
Polarizing Macrophage Functional Phenotype to Foster Cardiac Regeneration

 , , and
, , and
Abstract
1. Introduction
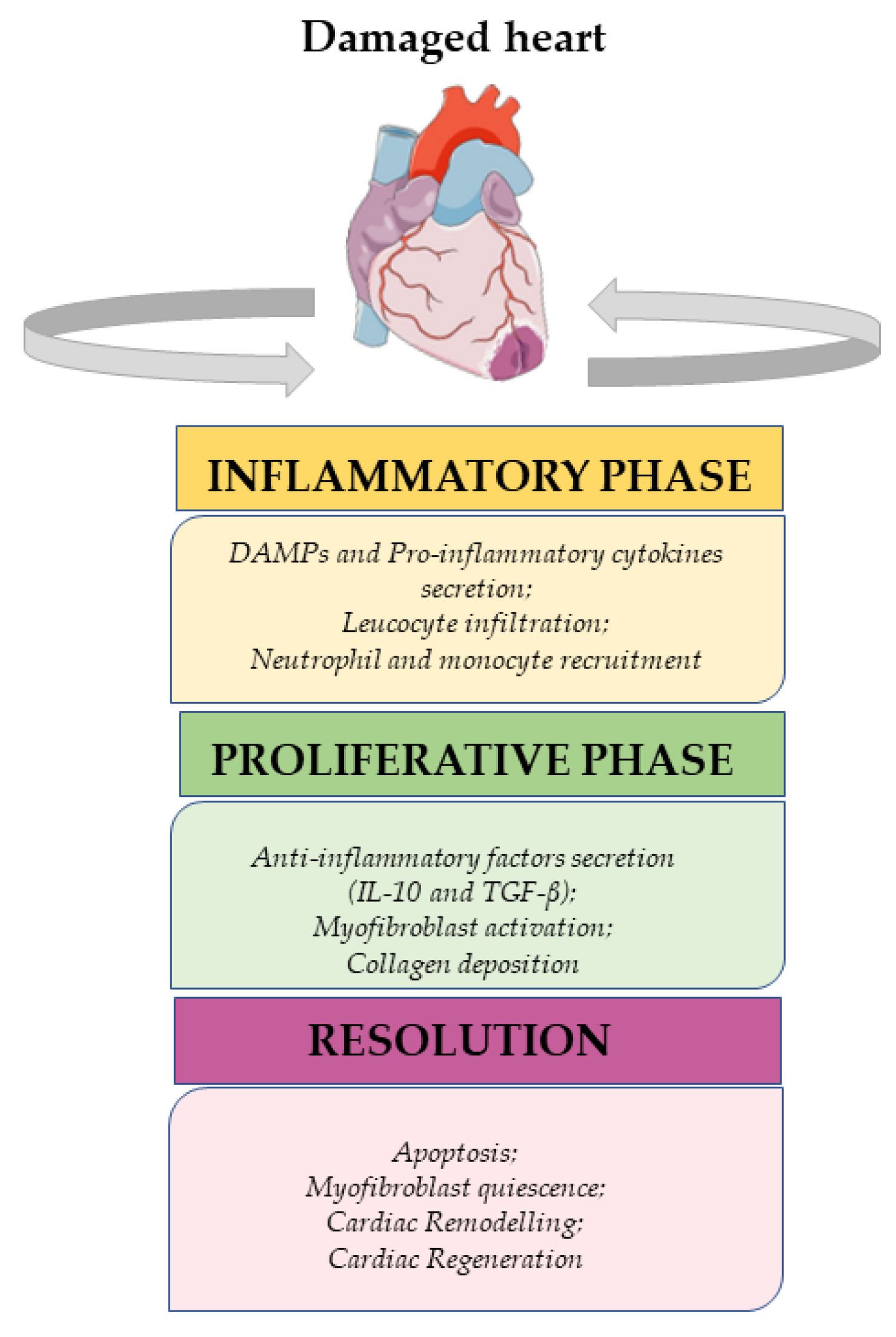
2. Innate Immunomodulation after Injury
2.1. Inflammatory Phase
2.2. Proliferative Phase
2.3. Resolutive Phase
3. Adaptive Immune Response
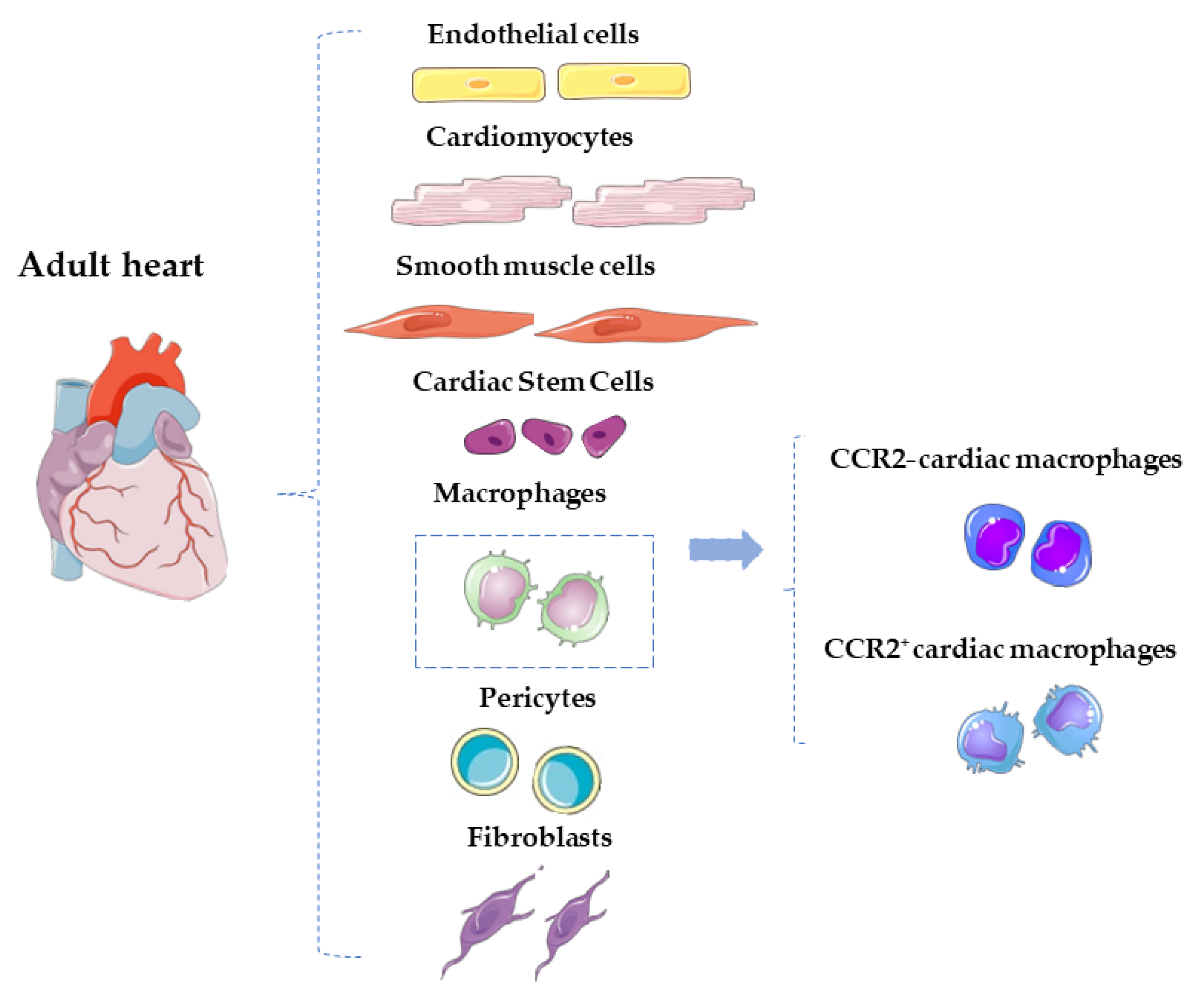
4. Distinct Cardiac Macrophage Subsets among the Adult Heart
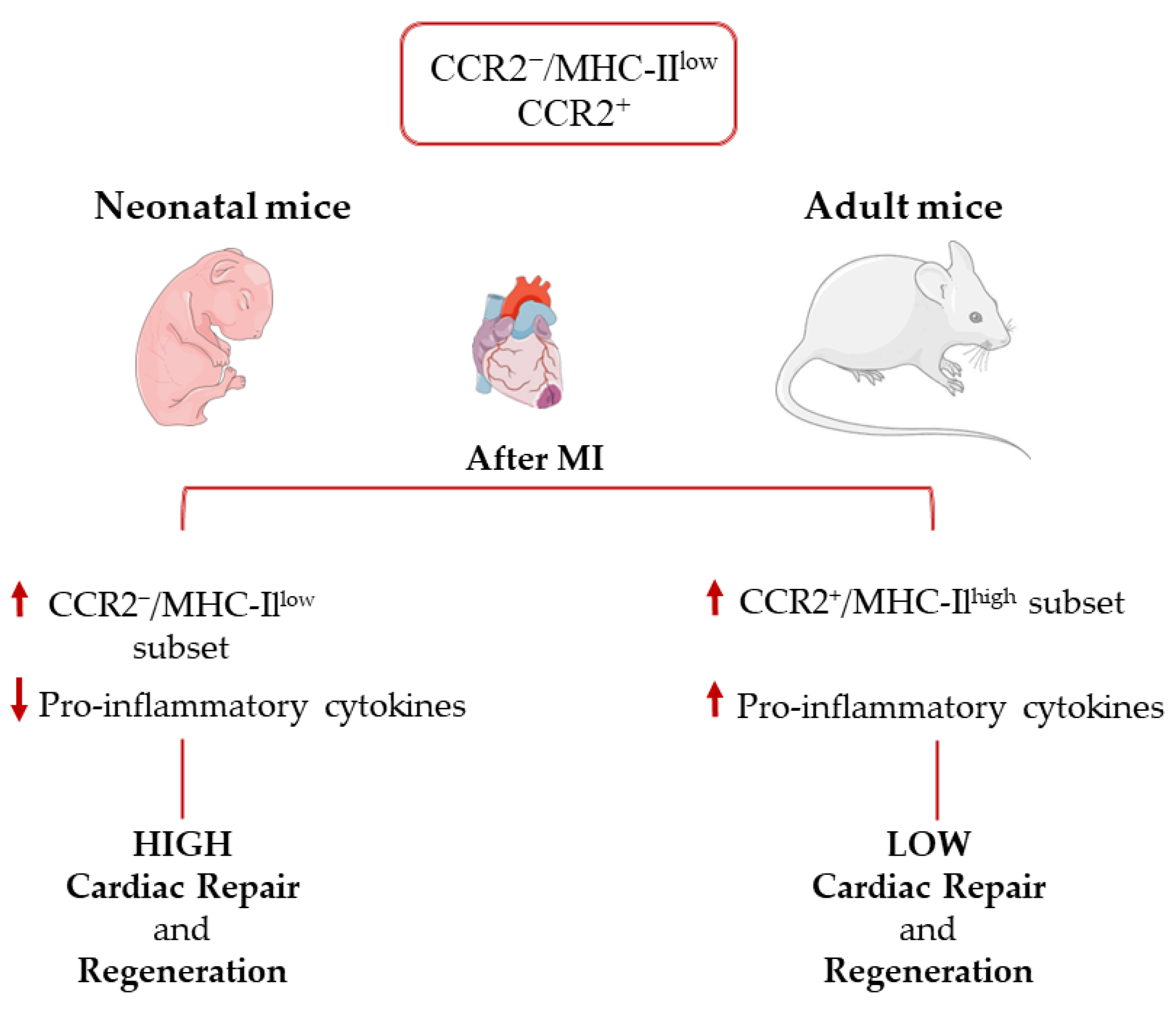
5. Cardiac Macrophage Recruitment following Myocardial Injury
6. Macrophages and Cardiac Tissue Regeneration
7. Macrophages and Tissue Degeneration during Aging
8. Therapeutic Perspectives
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Alonso, A.; Beaton, A.Z.; Bittencourt, M.S.; Boehme, A.K.; Buxton, A.E.; Carson, A.P.; Commodore-Mensah, Y.; et al. Heart Disease and Stroke Statistics-2022 Update: A Report from the American Heart Association. Circulation 2022, 145, e153–e639. [Google Scholar] [CrossRef] [PubMed]
- Smolina, K.; Wright, F.L.; Rayner, M.; Goldacre, M.J. Long-term survival and recurrence after acute myocardial infarction in England, 2004 to 2010. Circ. Cardiovasc. Qual. Outcomes 2012, 5, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol. Asp. Med. 2019, 65, 70–99. [Google Scholar] [CrossRef] [PubMed]
- Bajpai, G.; Bredemeyer, A.; Li, W.; Zaitsev, K.; Koenig, A.L.; Lokshina, I.; Mohan, J.; Ivey, B.; Hsiao, H.M.; Weinheimer, C.; et al. Tissue Resident CCR2− and CCR2+ Cardiac Macrophages Differentially Orchestrate Monocyte Recruitment and Fate Specification Following Myocardial Injury. Circ. Res. 2019, 124, 263–278. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnabé-Heider, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; et al. Evidence for cardiomyocyte renewal in humans. Science 2009, 324, 98–102. [Google Scholar] [CrossRef]
- Nadal-Ginard, B.; Ellison, G.M.; Torella, D. The cardiac stem cell compartment is indispensable for myocardial cell homeostasis, repair and regeneration in the adult. Stem Cell Res. 2014, 13, 615–630. [Google Scholar] [CrossRef]
- Salerno, N.; Marino, F.; Scalise, M.; Salerno, L.; Molinaro, C.; Filardo, A.; Chiefalo, A.; Panuccio, G.; De Angelis, A.; Urbanek, K.; et al. Pharmacological clearance of senescent cells improves cardiac remodeling and function after myocardial infarction in female aged mice. Mech. Ageing Dev. 2022, 208, 111740. [Google Scholar] [CrossRef]
- Scalise, M.; Marino, F.; Salerno, L.; Mancuso, T.; Cappetta, D.; Barone, A.; Parrotta, E.I.; Torella, A.; Palumbo, D.; Veltri, P.; et al. In vitro CSC-derived cardiomyocytes exhibit the typical microRNA-mRNA blueprint of endogenous cardiomyocytes. Commun. Biol. 2021, 4, 1146. [Google Scholar] [CrossRef]
- Cianflone, E.; Scalise, M.; Marino, F.; Salerno, L.; Salerno, N.; Urbanek, K.; Torella, D. The negative regulation of gene expression by microRNAs as key driver of inducers and repressors of cardiomyocyte differentiation. Clin. Sci. 2022, 136, 1179–1203. [Google Scholar] [CrossRef]
- Marino, F.; Scalise, M.; Salerno, N.; Salerno, L.; Molinaro, C.; Cappetta, D.; Torella, M.; Greco, M.; Foti, D.; Sasso, F.C.; et al. Diabetes-Induced Cellular Senescence and Senescence-Associated Secretory Phenotype Impair Cardiac Regeneration and Function Independently of Age. Diabetes 2022, 71, 1081–1098. [Google Scholar] [CrossRef]
- Molinaro, C.; Salerno, L.; Marino, F.; Scalise, M.; Salerno, N.; Pagano, L.; De Angelis, A.; Cianflone, E.; Torella, D.; Urbanek, K. Unraveling and Targeting Myocardial Regeneration Deficit in Diabetes. Antioxidants 2022, 11, 208. [Google Scholar] [CrossRef]
- Torella, D.; Iaconetti, C.; Tarallo, R.; Marino, F.; Giurato, G.; Veneziano, C.; Aquila, I.; Scalise, M.; Mancuso, T.; Cianflone, E.; et al. miRNA Regulation of the Hyperproliferative Phenotype of Vascular Smooth Muscle Cells in Diabetes. Diabetes 2018, 67, 2554–2568. [Google Scholar] [CrossRef]
- Marino, F.; Salerno, N.; Scalise, M.; Salerno, L.; Torella, A.; Molinaro, C.; Chiefalo, A.; Filardo, A.; Siracusa, C.; Panuccio, G.; et al. Streptozotocin-Induced Type 1 and 2 Diabetes Mellitus Mouse Models Show Different Functional, Cellular and Molecular Patterns of Diabetic Cardiomyopathy. Int. J. Mol. Sci. 2023, 24, 1132. [Google Scholar] [CrossRef]
- Cianflone, E.; Torella, M.; Chimenti, C.; De Angelis, A.; Beltrami, A.P.; Urbanek, K.; Rota, M.; Torella, D. Adult Cardiac Stem Cell Aging: A Reversible Stochastic Phenomenon? Oxid. Med. Cell. Longev. 2019, 2019, 5813147. [Google Scholar] [CrossRef]
- Costamagna, D.; Berardi, E.; Ceccarelli, G.; Sampaolesi, M. Adult Stem Cells and Skeletal Muscle Regeneration. Curr. Gene Ther. 2015, 15, 348–363. [Google Scholar] [CrossRef]
- Cianflone, E.; Torella, M.; Biamonte, F.; De Angelis, A.; Urbanek, K.; Costanzo, F.S.; Rota, M.; Ellison-Hughes, G.M.; Torella, D. Targeting Cardiac Stem Cell Senescence to Treat Cardiac Aging and Disease. Cells 2020, 9, 1558. [Google Scholar] [CrossRef]
- Salerno, N.; Salerno, L.; Marino, F.; Scalise, M.; Chiefalo, A.; Panuccio, G.; De Angelis, A.; Cianflone, E.; Urbanek, K.; Torella, D. Myocardial regeneration protocols towards the routine clinical scenario: An unseemly path from bench to bedside. eClinicalMedicine 2022, 50, 101530. [Google Scholar] [CrossRef]
- Mancuso, A.; Cianflone, E.; Cristiano, M.C.; Salerno, N.; Tarsitano, M.; Marino, F.; Molinaro, C.; Fresta, M.; Torella, D.; Paolino, D. Lyotropic Liquid Crystals: A Biocompatible and Safe Material for Local Cardiac Application. Pharmaceutics 2022, 14, 452. [Google Scholar] [CrossRef]
- Lafuse, W.P.; Wozniak, D.J.; Rajaram, M.V.S. Role of Cardiac Macrophages on Cardiac Inflammation, Fibrosis and Tissue Repair. Cells 2020, 10, 51. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef]
- Lavine, K.J.; Epelman, S.; Uchida, K.; Weber, K.J.; Nichols, C.G.; Schilling, J.D.; Ornitz, D.M.; Randolph, G.J.; Mann, D.L. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc. Natl. Acad. Sci. USA 2014, 111, 16029–16034. [Google Scholar] [CrossRef] [PubMed]
- Rőszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediat. Inflamm. 2015, 2015, 816460. [Google Scholar] [CrossRef] [PubMed]
- Aquila, I.; Cianflone, E.; Scalise, M.; Marino, F.; Mancuso, T.; Filardo, A.; Smith, A.J.; Cappetta, D.; De Angelis, A.; Urbanek, K.; et al. c-kit Haploinsufficiency impairs adult cardiac stem cell growth, myogenicity and myocardial regeneration. Cell Death Dis. 2019, 10, 436. [Google Scholar] [CrossRef] [PubMed]
- Farache Trajano, L.; Smart, N. Immunomodulation for optimal cardiac regeneration: Insights from comparative analyses. NPJ Regen. Med. 2021, 6, 8. [Google Scholar] [CrossRef]
- Relja, B.; Land, W.G. Damage-associated molecular patterns in trauma. Eur. J. Trauma Emerg. Surg. 2020, 46, 751–775. [Google Scholar] [CrossRef]
- Toldo, S.; Mauro, A.G.; Cutter, Z.; Abbate, A. Inflammasome, pyroptosis, and cytokines in myocardial ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1553–H1568. [Google Scholar] [CrossRef]
- Carter, A.M. Complement activation: An emerging player in the pathogenesis of cardiovascular disease. Scientifica 2012, 2012, 402783. [Google Scholar] [CrossRef]
- Granger, C.B.; Mahaffey, K.W.; Weaver, W.D.; Theroux, P.; Hochman, J.S.; Filloon, T.G.; Rollins, S.; Todaro, T.G.; Nicolau, J.C.; Ruzyllo, W.; et al. Pexelizumab, an anti-C5 complement antibody, as adjunctive therapy to primary percutaneous coronary intervention in acute myocardial infarction—The COMplement inhibition in myocardial infarction treated with angioplasty (COMMA) trial. Circulation 2003, 108, 1184–1190. [Google Scholar] [CrossRef]
- Yan, X.; Anzai, A.; Katsumata, Y.; Matsuhashi, T.; Ito, K.; Endo, J.; Yamamoto, T.; Takeshima, A.; Shinmura, K.; Shen, W.; et al. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J. Mol. Cell. Cardiol. 2013, 62, 24–35. [Google Scholar] [CrossRef]
- Cassatella, M.A. On the production of TNF-related apoptosis-inducing ligand (TRAIL/Apo-2L) by human neutrophils. J. Leukoc. Biol. 2006, 79, 1140–1149. [Google Scholar] [CrossRef]
- Mayadas, T.N.; Cullere, X.; Lowell, C.A. The Multifaceted Functions of Neutrophils. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 181–218. [Google Scholar] [CrossRef]
- Vasilyev, N.; Williams, T.; Brennan, M.L.; Unzek, S.; Zhou, X.; Heinecke, J.W.; Spitz, D.R.; Topol, E.J.; Hazen, S.L.; Penn, M.S. Myeloperoxidase-generated oxidants modulate left ventricular remodeling but not infarct size after myocardial infarction. Circulation 2005, 112, 2812–2820. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, S.; Weng, X.; Zeng, S.; Yu, L.; Guo, J.; Xu, Y. Brg1 deficiency in vascular endothelial cells blocks neutrophil recruitment and ameliorates cardiac ischemia-reperfusion injury in mice. Int. J. Cardiol. 2018, 269, 250–258. [Google Scholar] [CrossRef]
- García-Prieto, J.; Villena-Gutiérrez, R.; Gómez, M.; Bernardo, E.; Pun-García, A.; García-Lunar, I.; Crainiciuc, G.; Fernández-Jiménez, R.; Sreeramkumar, V.; Bourio-Martínez, R.; et al. Neutrophil stunning by metoprolol reduces infarct size. Nat. Commun. 2017, 8, 14780. [Google Scholar] [CrossRef]
- Ma, Y.; Chiao, Y.A.; Clark, R.; Flynn, E.R.; Yabluchanskiy, A.; Ghasemi, O.; Zouein, F.; Lindsey, M.L.; Jin, Y.F. Deriving a cardiac ageing signature to reveal MMP-9-dependent inflammatory signalling in senescence. Cardiovasc. Res. 2015, 106, 421–431. [Google Scholar] [CrossRef]
- Ma, Y.; Mouton, A.J.; Lindsey, M.L. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl. Res. 2018, 191, 15–28. [Google Scholar] [CrossRef]
- Kiczak, L.; Tomaszek, A.; Bania, J.; Paslawska, U.; Zacharski, M.; Noszczyk-Nowak, A.; Janiszewski, A.; Skrzypczak, P.; Ardehali, H.; Jankowska, E.A.; et al. Expression and complex formation of MMP9, MMP2, NGAL, and TIMP1 in porcine myocardium but not in skeletal muscles in male pigs with tachycardia-induced systolic heart failure. BioMed Res. Int. 2013, 2013, 283856. [Google Scholar] [CrossRef]
- Kotwal, G.J.; Chien, S. Macrophage Differentiation in Normal and Accelerated Wound Healing. Results Probl. Cell Differ. 2017, 62, 353–364. [Google Scholar] [CrossRef]
- Gordon, S.; Plüddemann, A.; Martinez Estrada, F. Macrophage heterogeneity in tissues: Phenotypic diversity and functions. Immunol. Rev. 2014, 262, 36–55. [Google Scholar] [CrossRef]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef]
- Sommerfeld, S.D.; Cherry, C.; Schwab, R.M.; Chung, L.; Maestas, D.R., Jr.; Laffont, P.; Stein, J.E.; Tam, A.; Ganguly, S.; Housseau, F.; et al. Interleukin-36γ-producing macrophages drive IL-17-mediated fibrosis. Sci. Immunol. 2019, 4, eaax4783. [Google Scholar] [CrossRef] [PubMed]
- Britto, D.D.; Wyroba, B.; Chen, W.; Lockwood, R.A.; Tran, K.B.; Shepherd, P.R.; Hall, C.J.; Crosier, K.E.; Crosier, P.S.; Astin, J.W. Macrophages enhance Vegfa-driven angiogenesis in an embryonic zebrafish tumour xenograft model. Dis. Model. Mech. 2018, 11, dmm035998. [Google Scholar] [CrossRef] [PubMed]
- Richardson, W.J.; Clarke, S.A.; Quinn, T.A.; Holmes, J.W. Physiological Implications of Myocardial Scar Structure. Compr. Physiol. 2015, 5, 1877–1909. [Google Scholar] [CrossRef] [PubMed]
- Ignotz, R.A.; Massague, J. Transforming Growth-Factor-Beta Stimulates the Expression of Fibronectin and Collagen and Their Incorporation into the Extracellular-Matrix. J. Biol. Chem. 1986, 261, 4337–4345. [Google Scholar] [CrossRef]
- Wynn, T.A.; Barron, L. Macrophages: Master regulators of inflammation and fibrosis. Semin. Liver Dis. 2010, 30, 245–257. [Google Scholar] [CrossRef]
- Czubryt, M.P. Common threads in cardiac fibrosis, infarct scar formation, and wound healing. Fibrogenesis Tissue Repair 2012, 5, 19. [Google Scholar] [CrossRef]
- Serhan, C.N.; Savill, J. Resolution of inflammation: The beginning programs the end. Nat. Immunol. 2005, 6, 1191–1197. [Google Scholar] [CrossRef]
- Henson, P.M. Dampening inflammation. Nat. Immunol. 2005, 6, 1179–1181. [Google Scholar] [CrossRef]
- Kono, H.; Rock, K.L. How dying cells alert the immune system to danger. Nat. Rev. Immunol. 2008, 8, 279–289. [Google Scholar] [CrossRef]
- Vanderlugt, C.L.; Miller, S.D. Epitope spreading in immune-mediated diseases: Implications for immunotherapy. Nat. Rev. Immunol. 2002, 2, 85–95. [Google Scholar] [CrossRef]
- Zouggari, Y.; Ait-Oufella, H.; Bonnin, P.; Simon, T.; Sage, A.P.; Guérin, C.; Vilar, J.; Caligiuri, G.; Tsiantoulas, D.; Laurans, L.; et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat. Med. 2013, 19, 1273–1280. [Google Scholar] [CrossRef]
- Nian, M.; Lee, P.; Khaper, N.; Liu, P. Inflammatory cytokines and postmyocardial infarction remodeling. Circ. Res. 2004, 94, 1543–1553. [Google Scholar] [CrossRef]
- Porter, K.E.; Turner, N.A.; O’Regan, D.J.; Ball, S.G. Tumor necrosis factor alpha induces human atrial myofibroblast proliferation, invasion and MMP-9 secretion: Inhibition by simvastatin. Cardiovasc. Res. 2004, 64, 507–515. [Google Scholar] [CrossRef]
- Hofmann, U.; Beyersdorf, N.; Weirather, J.; Podolskaya, A.; Bauersachs, J.; Ertl, G.; Kerkau, T.; Frantz, S. Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation 2012, 125, 1652–1663. [Google Scholar] [CrossRef]
- Matsumoto, K.; Ogawa, M.; Suzuki, J.; Hirata, Y.; Nagai, R.; Isobe, M. Regulatory T lymphocytes attenuate myocardial infarction-induced ventricular remodeling in mice. Int. Heart J. 2011, 52, 382–387. [Google Scholar] [CrossRef]
- Tang, T.T.; Yuan, J.; Zhu, Z.F.; Zhang, W.C.; Xiao, H.; Xia, N.; Yan, X.X.; Nie, S.F.; Liu, J.; Zhou, S.F.; et al. Regulatory T cells ameliorate cardiac remodeling after myocardial infarction. Basic Res. Cardiol. 2012, 107, 232. [Google Scholar] [CrossRef]
- Epelman, S.; Lavine, K.J.; Randolph, G.J. Origin and functions of tissue macrophages. Immunity 2014, 41, 21–35. [Google Scholar] [CrossRef]
- Epelman, S.; Lavine, K.J.; Beaudin, A.E.; Sojka, D.K.; Carrero, J.A.; Calderon, B.; Brija, T.; Gautier, E.L.; Ivanov, S.; Satpathy, A.T.; et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014, 40, 91–104. [Google Scholar] [CrossRef]
- Motwani, M.P.; Gilroy, D.W. Macrophage development and polarization in chronic inflammation. Semin. Immunol. 2015, 27, 257–266. [Google Scholar] [CrossRef]
- Parisi, L.; Gini, E.; Baci, D.; Tremolati, M.; Fanuli, M.; Bassani, B.; Farronato, G.; Bruno, A.; Mortara, L. Macrophage Polarization in Chronic Inflammatory Diseases: Killers or Builders? J. Immunol. Res. 2018, 2018, 8917804. [Google Scholar] [CrossRef]
- Aurora, A.B.; Porrello, E.R.; Tan, W.; Mahmoud, A.I.; Hill, J.A.; Bassel-Duby, R.; Sadek, H.A.; Olson, E.N. Macrophages are required for neonatal heart regeneration. J. Clin. Investig. 2014, 124, 1382–1392. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS−) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef] [PubMed]
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; de Bruijn, M.F.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015, 518, 547–551. [Google Scholar] [CrossRef]
- Schulz, C.; Gomez Perdiguero, E.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.; Pollard, J.W.; et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef]
- Edholm, E.S.; Rhoo, K.H.; Robert, J. Evolutionary Aspects of Macrophages Polarization. Results Probl. Cell Differ. 2017, 62, 3–22. [Google Scholar] [CrossRef]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’Antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.A.; et al. Revisiting Cardiac Cellular Composition. Circ. Res. 2016, 118, 400–409. [Google Scholar] [CrossRef]
- Leid, J.; Carrelha, J.; Boukarabila, H.; Epelman, S.; Jacobsen, S.E.; Lavine, K.J. Primitive Embryonic Macrophages are Required for Coronary Development and Maturation. Circ. Res. 2016, 118, 1498–1511. [Google Scholar] [CrossRef]
- Hoyer, F.F.; Naxerova, K.; Schloss, M.J.; Hulsmans, M.; Nair, A.V.; Dutta, P.; Calcagno, D.M.; Herisson, F.; Anzai, A.; Sun, Y.; et al. Tissue-Specific Macrophage Responses to Remote Injury Impact the Outcome of Subsequent Local Immune Challenge. Immunity 2019, 51, 899–914.e7. [Google Scholar] [CrossRef]
- Epelman, S.; Liu, P.P.; Mann, D.L. Role of innate and adaptive immune mechanisms in cardiac injury and repair. Nat. Rev. Immunol. 2015, 15, 117–129. [Google Scholar] [CrossRef]
- Dick, S.A.; Macklin, J.A.; Nejat, S.; Momen, A.; Clemente-Casares, X.; Althagafi, M.G.; Chen, J.; Kantores, C.; Hosseinzadeh, S.; Aronoff, L.; et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat. Immunol. 2019, 20, 29–39. [Google Scholar] [CrossRef]
- Bajpai, G.; Schneider, C.; Wong, N.; Bredemeyer, A.; Hulsmans, M.; Nahrendorf, M.; Epelman, S.; Kreisel, D.; Liu, Y.; Itoh, A.; et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat. Med. 2018, 24, 1234–1245. [Google Scholar] [CrossRef]
- Heidt, T.; Courties, G.; Dutta, P.; Sager, H.B.; Sebas, M.; Iwamoto, Y.; Sun, Y.; Da Silva, N.; Panizzi, P.; van der Laan, A.M.; et al. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ. Res. 2014, 115, 284–295. [Google Scholar] [CrossRef]
- Nahrendorf, M.; Swirski, F.K.; Aikawa, E.; Stangenberg, L.; Wurdinger, T.; Figueiredo, J.L.; Libby, P.; Weissleder, R.; Pittet, M.J. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 2007, 204, 3037–3047. [Google Scholar] [CrossRef]
- Peet, C.; Ivetic, A.; Bromage, D.I.; Shah, A.M. Cardiac monocytes and macrophages after myocardial infarction. Cardiovasc. Res. 2020, 116, 1101–1112. [Google Scholar] [CrossRef]
- Ingersoll, M.A.; Spanbroek, R.; Lottaz, C.; Gautier, E.L.; Frankenberger, M.; Hoffmann, R.; Lang, R.; Haniffa, M.; Collin, M.; Tacke, F.; et al. Comparison of gene expression profiles between human and mouse monocyte subsets. Blood 2010, 115, e10–e19. [Google Scholar] [CrossRef]
- Hilgendorf, I.; Gerhardt, L.M.; Tan, T.C.; Winter, C.; Holderried, T.A.; Chousterman, B.G.; Iwamoto, Y.; Liao, R.; Zirlik, A.; Scherer-Crosbie, M.; et al. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ. Res. 2014, 114, 1611–1622. [Google Scholar] [CrossRef]
- Frangogiannis, N.G.; Dewald, O.; Xia, Y.; Ren, G.; Haudek, S.; Leucker, T.; Kraemer, D.; Taffet, G.; Rollins, B.J.; Entman, M.L. Critical role of monocyte chemoattractant protein-1/CC chemokine ligand 2 in the pathogenesis of ischemic cardiomyopathy. Circulation 2007, 115, 584–592. [Google Scholar] [CrossRef]
- Le Grand, F.; Rudnicki, M.A. Skeletal muscle satellite cells and adult myogenesis. Curr. Opin. Cell Biol. 2007, 19, 628–633. [Google Scholar] [CrossRef]
- Lepper, C.; Partridge, T.A.; Fan, C.M. An absolute requirement for Pax7-positive satellite cells in acute injury-induced skeletal muscle regeneration. Development 2011, 138, 3639–3646. [Google Scholar] [CrossRef] [PubMed]
- Saclier, M.; Yacoub-Youssef, H.; Mackey, A.L.; Arnold, L.; Ardjoune, H.; Magnan, M.; Sailhan, F.; Chelly, J.; Pavlath, G.K.; Mounier, R.; et al. Differentially activated macrophages orchestrate myogenic precursor cell fate during human skeletal muscle regeneration. Stem Cells 2013, 31, 384–396. [Google Scholar] [CrossRef] [PubMed]
- McLennan, I.S. Resident macrophages (ED2- and ED3-positive) do not phagocytose degenerating rat skeletal muscle fibres. Cell Tissue Res. 1993, 272, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Arnold, L.; Henry, A.; Poron, F.; Baba-Amer, Y.; van Rooijen, N.; Plonquet, A.; Gherardi, R.K.; Chazaud, B. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J. Exp. Med. 2007, 204, 1057–1069. [Google Scholar] [CrossRef]
- Varga, T.; Mounier, R.; Horvath, A.; Cuvellier, S.; Dumont, F.; Poliska, S.; Ardjoune, H.; Juban, G.; Nagy, L.; Chazaud, B. Highly Dynamic Transcriptional Signature of Distinct Macrophage Subsets during Sterile Inflammation, Resolution, and Tissue Repair. J. Immunol. 2016, 196, 4771–4782. [Google Scholar] [CrossRef]
- Geissmann, F.; Jung, S.; Littman, D.R. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 2003, 19, 71–82. [Google Scholar] [CrossRef]
- St Pierre, B.A.; Tidball, J.G. Differential response of macrophage subpopulations to soleus muscle reloading after rat hindlimb suspension. J. Appl. Physiol. 1994, 77, 290–297. [Google Scholar] [CrossRef]
- Duelen, R.; Sampaolesi, M. Stem Cell Technology in Cardiac Regeneration: A Pluripotent Stem Cell Promise. eBioMedicine 2017, 16, 30–40. [Google Scholar] [CrossRef]
- Scalise, M.; Marino, F.; Cianflone, E.; Mancuso, T.; Marotta, P.; Aquila, I.; Torella, M.; Nadal-Ginard, B.; Torella, D. Heterogeneity of Adult Cardiac Stem Cells. Adv. Exp. Med. Biol. 2019, 1169, 141–178. [Google Scholar] [CrossRef]
- Wysoczynski, M.; Bolli, R. A realistic appraisal of the use of embryonic stem cell-based therapies for cardiac repair. Eur. Heart J. 2020, 41, 2397–2404. [Google Scholar] [CrossRef]
- Venkatachalam, K.; Venkatesan, B.; Valente, A.J.; Melby, P.C.; Nandish, S.; Reusch, J.E.; Clark, R.A.; Chandrasekar, B. WISP1, a pro-mitogenic, pro-survival factor, mediates tumor necrosis factor-alpha (TNF-alpha)-stimulated cardiac fibroblast proliferation but inhibits TNF-alpha-induced cardiomyocyte death. J. Biol. Chem. 2009, 284, 14414–14427. [Google Scholar] [CrossRef]
- Suzuki, K.; Murtuza, B.; Beauchamp, J.R.; Brand, N.J.; Barton, P.J.; Varela-Carver, A.; Fukushima, S.; Coppen, S.R.; Partridge, T.A.; Yacoub, M.H. Role of interleukin-1beta in acute inflammation and graft death after cell transplantation to the heart. Circulation 2004, 110, II219–II224. [Google Scholar] [CrossRef]
- Hamid, T.; Xu, Y.; Ismahil, M.A.; Li, Q.; Jones, S.P.; Bhatnagar, A.; Bolli, R.; Prabhu, S.D. TNF receptor signaling inhibits cardiomyogenic differentiation of cardiac stem cells and promotes a neuroadrenergic-like fate. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H1189–H1201. [Google Scholar] [CrossRef]
- Vagnozzi, R.J.; Maillet, M.; Sargent, M.A.; Khalil, H.; Johansen, A.K.Z.; Schwanekamp, J.A.; York, A.J.; Huang, V.; Nahrendorf, M.; Sadayappan, S.; et al. An acute immune response underlies the benefit of cardiac stem cell therapy. Nature 2020, 577, 405–409. [Google Scholar] [CrossRef]
- Marino, F.; Scalise, M.; Cianflone, E.; Salerno, L.; Cappetta, D.; Salerno, N.; De Angelis, A.; Torella, D.; Urbanek, K. Physical Exercise and Cardiac Repair: The Potential Role of Nitric Oxide in Boosting Stem Cell Regenerative Biology. Antioxidants 2021, 10, 1002. [Google Scholar] [CrossRef]
- Simões, F.C.; Cahill, T.J.; Kenyon, A.; Gavriouchkina, D.; Vieira, J.M.; Sun, X.; Pezzolla, D.; Ravaud, C.; Masmanian, E.; Weinberger, M.; et al. Macrophages directly contribute collagen to scar formation during zebrafish heart regeneration and mouse heart repair. Nat. Commun. 2020, 11, 600. [Google Scholar] [CrossRef]
- Sadek, H.; Olson, E.N. Toward the Goal of Human Heart Regeneration. Cell Stem Cell 2020, 26, 7–16. [Google Scholar] [CrossRef]
- Hirose, K.; Payumo, A.Y.; Cutie, S.; Hoang, A.; Zhang, H.; Guyot, R.; Lunn, D.; Bigley, R.B.; Yu, H.; Wang, J.; et al. Evidence for hormonal control of heart regenerative capacity during endothermy acquisition. Science 2019, 364, 184–188. [Google Scholar] [CrossRef]
- Ashour, D.; Rebs, S.; Arampatzi, P.; Saliba, A.E.; Dudek, J.; Schulz, R.; Hofmann, U.; Frantz, S.; Cochain, C.; Streckfuß-Bömeke, K.; et al. An interferon gamma response signature links myocardial aging and immunosenescence. Cardiovasc. Res. 2023, cvad068. [Google Scholar] [CrossRef]
- Dolejsi, T.; Delgobo, M.; Schuetz, T.; Tortola, L.; Heinze, K.G.; Hofmann, U.; Frantz, S.; Bauer, A.; Ruschitzka, F.; Penninger, J.M.; et al. Adult T-cells impair neonatal cardiac regeneration. Eur. Heart J. 2022, 43, 2698–2709. [Google Scholar] [CrossRef]
- Chazaud, B. Inflammation and Skeletal Muscle Regeneration: Leave It to the Macrophages! Trends Immunol. 2020, 41, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Ponnappan, S.; Ponnappan, U. Aging and immune function: Molecular mechanisms to interventions. Antioxid. Redox Signal. 2011, 14, 1551–1585. [Google Scholar] [CrossRef] [PubMed]
- Gude, N.A.; Broughton, K.M.; Firouzi, F.; Sussman, M.A. Cardiac ageing: Extrinsic and intrinsic factors in cellular renewal and senescence. Nat. Rev. Cardiol. 2018, 15, 523–542. [Google Scholar] [CrossRef] [PubMed]
- Schultz, M.B.; Sinclair, D.A. When stem cells grow old: Phenotypes and mechanisms of stem cell aging. Development 2016, 143, 3–14. [Google Scholar] [CrossRef]
- Martínez-Zamudio, R.I.; Robinson, L.; Roux, P.F.; Bischof, O. SnapShot: Cellular Senescence Pathways. Cell 2017, 170, 816–816.e1. [Google Scholar] [CrossRef]
- Jackaman, C.; Radley-Crabb, H.G.; Soffe, Z.; Shavlakadze, T.; Grounds, M.D.; Nelson, D.J. Targeting macrophages rescues age-related immune deficiencies in C57BL/6J geriatric mice. Aging Cell 2013, 12, 345–357. [Google Scholar] [CrossRef]
- Mahbub, S.; Deburghgraeve, C.R.; Kovacs, E.J. Advanced age impairs macrophage polarization. J. Interferon Cytokine Res. 2012, 32, 18–26. [Google Scholar] [CrossRef]
- Savoia, C.; Schiffrin, E.L. Inflammation in hypertension. Curr. Opin. Nephrol. Hypertens. 2006, 15, 152–158. [Google Scholar] [CrossRef]
- Martín-Timón, I.; Sevillano-Collantes, C.; Segura-Galindo, A.; Del Cañizo-Gómez, F.J. Type 2 diabetes and cardiovascular disease: Have all risk factors the same strength? World J. Diabetes 2014, 5, 444–470. [Google Scholar] [CrossRef]
- Halade, G.V.; Kain, V.; Black, L.M.; Prabhu, S.D.; Ingle, K.A. Aging dysregulates D- and E-series resolvins to modulate cardiosplenic and cardiorenal network following myocardial infarction. Aging 2016, 8, 2611–2634. [Google Scholar] [CrossRef]
- Molawi, K.; Wolf, Y.; Kandalla, P.K.; Favret, J.; Hagemeyer, N.; Frenzel, K.; Pinto, A.R.; Klapproth, K.; Henri, S.; Malissen, B.; et al. Progressive replacement of embryo-derived cardiac macrophages with age. J. Exp. Med. 2014, 211, 2151–2158. [Google Scholar] [CrossRef]
- Lujambio, A.; Akkari, L.; Simon, J.; Grace, D.; Tschaharganeh, D.F.; Bolden, J.E.; Zhao, Z.; Thapar, V.; Joyce, J.A.; Krizhanovsky, V.; et al. Non-cell-autonomous tumor suppression by p53. Cell 2013, 153, 449–460. [Google Scholar] [CrossRef]
- Lin, I.H.; Chang, J.L.; Hua, K.; Huang, W.C.; Hsu, M.T.; Chen, Y.F. Skeletal muscle in aged mice reveals extensive transformation of muscle gene expression. BMC Genet. 2018, 19, 55. [Google Scholar] [CrossRef]
- Wang, Y.; Wehling-Henricks, M.; Welc, S.S.; Fisher, A.L.; Zuo, Q.; Tidball, J.G. Aging of the immune system causes reductions in muscle stem cell populations, promotes their shift to a fibrogenic phenotype, and modulates sarcopenia. FASEB J. 2019, 33, 1415–1427. [Google Scholar] [CrossRef]
- Wang, M.; Shah, A.M. Age-associated pro-inflammatory remodeling and functional phenotype in the heart and large arteries. J. Mol. Cell. Cardiol. 2015, 83, 101–111. [Google Scholar] [CrossRef]
- Wang, Y.; Wehling-Henricks, M.; Samengo, G.; Tidball, J.G. Increases of M2a macrophages and fibrosis in aging muscle are influenced by bone marrow aging and negatively regulated by muscle-derived nitric oxide. Aging Cell 2015, 14, 678–688. [Google Scholar] [CrossRef]
- Przybyla, B.; Gurley, C.; Harvey, J.F.; Bearden, E.; Kortebein, P.; Evans, W.J.; Sullivan, D.H.; Peterson, C.A.; Dennis, R.A. Aging alters macrophage properties in human skeletal muscle both at rest and in response to acute resistance exercise. Exp. Gerontol. 2006, 41, 320–327. [Google Scholar] [CrossRef]
- Harel-Adar, T.; Ben Mordechai, T.; Amsalem, Y.; Feinberg, M.S.; Leor, J.; Cohen, S. Modulation of cardiac macrophages by phosphatidylserine-presenting liposomes improves infarct repair. Proc. Natl. Acad. Sci. USA 2011, 108, 1827–1832. [Google Scholar] [CrossRef]
- Ben-Mordechai, T.; Holbova, R.; Landa-Rouben, N.; Harel-Adar, T.; Feinberg, M.S.; Abd Elrahman, I.; Blum, G.; Epstein, F.H.; Silman, Z.; Cohen, S.; et al. Macrophage subpopulations are essential for infarct repair with and without stem cell therapy. J. Am. Coll. Cardiol. 2013, 62, 1890–1901. [Google Scholar] [CrossRef]
- Ibrahim, A.G.; Cheng, K.; Marbán, E. Exosomes as critical agents of cardiac regeneration triggered by cell therapy. Stem Cell Rep. 2014, 2, 606–619. [Google Scholar] [CrossRef]
- Barile, L.; Lionetti, V.; Cervio, E.; Matteucci, M.; Gherghiceanu, M.; Popescu, L.M.; Torre, T.; Siclari, F.; Moccetti, T.; Vassalli, G. Extracellular vesicles from human cardiac progenitor cells inhibit cardiomyocyte apoptosis and improve cardiac function after myocardial infarction. Cardiovasc. Res. 2014, 103, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Cambier, L.; de Couto, G.; Ibrahim, A.; Echavez, A.K.; Valle, J.; Liu, W.; Kreke, M.; Smith, R.R.; Marbán, L.; Marbán, E. Y RNA fragment in extracellular vesicles confers cardioprotection via modulation of IL-10 expression and secretion. EMBO Mol. Med. 2017, 9, 337–352. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, T.; Barone, A.; Salatino, A.; Molinaro, C.; Marino, F.; Scalise, M.; Torella, M.; De Angelis, A.; Urbanek, K.; Torella, D.; et al. Unravelling the Biology of Adult Cardiac Stem Cell-Derived Exosomes to Foster Endogenous Cardiac Regeneration and Repair. Int. J. Mol. Sci. 2020, 21, 3725. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef] [PubMed]
- de Couto, G.; Gallet, R.; Cambier, L.; Jaghatspanyan, E.; Makkar, N.; Dawkins, J.F.; Berman, B.P.; Marbán, E. Exosomal MicroRNA Transfer Into Macrophages Mediates Cellular Postconditioning. Circulation 2017, 136, 200–214. [Google Scholar] [CrossRef] [PubMed]
- Thum, T.; Bauersachs, J.; Poole-Wilson, P.A.; Volk, H.D.; Anker, S.D. The dying stem cell hypothesis: Immune modulation as a novel mechanism for progenitor cell therapy in cardiac muscle. J. Am. Coll. Cardiol. 2005, 46, 1799–1802. [Google Scholar] [CrossRef]
- Ludman, A.; Venugopal, V.; Yellon, D.M.; Hausenloy, D.J. Statins and cardioprotection--more than just lipid lowering? Pharmacol. Ther. 2009, 122, 30–43. [Google Scholar] [CrossRef]
- Cianflone, E.; Cappetta, D.; Mancuso, T.; Sabatino, J.; Marino, F.; Scalise, M.; Albanese, M.; Salatino, A.; Parrotta, E.I.; Cuda, G.; et al. Statins Stimulate New Myocyte Formation After Myocardial Infarction by Activating Growth and Differentiation of the Endogenous Cardiac Stem Cells. Int. J. Mol. Sci. 2020, 21, 7927. [Google Scholar] [CrossRef]
- Kwak, B.; Mulhaupt, F.; Myit, S.; Mach, F. Statins as a newly recognized type of immunomodulator. Nat. Med. 2000, 6, 1399–1402. [Google Scholar] [CrossRef]
- Ghittoni, R.; Napolitani, G.; Benati, D.; Ulivieri, C.; Patrussi, L.; Laghi Pasini, F.; Lanzavecchia, A.; Baldari, C.T. Simvastatin inhibits the MHC class II pathway of antigen presentation by impairing Ras superfamily GTPases. Eur. J. Immunol. 2006, 36, 2885–2893. [Google Scholar] [CrossRef]
- Bernstein, K.E.; Ong, F.S.; Blackwell, W.L.; Shah, K.H.; Giani, J.F.; Gonzalez-Villalobos, R.A.; Shen, X.Z.; Fuchs, S.; Touyz, R.M. A modern understanding of the traditional and nontraditional biological functions of angiotensin-converting enzyme. Pharmacol. Rev. 2013, 65, 1–46. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Cell Types | Phenotype | Activity | Active Molecules |
|---|---|---|---|
| N1-Neutrophilis | CD11b CD16 CD15 CD87 | Degranulation Phagocytosis Apoptosis | MPO; ROS |
| N2-Neutrophilis | CD11b CD206 | Macrophage polarization Angiogenesis Apoptosis | MMP-9; MMP-12, vEGF |
| Inflammatory Eosinophils | CD62L− CD49d CD101high | Degranulation Oxidative stress | Il-5; ROS; EPO |
| Regulatory Eosinophils | CD62L+ CD101low Sigle-8+ | Macrophage polarization and angiogenesis | Il-4; vEGF; FGF; TGF-β |
| Monocytes | Cd14+CD16−/Ly-6Clow CXC3R1 | Secretion of anti-inflammatory and angiogenic cytokines | IL-1β; IL-6; TNFa; NO; TGF-β; vEGF |
| M1 Macrophages | CCR2 CD68 MHC II CD86 | Phagocytosis Secretion of inflammatory cytokines | IL-12, IL-23; ROS, NO |
| M2 Macrophages | CD206 IL-1Ra TGFβ vEGF | Secretion of anti-inflammatory and angiogenic cytokines | IL-10; IL.1Ra; vEGF; TGF-β |
| Mast Cells | CD64 CD117 | Secretion of inflammatory cytokines Degranulation | Histamine; INF-α; IL-6, IFN-γ |
| Dendritic Cells | MHC II CD80/CD86 | Antigen presentation | IL-23 IL-10 |
| B1- and B2-Lymphocytes | CD19 | Secretion of anti-inflammatory cytokines | IgM, IgG |
| Regulatory B-Lymphocytes | Tim-1 | Chemokine production | IL-10 |
| NK-Cells | CD69 | Secretion of inflammatory cytokines | CCL7 IFN-γ |
| T-Lymphocytes | CD4+ T-helper CD8+ T-cytotoxic CTLA-4 Tregs | Autoreactivity Secretion of inflammatory cytokines | IFN-γ IL-17 TGF-β |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molinaro, C.; Scalise, M.; Leo, I.; Salerno, L.; Sabatino, J.; Salerno, N.; De Rosa, S.; Torella, D.; Cianflone, E.; Marino, F. Polarizing Macrophage Functional Phenotype to Foster Cardiac Regeneration. Int. J. Mol. Sci. 2023, 24, 10747. https://doi.org/10.3390/ijms241310747
Molinaro C, Scalise M, Leo I, Salerno L, Sabatino J, Salerno N, De Rosa S, Torella D, Cianflone E, Marino F. Polarizing Macrophage Functional Phenotype to Foster Cardiac Regeneration. International Journal of Molecular Sciences. 2023; 24(13):10747. https://doi.org/10.3390/ijms241310747
Chicago/Turabian StyleMolinaro, Claudia, Mariangela Scalise, Isabella Leo, Luca Salerno, Jolanda Sabatino, Nadia Salerno, Salvatore De Rosa, Daniele Torella, Eleonora Cianflone, and Fabiola Marino. 2023. "Polarizing Macrophage Functional Phenotype to Foster Cardiac Regeneration" International Journal of Molecular Sciences 24, no. 13: 10747. https://doi.org/10.3390/ijms241310747
APA StyleMolinaro, C., Scalise, M., Leo, I., Salerno, L., Sabatino, J., Salerno, N., De Rosa, S., Torella, D., Cianflone, E., & Marino, F. (2023). Polarizing Macrophage Functional Phenotype to Foster Cardiac Regeneration. International Journal of Molecular Sciences, 24(13), 10747. https://doi.org/10.3390/ijms241310747