screening. Cells 2021, 10, 2319. [Google Scholar] [CrossRef]
- Kleinsorge, M.; Cyganek, L. Subtype-Directed Differentiation of Human iPSCs into Atrial and Ventricular Cardiomyocytes. STAR Protoc. 2020, 1, 100026. [Google Scholar] [CrossRef]
- Voigt, N.; Dobrev, D. Atrial-Selective Potassium Channel Blockers. Card. Electrophysiol. Clin. 2016, 8, 411–421. [Google Scholar] [CrossRef]
- Anderson, A.; Vo, B.N.; de Velasco, E.M.F.; Hopkins, C.R.; Weaver, C.D.; Wickman, K. Characterization of VU0468554, a New Selective Inhibitor of Cardiac G-protein-Gated Inwardly Rectifying K+ Channels. Mol. Pharmacol. 2021, 100, 540–547. [Google Scholar] [CrossRef]
- Cui, M.; Alhamshari, Y.; Cantwell, L.; Ei-Haou, S.; Eptaminitaki, G.C.; Chang, M.; Abou-Assali, O.; Tan, H.; Xu, K.; Masotti, M.; et al. A benzopyran with antiarrhythmic activity is an inhibitor of Kir3.1-containing potassium channels. J. Biol. Chem. 2021, 296, 100535. [Google Scholar] [CrossRef]
- Fenner, M.F.; Carstensen, H.; Dalgas Nissen, S.; Melis Hesselkilde, E.; Scott Lunddahl, C.; Adler Hess Jensen, M.; Loft-Andersen, A.V.; Sattler, S.M.; Platonov, P.; El-Haou, S.; et al. Effect of selective IK,ACh inhibition by XAF-1407 in an equine model of tachypacing-induced persistent atrial fibrillation. Br. J. Pharmacol. 2020, 177, 3778–3794. [Google Scholar] [CrossRef]
- Sobota, V.; Gatta, G.; van Hunnik, A.; van Tuijn, I.; Kuiper, M.; Milnes, J.; Jespersen, T.; Schotten, U.; Verheule, S. The Acetylcholine-Activated Potassium Current Inhibitor XAF-1407 Terminates Persistent Atrial Fibrillation in Goats. Front. Pharmacol. 2020, 11, 608410. [Google Scholar] [CrossRef]
- Jin, W.; Lu, Z. A novel high-affinity inhibitor for inward-rectifier K+ channels. Biochemistry 1998, 37, 13291–13299. [Google Scholar] [CrossRef]
- Hashimoto, N.; Yamashita, T.; Tsuruzoe, N. Tertiapin, a selective IKACh blocker, terminates atrial fibrillation with selective atrial effective refractory period prolongation. Pharmacol. Res. 2006, 54, 136–141. [Google Scholar] [CrossRef]
- Liu, X.; Yang, J.; Shang, F.; Hong, C.; Guo, W.; Wang, B.; Zheng, Q. Silencing GIRK4 expression in human atrial myocytes by adenovirus-delivered small hairpin RNA. Mol. Biol. Rep. 2009, 36, 1345–1352. [Google Scholar] [CrossRef]
- Cao, G.; Xuan, X.; Zhang, R.; Hu, J.; Dong, H. Gene Therapy for Cardiovascular Disease: Basic Research and Clinical Prospects. Front. Cardiovasc. Med. 2021, 8, 760140. [Google Scholar] [CrossRef]
- Zhao, Y.; Gameiro-Ros, I.; Glaaser, I.W.; Slesinger, P.A. Advances in Targeting GIRK Channels in Disease. Trends Pharmacol. Sci. 2021, 42, 203–215. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meyer, K.M.; Malhotra, N.; Kwak, J.s.; El Refaey, M. Relevance of KCNJ5 in Pathologies of Heart Disease. Int. J. Mol. Sci. 2023, 24, 10849. https://doi.org/10.3390/ijms241310849
Meyer KM, Malhotra N, Kwak Js, El Refaey M. Relevance of KCNJ5 in Pathologies of Heart Disease. International Journal of Molecular Sciences. 2023; 24(13):10849. https://doi.org/10.3390/ijms241310849
Chicago/Turabian StyleMeyer, Karisa M., Nipun Malhotra, Jung seo Kwak, and Mona El Refaey. 2023. "Relevance of KCNJ5 in Pathologies of Heart Disease" International Journal of Molecular Sciences 24, no. 13: 10849. https://doi.org/10.3390/ijms241310849
APA StyleMeyer, K. M., Malhotra, N., Kwak, J. s., & El Refaey, M. (2023). Relevance of KCNJ5 in Pathologies of Heart Disease. International Journal of Molecular Sciences, 24(13), 10849. https://doi.org/10.3390/ijms241310849