
Rapid Identification by Resequencing-Based QTL Mapping of a Novel Allele RGA1-FH Decreasing Grain Length in a Rice Restorer Line ‘Fuhui212’

 ,
, {kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
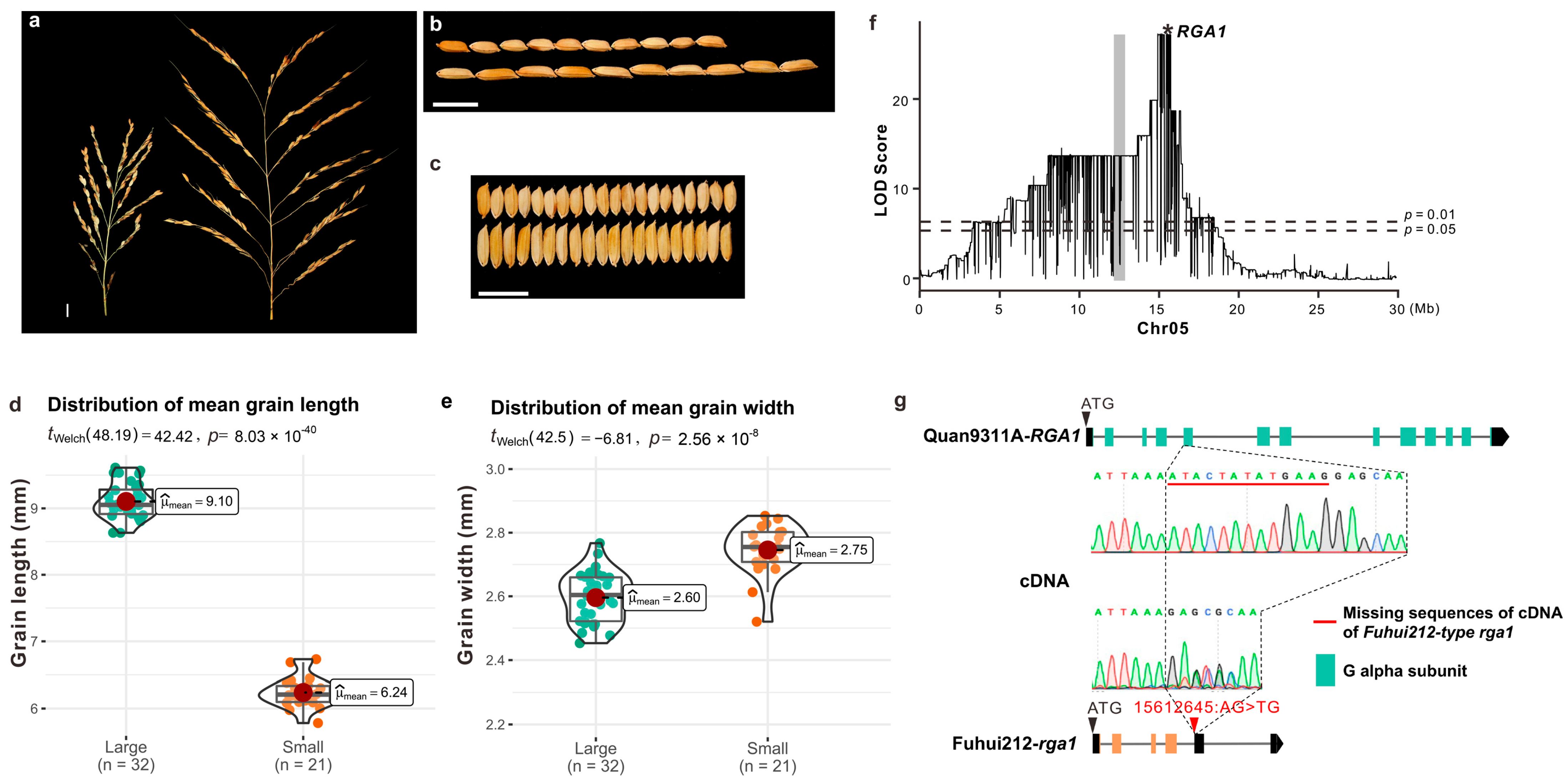
2.1. Phenotyping Analysis of Grain Size in the Cross of ‘Fuhui212’ and ‘Quan9311A’
2.2. Resequencing and Genotyping the F2 Generation Individuals
2.3. QTL Mapping Identifies the Strongest Signals in the Regions of Chromosome 5
2.4. Identification of a Causal Variant and Target Gene
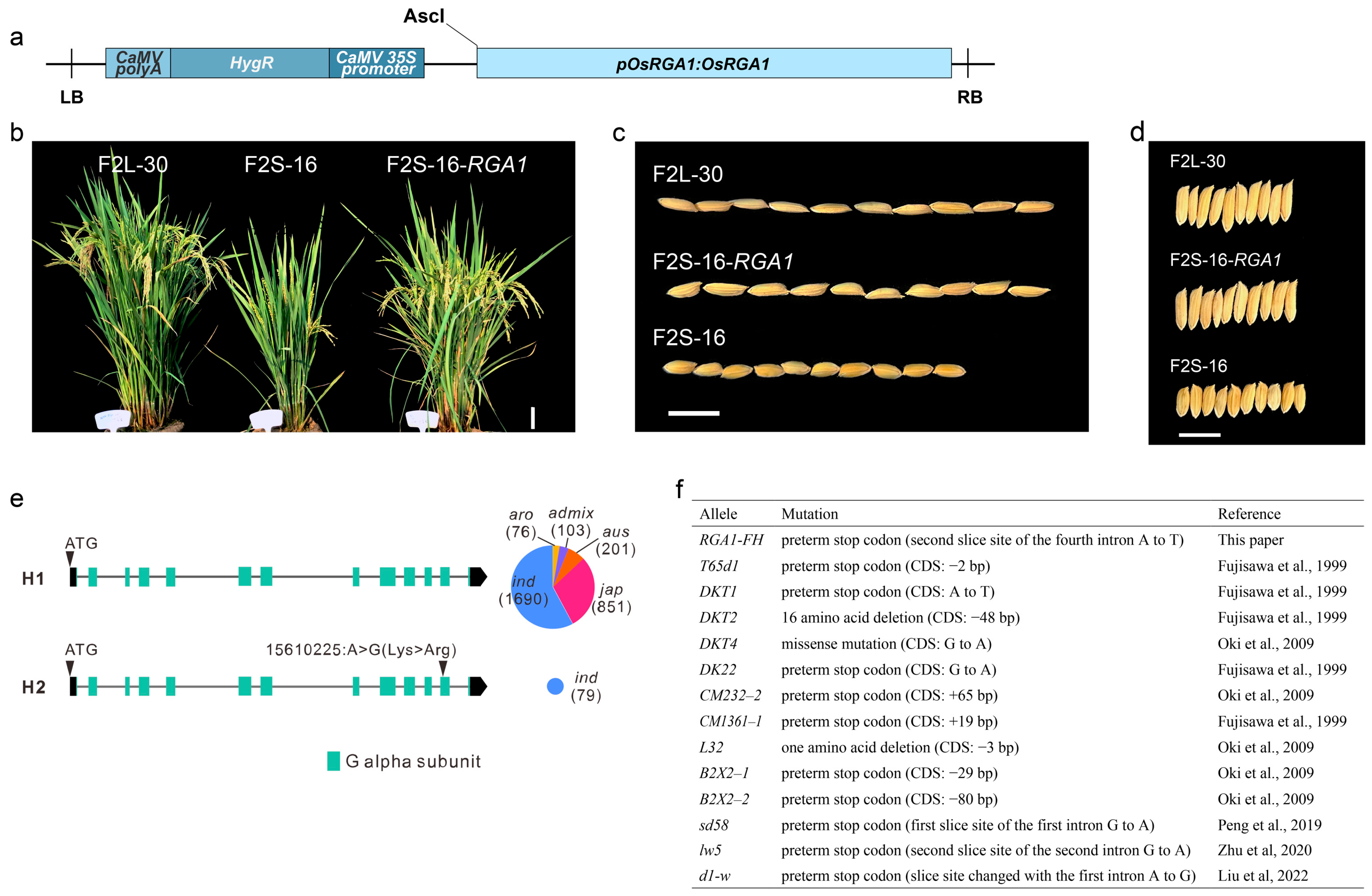
2.5. Complementation Test Confirmed RGA1 as the Dominant Locus
3. Discussion
4. Materials and Methods
4.1. Sampling and Whole-Genome Sequencing
4.2. Sequencing Data Process and Variants Calling
4.3. QTL Mapping
4.4. cDNA Analysis of RGA1
4.5. Genetic Complementation of RGA1
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zuo, J.; Li, J. Molecular Genetic Dissection of Quantitative Trait Loci Regulating Rice Grain Size. Annu. Rev. Genet. 2014, 48, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Zhang, A.; Liu, X.; Chen, J. Grain Size Associated Genes and the Molecular Regulatory Mechanism in Rice. Int. J. Mol. Sci. 2022, 23, 3169. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, S.; Liu, Q.; Wu, K.; Zhang, J.; Wang, S.; Wang, Y.; Chen, X.; Zhang, Y.; Gao, C.; et al. The OsSPL16-GW7 Regulatory Module Determines Grain Shape and Simultaneously Improves Rice Yield and Grain Quality. Nat. Genet. 2015, 47, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Si, L.; Chen, J.; Huang, X.; Gong, H.; Luo, J.; Hou, Q.; Zhou, T.; Lu, T.; Zhu, J.; Shangguan, Y.; et al. OsSPL13 Controls Grain Size in Cultivated Rice. Nat. Genet. 2016, 48, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Song, X.-J.; Huang, W.; Shi, M.; Zhu, M.-Z.; Lin, H.-X. A QTL for Rice Grain Width and Weight Encodes a Previously Unknown RING-Type E3 Ubiquitin Ligase. Nat. Genet. 2007, 39, 623–630. [Google Scholar] [CrossRef]
- Sun, S.; Wang, L.; Mao, H.; Shao, L.; Li, X.; Xiao, J.; Ouyang, Y.; Zhang, Q. A G-Protein Pathway Determines Grain Size in Rice. Nat. Commun. 2018, 9, 851. [Google Scholar] [CrossRef]
- Fujisawa, Y.; Kato, T.; Ohki, S.; Ishikawa, A.; Kitano, H.; Sasaki, T.; Asahi, T.; Iwasaki, Y. Suppression of the Heterotrimeric G Protein Causes Abnormal Morphology, Including Dwarfism, in Rice. Proc. Natl. Acad. Sci. USA 1999, 96, 7575–7580. [Google Scholar] [CrossRef]
- Ashikari, M.; Wu, J.; Yano, M.; Sasaki, T.; Yoshimura, A. Rice Gibberellin-Insensitive Dwarf Mutant Gene Dwarf 1 Encodes the α-Subunit of GTP-Binding Protein. Proc. Natl. Acad. Sci. USA 1999, 96, 10284–10289. [Google Scholar] [CrossRef]
- Utsunomiya, Y.; Samejima, C.; Takayanagi, Y.; Izawa, Y.; Yoshida, T.; Sawada, Y.; Fujisawa, Y.; Kato, H.; Iwasaki, Y. Suppression of the Rice Heterotrimeric G Protein β-Subunit Gene, RGB1, Causes Dwarfism and Browning of Internodes and Lamina Joint Regions. Plant J. 2011, 67, 907–916. [Google Scholar] [CrossRef]
- Liu, Q.; Han, R.; Wu, K.; Zhang, J.; Ye, Y.; Wang, S.; Chen, J.; Pan, Y.; Li, Q.; Xu, X.; et al. G-Protein Βγ Subunits Determine Grain Size through Interaction with MADS-Domain Transcription Factors in Rice. Nat. Commun. 2018, 9, 852. [Google Scholar] [CrossRef]
- Oki, K.; Inaba, N.; Kitano, H.; Takahashi, S.; Fujisawa, Y.; Kato, H.; Iwasaki, Y. Study of Novel D1 Alleles, Defective Mutants of the α Subunit of Heterotrimeric G-Protein in Rice. Genes Genet. Syst. 2009, 84, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Qian, Q.; Wu, K.; Luo, J.; Wang, S.; Zhang, C.; Ma, Y.; Liu, Q.; Huang, X.; Yuan, Q.; et al. Heterotrimeric G Proteins Regulate Nitrogen-Use Efficiency in Rice. Nat. Genet. 2014, 46, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Agetsuma, M.; Kitano, H.; Yoshimura, A.; Matsuoka, M.; Jacobsen, S.E.; Ashikari, M. A Metastable DWARF1 Epigenetic Mutant Affecting Plant Stature in Rice. Proc. Natl. Acad. Sci. USA 2009, 106, 11218–11223. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wang, H.; You, Q.; Zhou, P.; Tu, S.; Zheng, F.; Dong, R.; Zheng, J.; Huang, T. Breeding of a New Three-line Hybrid Rice Variety M76 You 212. Fujian J. Agric. Sci. 2020, 35, 1289–1295. [Google Scholar] [CrossRef]
- Qin, Y.; Cheng, P.; Cheng, Y.; Feng, Y.; Huang, D.; Huang, T.; Song, X.; Ying, J. QTL-Seq Identified a Major QTL for Grain Length and Weight in Rice Using Near Isogenic F2 Population. Rice Sci. 2018, 25, 121–131. [Google Scholar] [CrossRef]
- Lv, Q.; Li, W.; Sun, Z.; Ouyang, N.; Jing, X.; He, Q.; Wu, J.; Zheng, J.; Zheng, J.; Tang, S.; et al. Resequencing of 1143 Indica Rice Accessions Reveals Important Genetic Variations and Different Heterosis Patterns. Nat. Commun. 2020, 11, 4778. [Google Scholar] [CrossRef]
- Broman, K.W.; Wu, H.; Sen, Ś.; Churchill, G.A. R/Qtl: QTL Mapping in Experimental Crosses. Bioinformatics 2003, 19, 889–890. [Google Scholar] [CrossRef]
- Ueguchi-Tanaka, M.; Fujisawa, Y.; Kobayashi, M.; Ashikari, M.; Iwasaki, Y.; Kitano, H.; Matsuoka, M. Rice Dwarf Mutant D1, Which Is Defective in the α Subunit of the Heterotrimeric G Protein, Affects Gibberellin Signal Transduction. Proc. Natl. Acad. Sci. USA 2000, 97, 11638–11643. [Google Scholar] [CrossRef]
- Bethke, P.C.; Hwang, Y.; Zhu, T.; Jones, R.L. Global Patterns of Gene Expression in the Aleurone of Wild-Type and Dwarf1 Mutant Rice. Plant Physiol. 2006, 140, 484–498. [Google Scholar] [CrossRef]
- Baker, K.E.; Parker, R. Nonsense-Mediated MRNA Decay: Terminating Erroneous Gene Expression. Curr. Opin. Cell Biol. 2004, 16, 293–299. [Google Scholar] [CrossRef]
- Peng, P.; Gao, Y.; Li, Z.; Yu, Y.; Qin, H.; Guo, Y.; Huang, R.; Wang, J. Proteomic Analysis of a Rice Mutant Sd58 Possessing a Novel D1 Allele of Heterotrimeric G Protein Alpha Subunit (RGA1) in Salt Stress with a Focus on ROS Scavenging. Int. J. Mol. Sci. 2019, 20, 167. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, T.; Xu, J.; Wang, J.; Wang, L.; Zou, W.; Zeng, D.; Zhu, L.; Chen, G.; Hu, J.; et al. Leaf Width Gene LW5/D1 Affects Plant Architecture and Yield in Rice by Regulating Nitrogen Utilization Efficiency. Plant Physiol. Biochem. 2020, 157, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ting, L.; Zhishu, J.; Chuihai, Z.; Rong, H.; Jiao, Q.; Xiaoli, L.; Limei, P.; Yongping, S.; Dahu, Z.; et al. Characterization of a Novel Weak Allele of RGA1/D1 and Its Potential Application in Rice Breeding. Rice Sci. 2022, 29, 522–534. [Google Scholar] [CrossRef]
- Wang, W.; Mauleon, R.; Hu, Z.; Chebotarov, D.; Tai, S.; Wu, Z.; Li, M.; Zheng, T.; Fuentes, R.R.; Zhang, F.; et al. Genomic Variation in 3010 Diverse Accessions of Asian Cultivated Rice. Nature 2018, 557, 43–49. [Google Scholar] [CrossRef]
- Ferrero-Serrano, Á.; Assmann, S.M. The α-Subunit of the Rice Heterotrimeric G Protein, RGA1, Regulates Drought Tolerance during the Vegetative Phase in the Dwarf Rice Mutant D1. J. Exp. Bot. 2016, 67, 3433–3443. [Google Scholar] [CrossRef]
- Ferrero-Serrano, Á.; Su, Z.; Assmann, S.M. Illuminating the Role of the Gα Heterotrimeric G Protein Subunit, RGA1, in Regulating Photoprotection and Photoavoidance in Rice. Plant Cell Environ. 2018, 41, 451–468. [Google Scholar] [CrossRef]
- Clarke, J.D. Cetyltrimethyl Ammonium Bromide (CTAB) DNA Miniprep for Plant DNA Isolation. Cold Spring Harb. Protoc. 2009, 2009, pdb.prot5177. [Google Scholar] [CrossRef]
- Kawahara, Y.; de la Bastide, M.; Hamilton, J.P.; Kanamori, H.; McCombie, W.R.; Ouyang, S.; Schwartz, D.C.; Tanaka, T.; Wu, J.; Zhou, S.; et al. Improvement of the Oryza Sativa Nipponbare Reference Genome Using next Generation Sequence and Optical Map Data. Rice 2013, 6, 4. [Google Scholar] [CrossRef]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Broman, K.W.; Sen, S. A Guide to QTL Mapping with R/Qtl; Statistics for Biology and Health; Springer: New York, NY, USA, 2009; ISBN 978-0-387-92124-2. [Google Scholar]
- De Baets, G.; Van Durme, J.; Reumers, J.; Maurer-Stroh, S.; Vanhee, P.; Dopazo, J.; Schymkowitz, J.; Rousseau, F. SNPeffect 4.0: On-Line Prediction of Molecular and Structural Effects of Protein-Coding Variants. Nucleic Acids Res. 2012, 40, D935–D939. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, S.; Zhong, Y.; Zheng, S.; He, Y.; Yang, S.; Wang, L.; Traw, M.B.; Zhang, Q.; Zhang, X. Rapid Identification by Resequencing-Based QTL Mapping of a Novel Allele RGA1-FH Decreasing Grain Length in a Rice Restorer Line ‘Fuhui212’. Int. J. Mol. Sci. 2023, 24, 10746. https://doi.org/10.3390/ijms241310746
Ma S, Zhong Y, Zheng S, He Y, Yang S, Wang L, Traw MB, Zhang Q, Zhang X. Rapid Identification by Resequencing-Based QTL Mapping of a Novel Allele RGA1-FH Decreasing Grain Length in a Rice Restorer Line ‘Fuhui212’. International Journal of Molecular Sciences. 2023; 24(13):10746. https://doi.org/10.3390/ijms241310746
Chicago/Turabian StyleMa, Shiying, Yifan Zhong, Shuyi Zheng, Ying He, Sihai Yang, Long Wang, Milton Brian Traw, Qijun Zhang, and Xiaohui Zhang. 2023. "Rapid Identification by Resequencing-Based QTL Mapping of a Novel Allele RGA1-FH Decreasing Grain Length in a Rice Restorer Line ‘Fuhui212’" International Journal of Molecular Sciences 24, no. 13: 10746. https://doi.org/10.3390/ijms241310746
APA StyleMa, S., Zhong, Y., Zheng, S., He, Y., Yang, S., Wang, L., Traw, M. B., Zhang, Q., & Zhang, X. (2023). Rapid Identification by Resequencing-Based QTL Mapping of a Novel Allele RGA1-FH Decreasing Grain Length in a Rice Restorer Line ‘Fuhui212’. International Journal of Molecular Sciences, 24(13), 10746. https://doi.org/10.3390/ijms241310746