
The Presence of T Allele (rs35705950) of the MUC5B Gene Predicts Lower Baseline Forced Vital Capacity and Its Subsequent Decline in Patients with Hypersensitivity Pneumonitis

 ,
,  ,
,  , , ,
, , ,  and
and 
Abstract
1. Introduction
2. Results
2.1. Baseline Characteristics of the Study Group
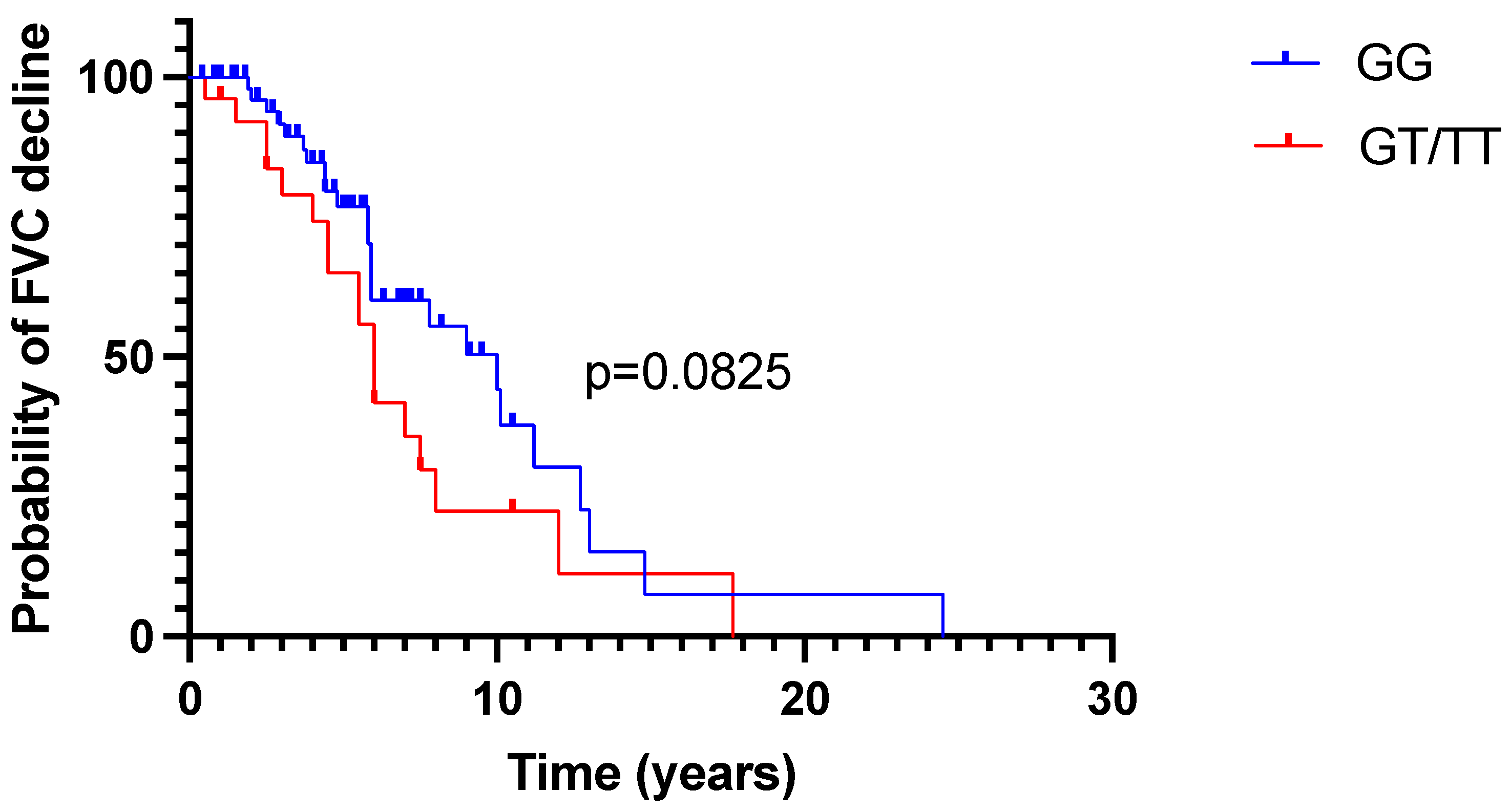
2.2. FVC Trajectory in Time
2.3. Treatment Effect Assessment According to MUC5B Genotype
3. Discussion
Study Limitations
4. Material and Methods
4.1. Regulatory Board Approval
4.2. Study Group
4.3. Study Procedures
4.4. Assessment of the Disease Course
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vasakova, M.; Morell, F.; Walsh, S.; Leslie, K.; Raghu, G. Hypersensitivity Pneumonitis: Perspectives in Diagnosis and Management. Am. J. Respir. Crit. Care Med. 2017, 196, 680–689. [Google Scholar] [CrossRef]
- Raghu, G.; Remy-Jardin, M.; Ryerson, C.J.; Myers, J.L.; Kreuter, M.; Vasakova, M.; Bargagli, E.; Chung, J.H.; Collins, B.F.; Bendstrup, E.; et al. Diagnosis of Hypersensitivity Pneumonitis in Adults. An Official ATS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2020, 202, e36–e69. [Google Scholar] [CrossRef]
- Salisbury, M.L.; Gu, T.; Murray, S.; Gross, B.H.; Chughtai, A.; Sayyouh, M.; Kazerooni, E.A.; Myers, J.L.; Lagstein, A.; Konopka, K.E.; et al. Hypersensitivity Pneumonitis: Radiologic Phenotypes Are Associated with Distinct Survival Time and Pulmonary Function Trajectory. Chest 2019, 155, 699–711. [Google Scholar] [CrossRef]
- Wijsenbeek, M.; Kreuter, M.; Olson, A.; Fischer, A.; Bendstrup, E.; Wells, C.D.; Denton, C.P.; Mounir, B.; Zouad-Lejour, L.; Quaresma, M.; et al. Progressive fibrosing interstitial lung diseases: Current practice in diagnosis and management. Curr. Med. Res. Opin. 2019, 35, 2015–2024. [Google Scholar] [CrossRef]
- Seibold, M.A.; Wise, A.L.; Speer, M.C.; Steele, M.P.; Brown, K.K.; Loyd, J.E.; Fingerlin, T.E.; Zhang, W.; Gudmundsson, G.; Groshong, S.D.; et al. A Common MUC5B Promoter Polymorphism and Pulmonary Fibrosis. N. Engl. J. Med. 2011, 364, 1503–1512. [Google Scholar] [CrossRef]
- Zhang, Y.; Noth, I.; Garcia, J.G.; Kaminski, N. A Variant in the Promoter of MUC5B and Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2011, 364, 1576–1577. [Google Scholar] [CrossRef]
- Fingerlin, T.E.; Murphy, E.; Zhang, W.; Peljto, A.L.; Brown, K.K.; Steele, M.P.; Loyd, J.E.; Cosgrove, G.P.; Lynch, D.; Groshong, S.; et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat. Genet. 2013, 45, 613–620. [Google Scholar] [CrossRef]
- Fingerlin, T.E.; Zhang, W.; Yang, I.V.; Ainsworth, H.C.; Russell, P.H.; Blumhagen, R.Z.; Schwarz, M.I.; Brown, K.K.; Steele, M.P.; Loyd, J.E.; et al. Genome-wide imputation study identifies novel HLA locus for pulmonary fibrosis and potential role for auto-immunity in fibrotic idiopathic interstitial pneumonia. BMC Genet. 2016, 17, 74. [Google Scholar] [CrossRef]
- Moore, C.; Blumhagen, R.Z.; Yang, I.V.; Walts, A.; Powers, J.; Walker, T.; Bishop, M.; Russell, P.; Vestal, B.; Cardwell, J.; et al. Resequencing Study Confirms That Host Defense and Cell Senescence Gene Variants Contribute to the Risk of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 200, 199–208. [Google Scholar] [CrossRef]
- Allen, R.J.; Porte, J.; Braybrooke, R.; Flores, C.; Fingerlin, T.E.; Oldham, J.M.; Guillen-Guio, B.; Ma, S.-F.; Okamoto, T.; John, A.E.; et al. Genetic variants associated with susceptibility to idiopathic pulmonary fibrosis in people of European ancestry: A genome-wide association study. Lancet Respir. Med. 2017, 5, 869–880. [Google Scholar] [CrossRef]
- Thornton, D.J.; Devine, P.L.; Hanski, C.; Howard, M.; Sheehan, J.K. Identification of two major populations of mucins in respiratory secretions. Am. J. Respir. Crit. Care Med. 1994, 150, 823–832. [Google Scholar] [CrossRef]
- Thornton, D.; Carlstedt, I.; Howard, M.; Devine, P.L.; Price, M.R.; Sheehan, J.K. Respiratory mucins: Identification of core proteins and glycoforms. Biochem. J. 1996, 316 Pt 3, 967–975. [Google Scholar] [CrossRef]
- Roy, M.G.; Livraghi-Butrico, A.; Fletcher, A.A.; McElwee, M.M.; Evans, S.E.; Boerner, R.M.; Alexander, S.N.; Bellinghausen, L.K.; Song, A.S.; Petrova, Y.M.; et al. Muc5b is required for airway defence. Nature 2014, 505, 412–416. [Google Scholar] [CrossRef]
- Hancock, L.A.; Hennessy, C.E.; Solomon, G.M.; Dobrinskikh, E.; Estrella, A.; Hara, N.; Hill, D.B.; Kissner, W.J.; Markovetz, M.R.; Villalon, D.E.G.; et al. Muc5b overexpression causes mucociliary dysfunction and enhances lung fibrosis in mice. Nat. Commun. 2018, 9, 5363. [Google Scholar] [CrossRef]
- Evans, C.M.; Fingerlin, T.E.; Schwarz, M.I.; Lynch, D.; Kurche, J.; Warg, L.; Yang, I.V.; Schwartz, D.A. Idiopathic Pulmonary Fibrosis: A Genetic Disease That Involves Mucociliary Dysfunction of the Peripheral Airways. Physiol. Rev. 2016, 96, 1567–1591. [Google Scholar] [CrossRef]
- Selman, M.; Pardo, A. Revealing the Pathogenic and Aging-related Mechanisms of the Enigmatic Idiopathic Pulmonary Fibrosis. An Integral Model. Am. J. Respir. Crit. Care Med. 2014, 189, 1161–1172. [Google Scholar] [CrossRef]
- Schwartz, D.A. Idiopathic Pulmonary Fibrosis Is a Complex Genetic Disorder. Trans. Am. Clin. Clim. Assoc. 2016, 127, 34–45. [Google Scholar]
- Schwartz, D.A. Idiopathic Pulmonary Fibrosis Is a Genetic Disease Involving Mucus and the Peripheral Airways. Ann. Am. Thorac. Soc. 2018, 15 (Suppl. 3), S192–S197. [Google Scholar] [CrossRef]
- Adegunsoye, A.; Vij, R.; Noth, I. Integrating Genomics into Management of Fibrotic Interstitial Lung Disease. Chest 2019, 155, 1026–1040. [Google Scholar] [CrossRef]
- Falfán-Valencia, R.; Camarena, Á.; Pineda, C.L.; Montaño, M.; Juárez, A.; Buendía-Roldán, I.; Pérez-Rubio, G.; Reséndiz-Hernández, J.M.; Páramo, I.; Vega, A.; et al. Genetic susceptibility to multicase hypersensitivity pneumonitis is associated with the TNF-238 GG genotype of the promoter region and HLA-DRB1*04 bearing HLA haplotypes. Respir. Med. 2014, 108, 211–217. [Google Scholar] [CrossRef]
- Ley, B.; Newton, C.; Arnould, I.; Elicker, B.M.; Henry, T.S.; Vittinghoff, E.; Golden, J.A.; Jones, K.D.; Batra, K.; Torrealba, J.; et al. The MUC5B promoter polymorphism and telomere length in patients with chronic hypersensitivity pneumonitis: An observational cohort-control study. Lancet Respir. Med. 2017, 5, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, H.; Peljto, A.L.; Walts, A.D.; Cardwell, J.; Molyneaux, P.L.; Lee, J.S.; Pérez, E.R.F.; Wolters, P.J.; Yang, I.V.; Schwartz, D.A. Common idiopathic pulmonary fibrosis risk variants are associated with hypersensitivity pneumonitis. Thorax 2022, 77, 508–510. [Google Scholar] [CrossRef] [PubMed]
- Lettieri, C.J.; Nathan, S.D.; Browning, R.F.; Barnett, S.D.; Ahmad, S.; Shorr, A.F. The distance-saturation product predicts mortality in idiopathic pulmonary fibrosis. Respir. Med. 2006, 100, 1734–1741. [Google Scholar] [CrossRef]
- Ley, B.; Ryerson, C.J.; Vittinghoff, E.; Ryu, J.; Tomassetti, S.; Lee, J.S.; Poletti, V.; Buccioli, M.; Elicker, B.M.; Jones, K.D.; et al. A Multidimensional Index and Staging System for Idiopathic Pulmonary Fibrosis. Ann. Intern. Med. 2012, 156, 684–691. [Google Scholar] [CrossRef] [PubMed]
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef]
- Vasakova, M.; Selman, M.; Morell, F.; Sterclova, M.; Molina-Molina, M.; Raghu, G. Hypersensitivity Pneumonitis: Current Concepts of Pathogenesis and Potential Targets for Treatment. Am. J. Respir. Crit. Care Med. 2019, 200, 301–308. [Google Scholar] [CrossRef]
- Morell, F.; Roger, À.; Reyes, L.; Cruz, M.J.; Murio, C.; Muñoz, X. Bird Fancier’s Lung: A Series of 86 Patients. Medicine 2008, 87, 110–130. [Google Scholar] [CrossRef]
- McSharry, C.; Banham, S.W.; Boyd, G. Effect of cigarette smoking on the antibody response to inhaled antigens and the prevalence of extrinsic allergic alveolitis among pigeon breeders. Clin. Exp. Allergy 1985, 15, 487–494. [Google Scholar] [CrossRef]
- Postawy Polaków Wobec Palenia Tytoniu–Raport 2019 r.–Główny Inspektorat Sanitarny–Portal Gov.pl. Available online: https://www.gov.pl/web/gis/postawy-polakow-wobec-palenia-tytoniu-raport-2017 (accessed on 27 May 2023).
- Horimasu, Y.; Ohshimo, S.; Bonella, F.; Tanaka, S.; Ishikawa, N.; Hattori, N.; Kohno, N.; Guzman, J.; Costabel, U. MUC5B promoter polymorphism in Japanese patients with idiopathic pulmonary fibrosis. Respirology 2015, 20, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhuang, Y.; Guo, W.; Cao, L.; Zhang, H.; Xu, L.; Fan, Y.; Zhang, D.; Wang, Y. Mucin 5B Promoter Polymorphism Is Associated with Susceptibility to Interstitial Lung Diseases in Chinese Males. PLoS ONE 2014, 9, e104919. [Google Scholar] [CrossRef]
- Peljto, A.L.; Zhang, Y.; Fingerlin, T.E.; Ma, S.-F.; Garcia, J.G.N.; Richards, T.J.; Silveira, L.J.; Lindell, K.O.; Steele, M.P.; Loyd, J.; et al. Association Between the MUC5B Promoter Polymorphism and Survival in Patients with Idiopathic Pulmonary Fibrosis. JAMA 2013, 309, 2232–2239. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, H.; Cardwell, J.H.; Okamoto, T.; Walts, A.D.; Konigsberg, I.R.; Kurche, J.S.; Bang, T.J.; Schwarz, M.I.; Brown, K.K.; Kropski, J.A.; et al. Chronic Hypersensitivity Pneumonitis, an Interstitial Lung Disease with Distinct Molecular Signatures. Am. J. Respir. Crit. Care Med. 2020, 202, 1430–1444. [Google Scholar] [CrossRef]
- Noth, I.; Zhang, Y.; Ma, S.-F.; Flores, C.; Barber, M.; Huang, Y.; Broderick, S.M.; Wade, M.S.; Hysi, P.; Scuirba, J.; et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: A genome-wide association study. Lancet Respir. Med. 2013, 1, 309–317. [Google Scholar] [CrossRef]
- Jiang, H.; Hu, Y.; Shang, L.; Li, Y.; Yang, L.; Chen, Y. Association between MUC5B polymorphism and susceptibility and severity of idiopathic pulmonary fibrosis. Int. J. Clin. Exp. Pathol. 2015, 8, 14953–14958. [Google Scholar] [PubMed]
- Szturmowicz, M.; Barańska, I.; Jędrych, M.E.; Bartoszuk, I.; Radwan-Roehrenschef, P.; Roży, A.; Bestry, I.; Chorostowska-Wynimko, J.; Langfort, R.; Kuś, J. Hypersensitivity Pneumonitis Recognised in a Single Pulmonary Unit, between 2005 and 2015: Comparison with Recently Proposed Diagnostic Criteria. Adv. Respir. Med. 2019, 87, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Lewandowska, K.B.; Barańska, I.; Sobiecka, M.; Radwan-Rohrenschef, P.; Dybowska, M.; Franczuk, M.; Roży, A.; Skoczylas, A.; Bestry, I.; Kuś, J.; et al. Factors Predictive for Immunomodulatory Therapy Response and Survival in Patients with Hypersensitivity Pneumonitis—Retrospective Cohort Analysis. Diagnostics 2022, 12, 2767. [Google Scholar] [CrossRef] [PubMed]
- Graham, B.L.; Brusasco, V.; Burgos, F.; Cooper, B.G.; Jensen, R.; Kendrick, A.; MacIntyre, N.R.; Thompson, B.R.; Wanger, J. 2017 ERS/ATS standards for single-breath carbon monoxide uptake in the lung. Eur. Respir. J. 2017, 49, 1600016. [Google Scholar] [CrossRef]
- ATS Committee on Proficiency Standards for Clinical Pulmonary Function Laboratories. ATS statement: Guidelines for the six-minute walk test. Am. J. Respir. Crit. Care Med. 2002, 166, 111–117. [Google Scholar] [CrossRef]
- Przybyłowski, T.; Tomalak, W.; Siergiejko, Z.; Jastrzębski, D.; Maskey-Warzęchowska, M.; Piorunek, T.; Wojda, E.; Boros, P. Polish Respiratory Society Guidelines for the Methodology and Interpretation of the 6 Minute Walk Test (6MWT). Pneumonol. Alergol. Pol. Wyd. Pol. 2015, 1, 9–25. (In Polish) [Google Scholar] [CrossRef]
- Lewandowska, K.B.; Sobiecka, M.; Boros, P.W.; Dybowska, M.; Barańska, I.; Jędrych, M.E.; Gładzka, A.; Tomkowski, W.Z.; Szturmowicz, M. New 6-Minute-Walking Test Parameter—Distance/Desaturation Index (DDI) Correctly Diagnoses Short-Term Response to Immunomodulatory Therapy in Hypersensitivity Pneumonitis. Diagnostics 2023, 13, 1109. [Google Scholar] [CrossRef]


{kind=link}
| Variable | Whole Group n = 86 | MUC5B GG Genotype n = 60 | MUC5B GT/TT Genotype n = 26 | p Value (GG vs. GT/TT) | |
|---|---|---|---|---|---|
| Age at diagnosis (y), median (range) | 58 (20–75) | 59 (23–75) | 53 (20–74) | 0.3454 | |
| Follow-up time (mth), median (range) | 62.4 (2.2–293.9) | 54.8 (2.2–293.9) | 67.7 (4.9–205.8) | 0.2794 | |
| Males, n (%) | 38 (44) | 29 (48.3) | 9 (34.6) | 0.3445 | |
| Ever smokers, n (%) | 27 (31.5) | 20 (33.3) | 7 (26.9) | 0.6207 | |
| FVC (L), median (range) | 2.6 (1.52–5.68) | 2.97 (1.52–5.68) | 2.48 (1.53–5.45) | 0.0610 | |
| FVC (%pred), mean (±SD) | 79.1 (±19.3) | 82.18 (±19.04) | 72.28 (±18.29) | 0.0315 | |
| TL,co (%pred), median (range) | 57 (28–114) | 54.5 (28–113) | 59 (32–114) | 0.8695 | |
| 6MWD (m), mean (±SD) | 509.5 (±101.4) | 505.6 (±113.5) | 518.0 (±69.96) | 0.6180 | |
| DSP, mean (±SD) | 459.4 (±101.0) | 457.7 (±113.6) | 463.1 (±66.16) | 0.6848 | |
| GAP stage (n = 83) | 1 | 65 (78.3) | 45 (77.6) | 20 (83.3) | 0.7748 |
| 2 | 15 (18.1) | 11 (19) | 3 (12.5) | ||
| 3 | 2 (3.6) | 2 (3.4) | 1 (4.2) | ||
| Fibrotic HP n (%) | 57 (66.3) | 36 (63.2) | 21 (82.8) | 0.0828 | |
| Immunomodulatory treatment n (%) | 75 (87.2) | 52 (86.7) | 23 (88.5) | 0.9999 | |
| Antifibrotic treatment n (%) | 11 (12.8) | 5 (8.2) | 6 (22.2) | 0.0853 | |
| FVC%Pred Trajectory | MUC5B GG Genotype, n (%) | MUC5B GT/TT Genotype, n (%) | Total, n (%) | p Value |
|---|---|---|---|---|
| FVC%pred increase | 32 (57.2) | 8 (30.8) | 40 (48.8) | 0.0336 |
| FVC%pred decrease | 24 (42.8) | 18 (69.2) | 42 (51.2) | |
| Total, n (%) | 56 (68.3) | 26 (31.7) | 82 (100) |
| Variable | OR | 95%CI | p Value |
|---|---|---|---|
| T allele (YES) | 3.598 | 1.299–10.87 | 0.0171 |
| Treatment (NO) | 0.2391 | 0.0397–1.130 | 0.0865 |
| Age at diagnosis | 1.003 | 0.9681–1.039 | 0.8867 |
| Baseline FVC%pred | 1.015 | 0.9885–1.044 | 0.2713 |
| Parameter | GG Genotype | p Value (GG Pre vs. GG Post) | GT/TT Genotype | p Value (GT/TT Pre vs. GT/TT Post) | ||
|---|---|---|---|---|---|---|
| Pretreatment | Posttreatment | Pretreatment | Posttreatment | |||
| FVC (l), median (range) | 2.655 (1.52–5.68) * | 2.995 (1.51–5.03) * | 0.0014 | 2.12 (1.23–5.45) @ | 2.21 (1.3–5.83) @ | 0.0671 |
| FVC (%), mean (±SD) | 78.87(± 19.93) # | 85.67(± 21.94) # | 0.0015 | 67.19 (±16.56) & | 72.46 (±19.08) & | 0.0082 |
| TL,co (%), median (range) | 51(28–113) # | 58.5 (33–114) # | 0.0004 | 55.5 (31–114) @ | 59 (31–119) @ | 0.0586 |
| 6MWD, mean (±SD) | 513 (±104.2) $ | 542.2 (±98.94) $ | 0.0106 | 524.8 (±78.95) @ | 529.1 (±92.20) @ | 0.8195 |
| DSP, mean (±SD) | 457.5 (±106.2) $ | 490 (±104.6) $ | 0.0029 | 465.5 (±79.57) @ | 465.6 (±101.7) @ | 0.8117 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lewandowska, K.B.; Szturmowicz, M.; Lechowicz, U.; Franczuk, M.; Błasińska, K.; Falis, M.; Błaszczyk, K.; Sobiecka, M.; Wyrostkiewicz, D.; Siemion-Szcześniak, I.; et al. The Presence of T Allele (rs35705950) of the MUC5B Gene Predicts Lower Baseline Forced Vital Capacity and Its Subsequent Decline in Patients with Hypersensitivity Pneumonitis. Int. J. Mol. Sci. 2023, 24, 10748. https://doi.org/10.3390/ijms241310748
Lewandowska KB, Szturmowicz M, Lechowicz U, Franczuk M, Błasińska K, Falis M, Błaszczyk K, Sobiecka M, Wyrostkiewicz D, Siemion-Szcześniak I, et al. The Presence of T Allele (rs35705950) of the MUC5B Gene Predicts Lower Baseline Forced Vital Capacity and Its Subsequent Decline in Patients with Hypersensitivity Pneumonitis. International Journal of Molecular Sciences. 2023; 24(13):10748. https://doi.org/10.3390/ijms241310748
Chicago/Turabian StyleLewandowska, Katarzyna B., Monika Szturmowicz, Urszula Lechowicz, Monika Franczuk, Katarzyna Błasińska, Maria Falis, Kamila Błaszczyk, Małgorzata Sobiecka, Dorota Wyrostkiewicz, Izabela Siemion-Szcześniak, and et al. 2023. "The Presence of T Allele (rs35705950) of the MUC5B Gene Predicts Lower Baseline Forced Vital Capacity and Its Subsequent Decline in Patients with Hypersensitivity Pneumonitis" International Journal of Molecular Sciences 24, no. 13: 10748. https://doi.org/10.3390/ijms241310748
APA StyleLewandowska, K. B., Szturmowicz, M., Lechowicz, U., Franczuk, M., Błasińska, K., Falis, M., Błaszczyk, K., Sobiecka, M., Wyrostkiewicz, D., Siemion-Szcześniak, I., Bartosiewicz, M., Radwan-Röhrenschef, P., Roży, A., Chorostowska-Wynimko, J., & Tomkowski, W. Z. (2023). The Presence of T Allele (rs35705950) of the MUC5B Gene Predicts Lower Baseline Forced Vital Capacity and Its Subsequent Decline in Patients with Hypersensitivity Pneumonitis. International Journal of Molecular Sciences, 24(13), 10748. https://doi.org/10.3390/ijms241310748