

Human Sinoatrial Node Pacemaker Activity: Role of the Slow Component of the Delayed Rectifier K+ Current, IKs



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
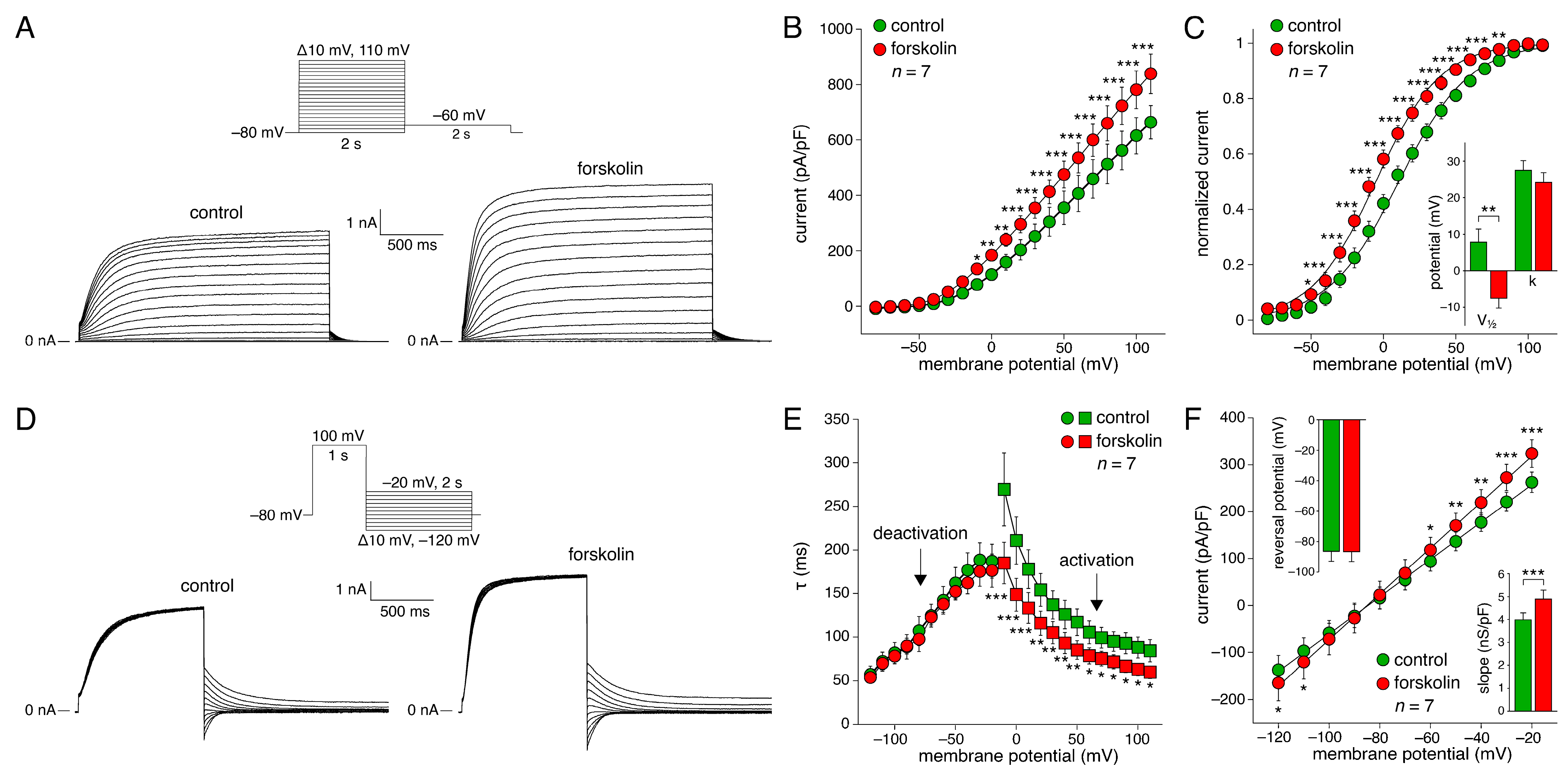
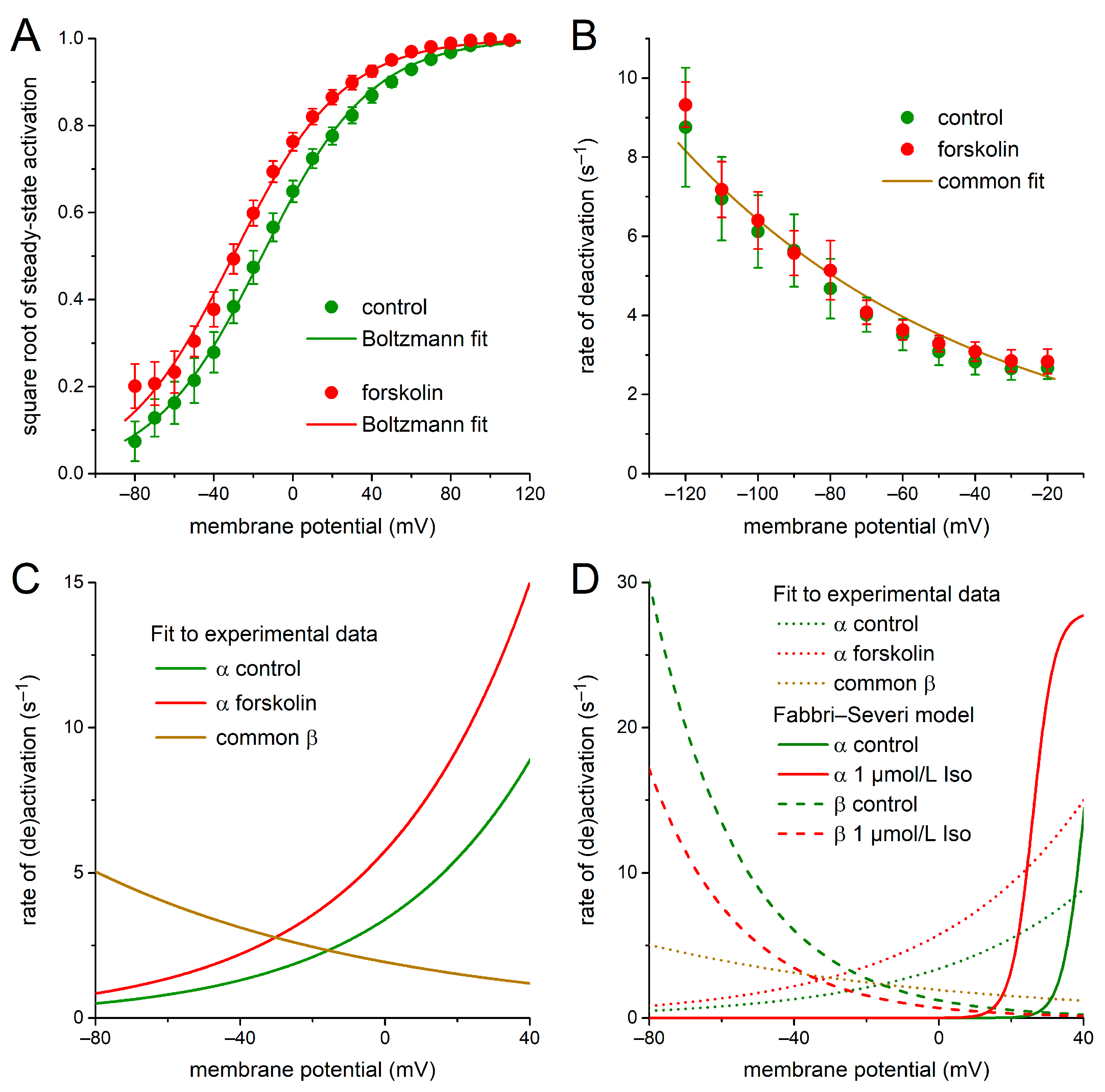
2.1. KCNQ1/KCNE1 Current in Response to Square Voltage Clamp Steps
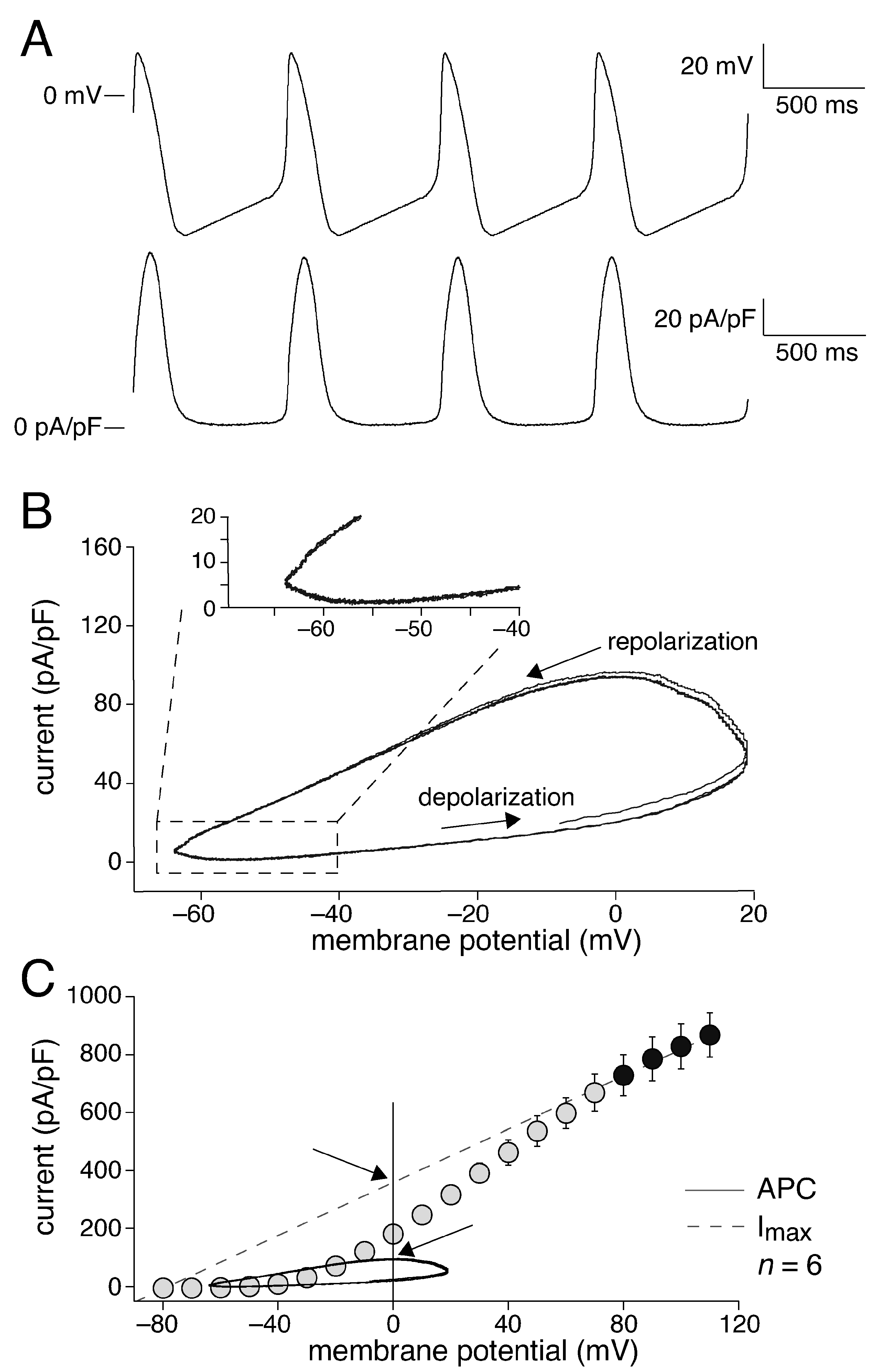
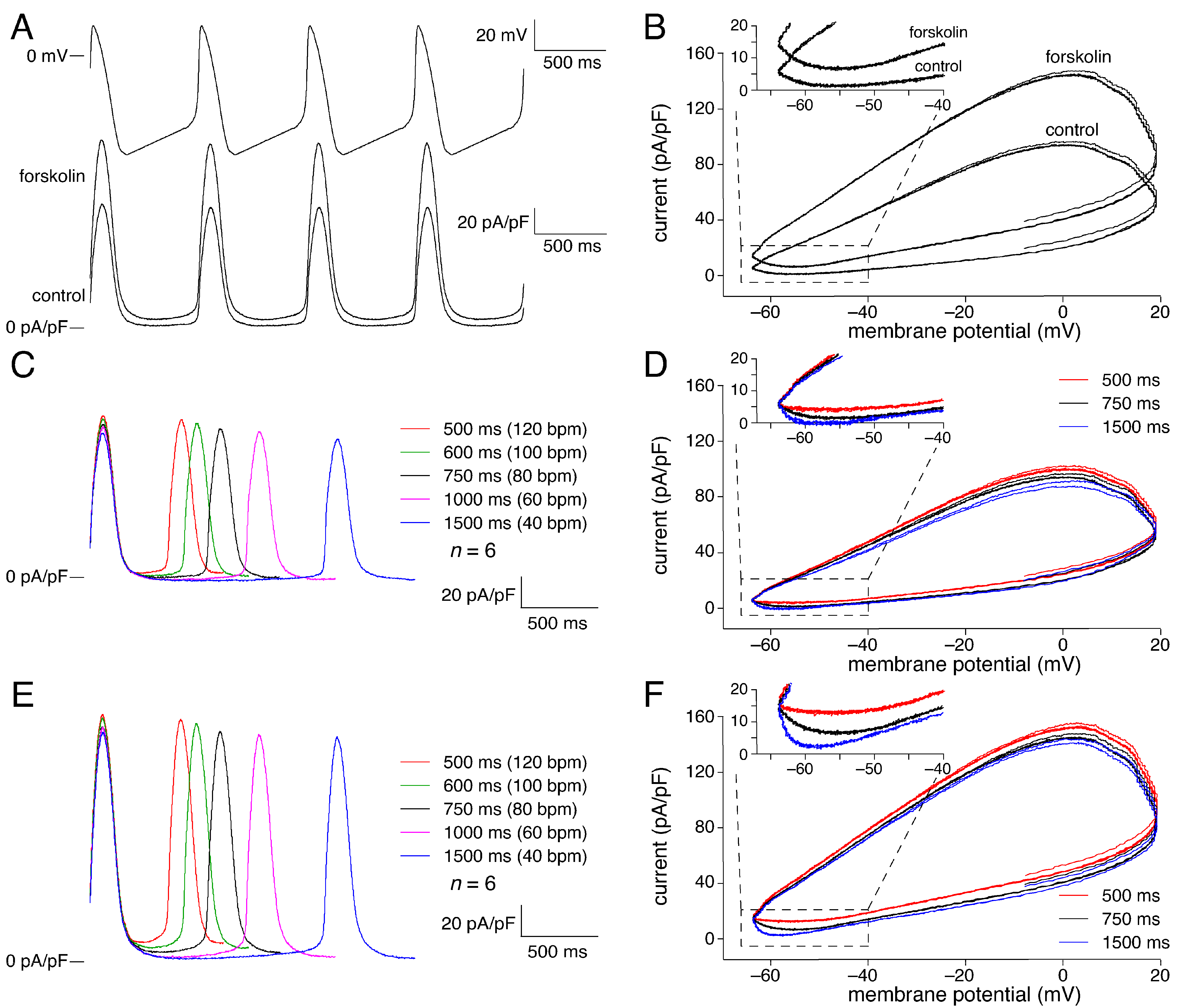
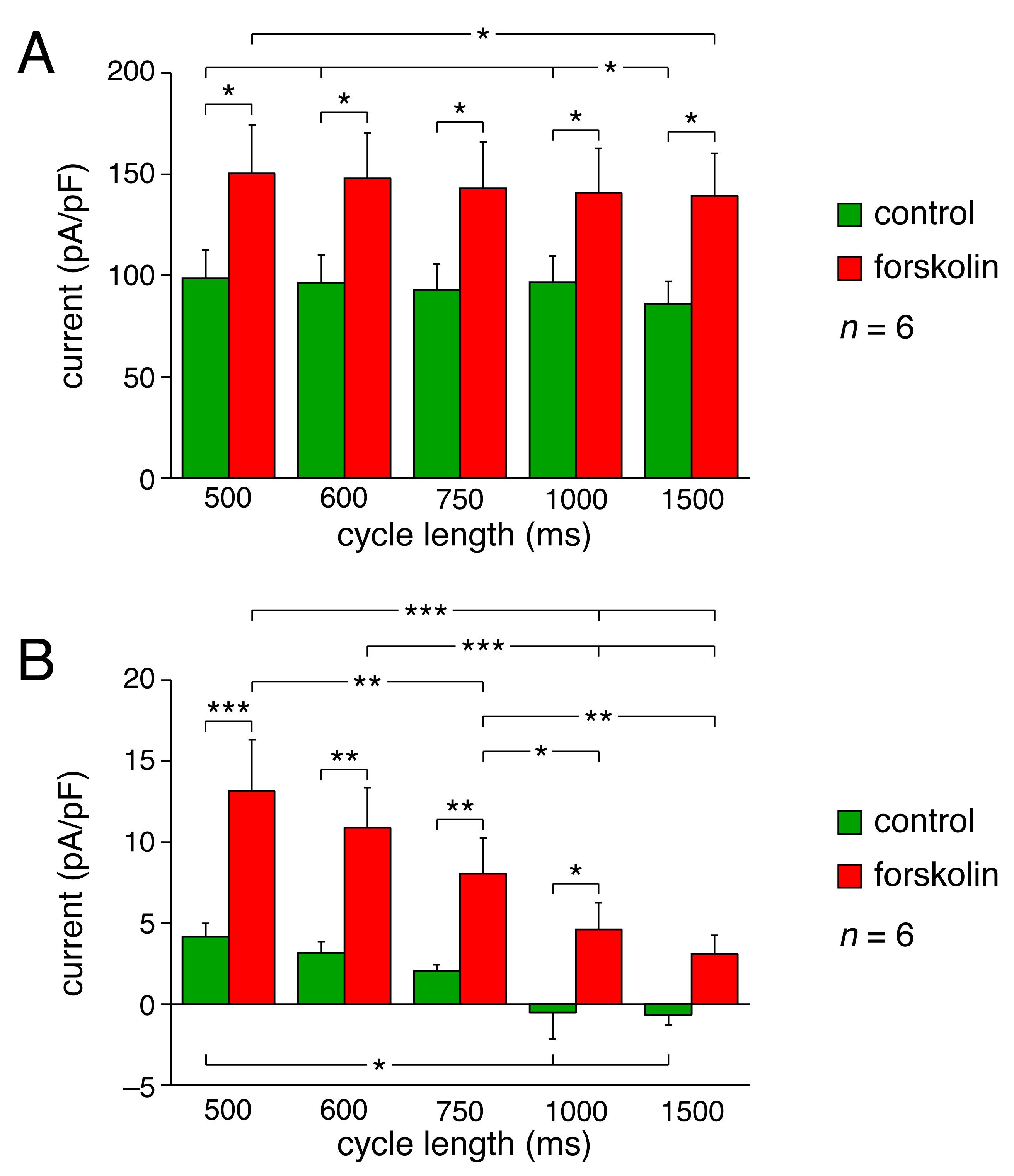
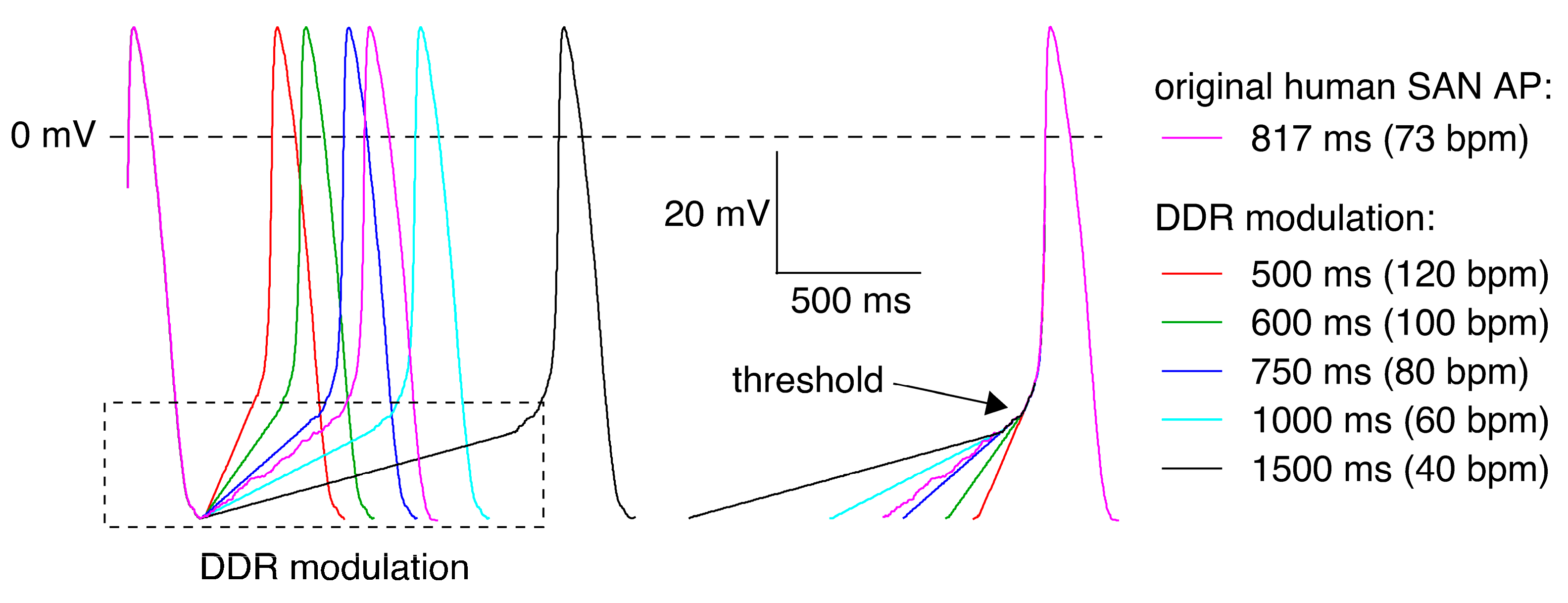
2.2. KCNQ1/KCNE1 Current in Response to Human SAN-like AP Waveforms
2.3. Data on KCNQ1/KCNE1 Current Incorporated into Human SAN Cell Model
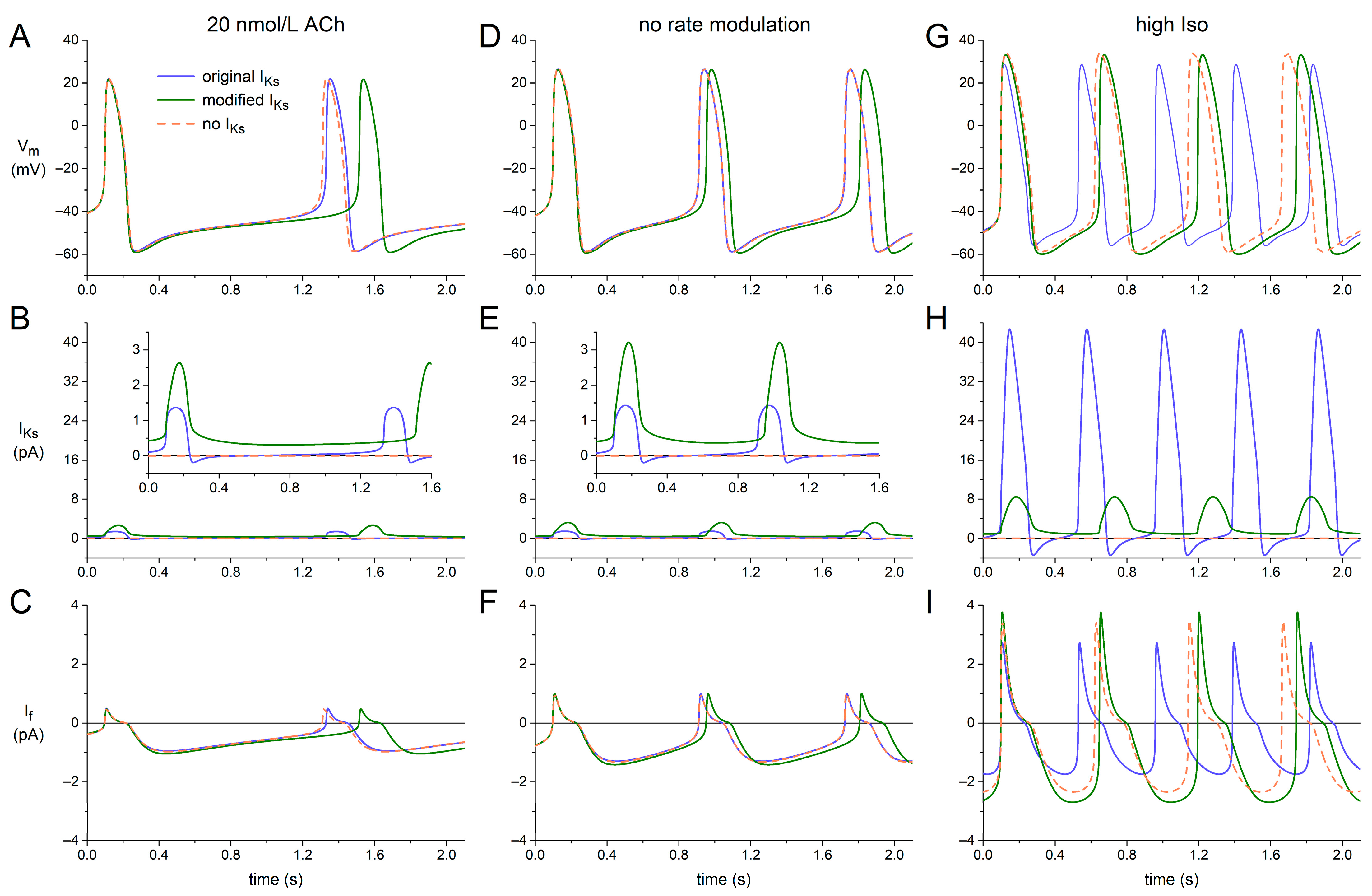
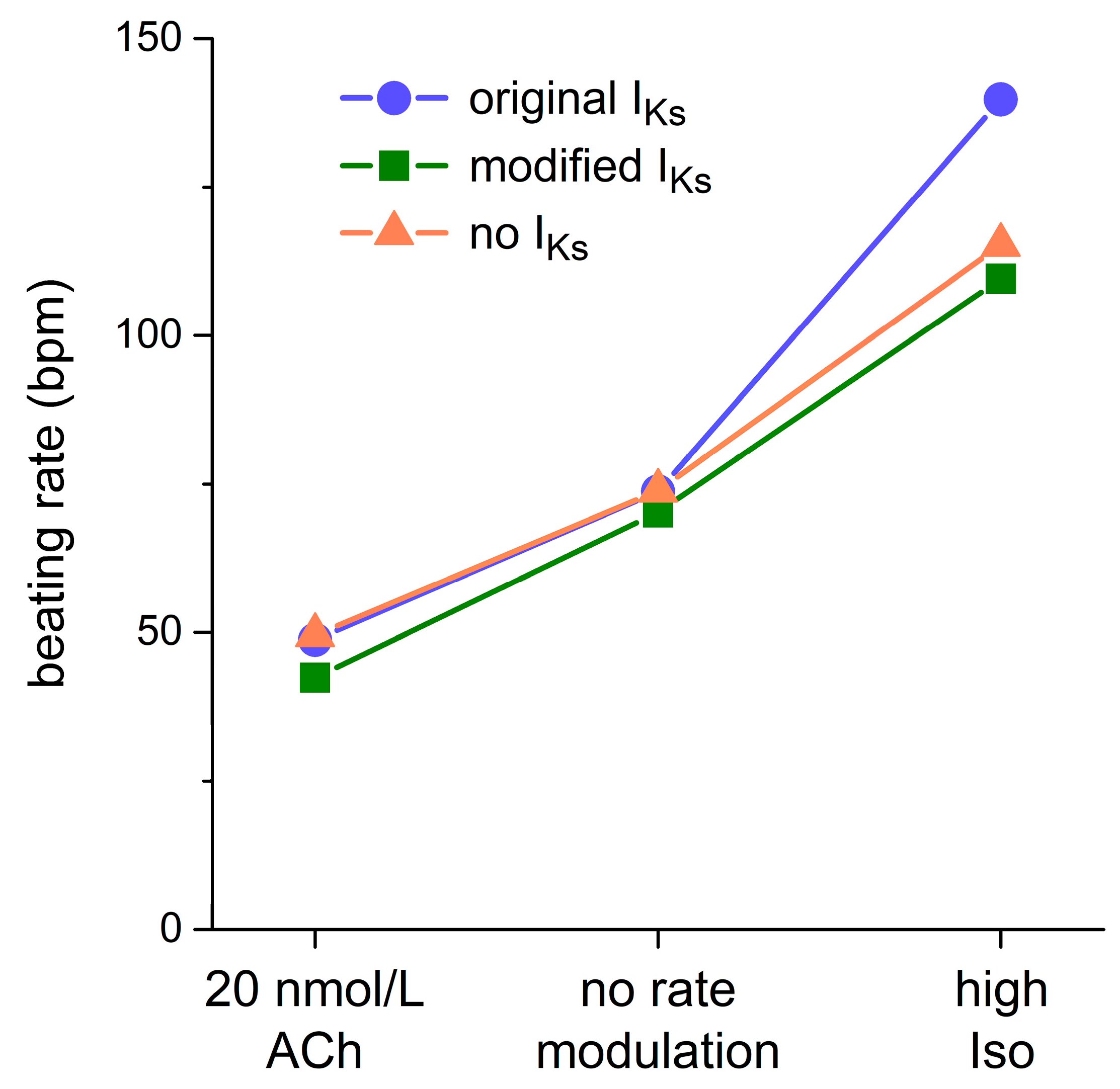
2.4. Dynamics of KCNQ1/KCNE1 Encoded IKs in Human SAN Cell Model
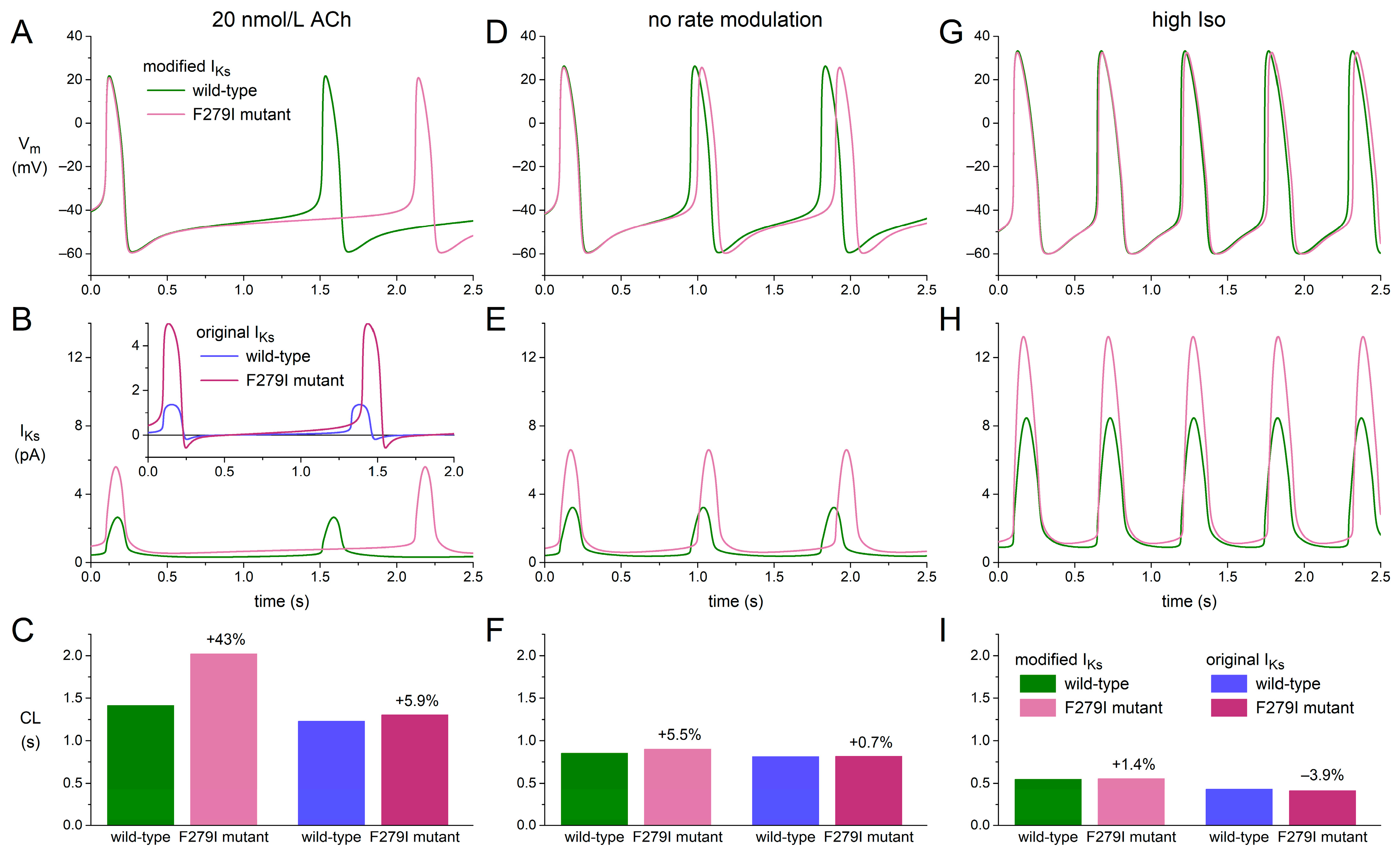
2.5. Bradycardic Effect of F279I Gain-of-Function Mutation in KCNQ1
3. Discussion
3.1. Overview
3.2. Computer Simulations with KCNQ1/KCNE1 Current Based IKs
3.3. Simulating Gain-of-Function Mutations in KCNQ1
3.4. Clinical Observations
3.5. Limitations
4. Materials and Methods
4.1. Cell Preparations
4.2. Data Acquisition
4.3. Square-Step Voltage Clamp Experiments
4.4. Action Potential Clamp Experiments
4.5. Drugs
4.6. Computer Simulations
4.7. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dobrzynski, H.; Boyett, M.R.; Anderson, R.H. New insights into pacemaker activity: Promoting understanding of sick sinus syndrome. Circulation 2007, 115, 1921–1932. [Google Scholar] [CrossRef]
- Mortola, J.P.; Lanthier, C. Scaling the amplitudes of the circadian pattern of resting oxygen consumption, body temperature and heart rate in mammals. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2004, 139, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Noujaim, S.F.; Lucca, E.; Muñoz, V.; Persaud, D.; Berenfeld, O.; Meijler, F.L.; Jalife, J. From mouse to whale: A universal scaling relation for the PR interval of the electrocardiogram of mammals. Circulation 2004, 110, 2802–2808. [Google Scholar] [CrossRef]
- Bassil, G.; Zarzoso, M.; Noujaim, S.F. Allometric scaling of electrical excitation and propagation in the mammalian heart. J. Theor. Biol. 2017, 419, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.O.; Van Ginneken, A.C.G.; Wilders, R. Pacemaker activity of the human sinoatrial node: Role of the hyperpolarization-activated current, If. Int. J. Cardiol. 2009, 132, 318–336. [Google Scholar] [CrossRef] [PubMed]
- Kalyanasundaram, A.; Li, N.; Hansen, B.J.; Zhao, J.; Fedorov, V.V. Canine and human sinoatrial node: Differences and similarities in the structure, function, molecular profiles, and arrhythmia. J. Vet. Cardiol. 2019, 22, 2–19. [Google Scholar] [CrossRef]
- Rodriguez, R.D.; Schocken, D.D. Update on sick sinus syndrome, a cardiac disorder of aging. Geriatrics 1990, 45, 26–30, 33–36. [Google Scholar]
- Morris, G.M.; Kalman, J.M. Fibrosis, electrics and genetics: Perspectives on sinoatrial node disease. Circ. J. 2014, 78, 1272–1282. [Google Scholar] [CrossRef] [PubMed]
- Boyett, M.R.; Yanni, J.; Tellez, J.; Bucchi, A.; Mesirca, P.; Cai, X.; Logantha, S.J.R.J.; Wilson, C.; Anderson, C.; Ariyaratnam, J.; et al. Regulation of sinus node pacemaking and atrioventricular node conduction by HCN channels in health and disease. Prog. Biophys. Mol. Biol. 2021, 166, 61–85. [Google Scholar] [CrossRef]
- Gatto, M.C.; Persi, A.; Tung, M.; Masi, R.; Canitano, S.; Kol, A. Bradyarrhythmias in patients with SARS-CoV-2 infection: A narrative review and a clinical report. Pacing Clin. Electrophysiol. 2021, 44, 1607–1615. [Google Scholar] [CrossRef]
- Al Kury, L.T.; Chacar, S.; Alefishat, E.; Khraibi, A.A.; Nader, M. Structural and electrical remodeling of the sinoatrial node in diabetes: New dimensions and perspectives. Front. Endocrinol. 2022, 13, 946313. [Google Scholar] [CrossRef]
- Lei, M.; Huang, C.L.-H.; Zhang, Y. Genetic Na+ channelopathies and sinus node dysfunction. Prog. Biophys. Mol. Biol. 2008, 98, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Faggioni, M.; Van der Werf, C.; Knollmann, B.C. Sinus node dysfunction in catecholaminergic polymorphic ventricular tachycardia: Risk factor and potential therapeutic target? Trends Cardiovasc. Med. 2014, 24, 273–278. [Google Scholar] [CrossRef]
- Wilders, R.; Verkerk, A.O. Long QT syndrome and sinus bradycardia—A mini review. Front. Cardiovasc. Med. 2018, 5, 106. [Google Scholar] [CrossRef]
- DiFrancesco, D. Funny channel gene mutations associated with arrhythmias. J. Physiol. 2013, 591, 4117–4124. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.O.; Wilders, R. Pacemaker activity of the human sinoatrial node: An update on the effects of mutations in HCN4 on the hyperpolarization-activated current. Int. J. Mol. Sci. 2015, 16, 3071–3094. [Google Scholar] [CrossRef]
- Yusuf, S.; Camm, A.J. Sinus tachyarrhythmias and the specific bradycardic agents: A marriage made in heaven? J. Cardiovasc. Pharmacol. Ther. 2003, 8, 89–105. [Google Scholar] [CrossRef] [PubMed]
- Minoia, C.; Giannoccaro, M.; Iacobazzi, A.; Santini, D.; Silvestris, N.; Fioretti, A.; Oliva, S.; Guarini, A. Antineoplastic drug-induced bradyarrhythmias. Expert Opin. Drug Saf. 2012, 11, 739–751. [Google Scholar] [CrossRef]
- Vogler, J.; Breithardt, G.; Eckardt, L. Bradyarrhythmias and conduction blocks. Rev. Esp. Cardiol. (Engl. Ed.) 2012, 65, 656–667. [Google Scholar] [CrossRef]
- Ishikawa, T.; Tsuji, Y.; Makita, N. Inherited bradyarrhythmia: A diverse genetic background. J. Arrhythm. 2016, 32, 352–358. [Google Scholar] [CrossRef]
- Kojima, A.; Matsuura, H. Ionic mechanisms of the action of anaesthetics on sinoatrial node automaticity. Eur. J. Pharmacol. 2017, 814, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Joung, B.; Chen, P.-S. Function and dysfunction of human sinoatrial node. Korean Circ. J. 2015, 45, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Chandler, N.J.; Greener, I.D.; Tellez, J.O.; Inada, S.; Musa, H.; Molenaar, P.; DiFrancesco, D.; Baruscotti, M.; Longhi, R.; Anderson, R.H.; et al. Molecular architecture of the human sinus node: Insights into the function of the cardiac pacemaker. Circulation 2009, 119, 1562–1575. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.O.; Wilders, R.; Van Borren, M.M.G.J.; Peters, R.J.G.; Broekhuis, E.; Lam, K.; Coronel, R.; De Bakker, J.M.T.; Tan, H.L. Pacemaker current (If) in the human sinoatrial node. Eur. Heart J. 2007, 28, 2472–2478. [Google Scholar] [CrossRef]
- Verkerk, A.O.; Wilders, R.; Van Borren, M.M.G.J.; Tan, H.L. Is sodium current present in human sinoatrial node cells? Int. J. Biol. Sci. 2009, 5, 201–204. [Google Scholar] [CrossRef]
- Verkerk, A.O.; Van Borren, M.M.G.J.; Wilders, R. Calcium transient and sodium-calcium exchange current in human versus rabbit sinoatrial node pacemaker cells. Sci. World J. 2013, 2013, 507872. [Google Scholar] [CrossRef]
- Tsutsui, K.; Monfredi, O.J.; Sirenko-Tagirova, S.G.; Maltseva, L.A.; Bychkov, R.; Kim, M.S.; Ziman, B.D.; Tarasov, K.V.; Tarasova, Y.S.; Zhang, J.; et al. A coupled-clock system drives the automaticity of human sinoatrial nodal pacemaker cells. Sci. Signal. 2018, 11, eaap7608. [Google Scholar] [CrossRef] [PubMed]
- Maltsev, V.A.; Lakatta, E.G. Synergism of coupled subsarcolemmal Ca2+ clocks and sarcolemmal voltage clocks confers robust and flexible pacemaker function in a novel pacemaker cell model. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H594–H615. [Google Scholar] [CrossRef]
- Lakatta, E.G.; Maltsev, V.A.; Vinogradova, T.M. A coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart’s pacemaker. Circ. Res. 2010, 106, 659–673. [Google Scholar] [CrossRef]
- Li, N.; Csepe, T.A.; Hansen, B.J.; Dobrzynski, H.; Higgins, R.S.D.; Kilic, A.; Mohler, P.J.; Janssen, P.M.L.; Rosen, M.R.; Biesiadecki, B.J.; et al. Molecular mapping of sinoatrial node HCN channel expression in the human heart. Circ. Arrhythm. Electrophysiol. 2015, 8, 1219–1227. [Google Scholar] [CrossRef]
- Sanguinetti, M.C.; Curran, M.E.; Zou, A.; Shen, J.; Spector, P.S.; Atkinson, D.L.; Keating, M.T. Coassembly of KvLQT1 and minK (IsK) proteins to form cardiac IKs potassium channel. Nature 1996, 384, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Barhanin, J.; Lesage, F.; Guillemare, E.; Fink, M.; Lazdunski, M.; Romey, G. KvLQT1 and IsK (minK) proteins associate to form the IKS cardiac potassium current. Nature 1996, 384, 78–80. [Google Scholar] [CrossRef] [PubMed]
- Charpentier, F.; Mérot, J.; Loussouarn, G.; Baró, I. Delayed rectifier K+ currents and cardiac repolarization. J. Mol. Cell. Cardiol. 2010, 48, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Jurkiewicz, N.K.; Sanguinetti, M.C. Rate-dependent prolongation of cardiac action potentials by a methanesulfonanilide class III antiarrhythmic agent: Specific block of rapidly activating delayed rectifier K+ current by dofetilide. Circ. Res. 1993, 72, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Stengl, M.; Volders, P.G.A.; Thomsen, M.B.; Spätjens, R.L.H.M.G.; Sipido, K.R.; Vos, M.A. Accumulation of slowly activating delayed rectifier potassium current (IKs) in canine ventricular myocytes. J. Physiol. 2003, 551, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Eldstrom, J.; Fedida, D. Gating and regulation of KCNQ1 and KCNQ1 + KCNE1 channel complexes. Front. Physiol. 2020, 11, 504. [Google Scholar] [CrossRef]
- Wu, X.; Larsson, H.P. Insights into cardiac IKs (KCNQ1/KCNE1) channels regulation. Int. J. Mol. Sci. 2020, 21, 9440. [Google Scholar] [CrossRef]
- Sanguinetti, M.C.; Seebohm, G. Physiological functions, biophysical properties, and regulation of KCNQ1 (KV7.1) potassium channels. Adv. Exp. Med. Biol. 2021, 1349, 335–353. [Google Scholar] [CrossRef]
- Sanguinetti, M.C.; Jurkiewicz, N.K.; Scott, A.; Siegl, P.K.S. Isoproterenol antagonizes prolongation of refractory period by the class III antiarrhythmic agent E-4031 in guinea pig myocytes: Mechanism of action. Circ. Res. 1991, 68, 77–84. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Ackerman, M.J.; Antzelevitch, C.; Bezzina, C.R.; Borggrefe, M.; Cuneo, B.F.; Wilde, A.A.M. Inherited cardiac arrhythmias. Nat. Rev. Dis. Primers 2020, 6, 58. [Google Scholar] [CrossRef]
- Fabbri, A.; Fantini, M.; Wilders, R.; Severi, S. Computational analysis of the human sinus node action potential: Model development and effects of mutations. J. Physiol. 2017, 595, 2365–2396. [Google Scholar] [CrossRef]
- Verkerk, A.O.; Wilders, R. Contribution of the slow delayed rectifier K+ current to pacemaker activity of the human sinoatrial node. Comp. Cardiol. 2022, 49, 91. [Google Scholar] [CrossRef]
- Hodgkin, A.L.; Huxley, A.F. A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. 1952, 117, 500–544. [Google Scholar] [CrossRef]
- Zhang, H.; Holden, A.V.; Kodama, I.; Honjo, H.; Lei, M.; Varghese, T.; Boyett, M.R. Mathematical models of action potentials in the periphery and center of the rabbit sinoatrial node. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H397–H421. [Google Scholar] [CrossRef] [PubMed]
- Kurata, Y.; Hisatome, I.; Imanishi, S.; Shibamoto, T. Dynamical description of sinoatrial node pacemaking: Improved mathematical model for primary pacemaker cell. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H2074–H2101. [Google Scholar] [CrossRef] [PubMed]
- Kharche, S.; Yu, J.; Lei, M.; Zhang, H. A mathematical model of action potentials of mouse sinoatrial node cells with molecular bases. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H945–H963. [Google Scholar] [CrossRef]
- Tao, T.; Paterson, D.J.; Smith, N.P. A model of cellular cardiac-neural coupling that captures the sympathetic control of sinoatrial node excitability in normotensive and hypertensive rats. Biophys. J. 2011, 101, 594–602. [Google Scholar] [CrossRef]
- Severi, S.; Fantini, M.; Charawi, L.A.; DiFrancesco, D. An updated computational model of rabbit sinoatrial action potential to investigate the mechanisms of heart rate modulation. J. Physiol. 2012, 590, 4483–4499. [Google Scholar] [CrossRef]
- Moreno, C.; Oliveras, A.; De la Cruz, A.; Bartolucci, C.; Muñoz, C.; Salar, E.; Gimeno, J.R.; Severi, S.; Comes, N.; Felipe, A.; et al. A new KCNQ1 mutation at the S5 segment that impairs its association with KCNE1 is responsible for short QT syndrome. Cardiovasc. Res. 2015, 107, 613–623. [Google Scholar] [CrossRef]
- Danielsson, C.; Brask, J.; Sköld, A.-C.; Genead, R.; Andersson, A.; Andersson, U.; Stockling, K.; Pehrson, R.; Grinnemo, K.-H.; Salari, S.; et al. Exploration of human, rat, and rabbit embryonic cardiomyocytes suggests K-channel block as a common teratogenic mechanism. Cardiovasc. Res. 2013, 97, 23–32. [Google Scholar] [CrossRef]
- Whittaker, D.G.; Colman, M.A.; Ni, H.; Hancox, J.C.; Zhang, H. Human atrial arrhythmogenesis and sinus bradycardia in KCNQ1-linked short QT syndrome: Insights from computational modelling. Front. Physiol. 2018, 9, 1402. [Google Scholar] [CrossRef] [PubMed]
- Britton, O.J.; Bueno-Orovio, A.; Van Ammel, K.; Lu, H.R.; Towart, R.; Gallacher, D.J.; Rodriguez, B. Experimentally calibrated population of models predicts and explains intersubject variability in cardiac cellular electrophysiology. Proc. Natl. Acad. Sci. USA 2013, 110, E2098–E2105. [Google Scholar] [CrossRef] [PubMed]
- Paci, M.; Passini, E.; Severi, S.; Hyttinen, J.; Rodriguez, B. Phenotypic variability in LQT3 human induced pluripotent stem cell-derived cardiomyocytes and their response to antiarrhythmic pharmacologic therapy: An in silico approach. Heart Rhythm 2017, 14, 1704–1712. [Google Scholar] [CrossRef]
- Henrion, U.; Zumhagen, S.; Steinke, K.; Strutz-Seebohm, N.; Stallmeyer, B.; Lang, F.; Schulze-Bahr, E.; Seebohm, G. Overlapping cardiac phenotype associated with a familial mutation in the voltage sensor of the KCNQ1 channel. Cell. Physiol. Biochem. 2012, 29, 809–818. [Google Scholar] [CrossRef]
- Ki, C.-S.; Jung, C.L.; Kim, H.-J.; Baek, K.-H.; Park, S.J.; On, Y.K.; Kim, K.-S.; Noh, S.J.; Youm, J.B.; Kim, J.S.; et al. A KCNQ1 mutation causes age-dependant bradycardia and persistent atrial fibrillation. Pflügers Arch. 2014, 466, 529–540. [Google Scholar] [CrossRef]
- Zhou, X.; Bueno-Orovio, A.; Schilling, R.J.; Kirkby, C.; Denning, C.; Rajamohan, D.; Burrage, K.; Tinker, A.; Rodriguez, B.; Harmer, S.C. Investigating the complex arrhythmic phenotype caused by the gain-of-function mutation KCNQ1-G229D. Front. Physiol. 2019, 10, 259. [Google Scholar] [CrossRef] [PubMed]
- Bellocq, C.; Van Ginneken, A.C.G.; Bezzina, C.R.; Alders, M.; Escande, D.; Mannens, M.M.A.M.; Baró, I.; Wilde, A.A.M. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation 2004, 109, 2394–2397. [Google Scholar] [CrossRef]
- Hasegawa, K.; Ohno, S.; Ashihara, T.; Itoh, H.; Ding, W.-G.; Toyoda, F.; Makiyama, T.; Aoki, H.; Nakamura, Y.; Delisle, B.P.; et al. A novel KCNQ1 missense mutation identified in a patient with juvenile-onset atrial fibrillation causes constitutively open IKs channels. Heart Rhythm 2014, 11, 67–75. [Google Scholar] [CrossRef]
- Hong, K.; Piper, D.R.; Diaz-Valdecantos, A.; Brugada, J.; Oliva, A.; Burashnikov, E.; Santos-de-Soto, J.; Grueso-Montero, J.; Diaz-Enfante, E.; Brugada, P.; et al. De novo KCNQ1 mutation responsible for atrial fibrillation and short QT syndrome in utero. Cardiovasc. Res. 2005, 68, 433–440. [Google Scholar] [CrossRef]
- Maltret, A.; Wiener-Vacher, S.; Denis, C.; Extramiana, F.; Morisseau-Durand, M.P.; Fressart, V.; Bonnet, D.; Chabbert, C. Type 2 short QT syndrome and vestibular dysfunction: Mirror of the Jervell and Lange-Nielsen syndrome? Int. J. Cardiol. 2014, 171, 291–293. [Google Scholar] [CrossRef]
- Villafañe, J.; Fischbach, P.; Gebauer, R. Short QT syndrome manifesting with neonatal atrial fibrillation and bradycardia. Cardiology 2014, 128, 236–240. [Google Scholar] [CrossRef]
- Harrell, D.T.; Ashihara, T.; Ishikawa, T.; Tominaga, I.; Mazzanti, A.; Takahashi, K.; Oginosawa, Y.; Abe, H.; Maemura, K.; Sumitomo, N.; et al. Genotype-dependent differences in age of manifestation and arrhythmia complications in short QT syndrome. Int. J. Cardiol. 2015, 190, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Sarquella-Brugada, G.; Campuzano, O.; Iglesias, A.; Grueso, J.; Bradley, D.J.; Kerst, G.; Shmorhun, D.; Brugada, J.; Brugada, R. Short QT and atrial fibrillation: A KCNQ1 mutation-specific disease. Late follow-up in three unrelated children. HeartRhythm Case Rep. 2015, 1, 193–197. [Google Scholar] [CrossRef]
- Righi, D.; Silvetti, M.S.; Drago, F. Sinus bradycardia, junctional rhythm, and low-rate atrial fibrillation in Short QT syndrome during 20 years of follow-up: Three faces of the same genetic problem. Cardiol. Young 2016, 26, 589–592. [Google Scholar] [CrossRef] [PubMed]
- Takechi, F.; Kato, K.; Makiyama, T.; Ohno, S.; Kabasawa, M.; Morishima, H.; Kawasoe, Y.; Okajima, Y.; Matsuo, K.; Horie, M.; et al. Ventricular fibrillation in a family with short QT syndrome type 2 carrying a heterozygous KCNQ1-V141M variant. J. Pediatr. Cardiol. Card. Surg. 2022, 6, 31–36. [Google Scholar] [CrossRef]
- Bendahhou, S.; Marionneau, C.; Haurogne, K.; Larroque, M.-M.; Derand, R.; Szuts, V.; Escande, D.; Demolombe, S.; Barhanin, J. In vitro molecular interactions and distribution of KCNE family with KCNQ1 in the human heart. Cardiovasc. Res. 2005, 67, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Lundquist, A.L.; Manderfield, L.J.; Vanoye, C.G.; Rogers, C.S.; Donahue, B.S.; Chang, P.A.; Drinkwater, D.C.; Murray, K.T.; George, A.L., Jr. Expression of multiple KCNE genes in human heart may enable variable modulation of IKs. J. Mol. Cell. Cardiol. 2005, 38, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Lundby, A.; Ravn, L.S.; Svendsen, J.H.; Olesen, S.-P.; Schmitt, N. KCNQ1 mutation Q147R is associated with atrial fibrillation and prolonged QT interval. Heart Rhythm 2007, 4, 1532–1541. [Google Scholar] [CrossRef]
- Tseng, G.-N. The phenotype of a KCNQ1 mutation depends on its KCNE partners: Is the cardiac slow delayed rectifier (IKs) channel more than a KCNQ1/KCNE1 complex? Heart Rhythm 2007, 4, 1542–1543. [Google Scholar] [CrossRef]
- Bartos, D.C.; Morotti, S.; Ginsburg, K.S.; Grandi, E.; Bers, D.M. Quantitative analysis of the Ca2+-dependent regulation of delayed rectifier K+ current IKs in rabbit ventricular myocytes. J. Physiol. 2017, 595, 2253–2268. [Google Scholar] [CrossRef]
- Horváth, B.; Kiss, D.; Dienes, C.; Hézső, T.; Kovács, Z.; Szentandrássy, N.; Almássy, J.; Magyar, J.; Bányász, T.; Nánási, P.P. Ion current profiles in canine ventricular myocytes obtained by the “onion peeling” technique. J. Mol. Cell. Cardiol. 2021, 158, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Zang, Y.; Zheng, D.; Xia, L.; Gong, Y. Role of CaMKII and PKA in early afterdepolarization of human ventricular myocardium cell: A computational model study. Comput. Math. Methods Med. 2016, 2016, 4576313. [Google Scholar] [CrossRef]
- Onal, B.; Gratz, D.; Hund, T.J. Ca2+/calmodulin-dependent kinase II-dependent regulation of atrial myocyte late Na+ current, Ca2+ cycling, and excitability: A mathematical modeling study. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H1227–H1239. [Google Scholar] [CrossRef] [PubMed]
- Zang, Y.; Marder, E. Interactions among diameter, myelination, and the Na/K pump affect axonal resilience to high-frequency spiking. Proc. Natl. Acad. Sci. USA 2021, 118, e2105795118. [Google Scholar] [CrossRef]
- Morotti, S.; Ni, H.; Peters, C.H.; Rickert, C.; Asgari-Targhi, A.; Sato, D.; Glukhov, A.V.; Proenza, C.; Grandi, E. Intracellular Na+ modulates pacemaking activity in murine sinoatrial node myocytes: An in silico analysis. Int. J. Mol. Sci. 2021, 22, 5645. [Google Scholar] [CrossRef]
- Veerman, C.C.; Verkerk, A.O.; Blom, M.T.; Klemens, C.A.; Langendijk, P.N.J.; Van Ginneken, A.C.G.; Wilders, R.; Tan, H.L. Slow delayed rectifier potassium current blockade contributes importantly to drug-induced long QT syndrome. Circ. Arrhythm. Electrophysiol. 2013, 6, 1002–1009. [Google Scholar] [CrossRef] [PubMed]
- Starzak, M.E.; Starzak, R.J. An action potential clamp to probe the effectiveness of space clamp in axons. IEEE Trans. Biomed. Eng. 1978, 25, 201–204. [Google Scholar] [CrossRef]
- Doerr, T.; Denger, R.; Trautwein, W. Calcium currents in single SA nodal cells of the rabbit heart studied with action potential clamp. Pflügers Arch. 1989, 413, 599–603. [Google Scholar] [CrossRef]
- Imredy, J.P.; Penniman, J.R.; Dech, S.J.; Irving, W.D.; Salata, J.J. Modeling of the adrenergic response of the human IKs current (hKCNQ1/hKCNE1) stably expressed in HEK-293 cells. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1867–H1881. [Google Scholar] [CrossRef]
- Lloyd, C.M.; Lawson, J.R.; Hunter, P.J.; Nielsen, P.F. The CellML Model Repository. Bioinformatics 2008, 24, 2122–2123. [Google Scholar] [CrossRef]
- Garny, A.; Kohl, P.; Noble, D. Cellular open resource (COR): A public CellML based environment for modelling biological function. Int. J. Bifurcat. Chaos 2003, 13, 3579–3590. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verkerk, A.O.; Wilders, R. Human Sinoatrial Node Pacemaker Activity: Role of the Slow Component of the Delayed Rectifier K+ Current, IKs. Int. J. Mol. Sci. 2023, 24, 7264. https://doi.org/10.3390/ijms24087264
Verkerk AO, Wilders R. Human Sinoatrial Node Pacemaker Activity: Role of the Slow Component of the Delayed Rectifier K+ Current, IKs. International Journal of Molecular Sciences. 2023; 24(8):7264. https://doi.org/10.3390/ijms24087264
Chicago/Turabian StyleVerkerk, Arie O., and Ronald Wilders. 2023. "Human Sinoatrial Node Pacemaker Activity: Role of the Slow Component of the Delayed Rectifier K+ Current, IKs" International Journal of Molecular Sciences 24, no. 8: 7264. https://doi.org/10.3390/ijms24087264
APA StyleVerkerk, A. O., & Wilders, R. (2023). Human Sinoatrial Node Pacemaker Activity: Role of the Slow Component of the Delayed Rectifier K+ Current, IKs. International Journal of Molecular Sciences, 24(8), 7264. https://doi.org/10.3390/ijms24087264