
Inhibition of Cancer Development by Natural Plant Polyphenols: Molecular Mechanisms


Abstract
1. Introduction
2. Structure and Properties of Plant Polyphenols
3. Genetic Instability
4. Epigenetic Reprogramming
5. Tumor-Promoting Inflammation
6. Interactions with Cancer-Associated Microbiota
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Breasted, J.H. The Edwin Smith Surgical Papyrus: Published in Facsimile and Hieroglyphic Transliteration with Translation and Commentary in Two Volumes; University of Chicago Oriental Institute Publications; University of Chicago Press: Chicago, IL, USA, 1991; ISBN 978-0-918986-73-3. [Google Scholar]
- Stratton, M.R. Exploring the Genomes of Cancer Cells: Progress and Promise. Science 2011, 331, 1553–1558. [Google Scholar] [CrossRef]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The Cancer Genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive Genomic Rearrangement Acquired in a Single Catastrophic Event during Cancer Development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer Genome Landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Vogelstein, B.; Kinzler, K.W. The Path to Cancer—Three Strikes and You’re Out. N. Engl. J. Med. 2015, 373, 1895–1898. [Google Scholar] [CrossRef]
- Loeb, L.A.; Harris, C.C. Advances in Chemical Carcinogenesis: A Historical Review and Prospective. Cancer Res. 2008, 68, 6863–6872. [Google Scholar] [CrossRef]
- Valastyan, S.; Weinberg, R.A. Tumor Metastasis: Molecular Insights and Evolving Paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef]
- Nagrath, S.; Sequist, L.V.; Maheswaran, S.; Bell, D.W.; Irimia, D.; Ulkus, L.; Smith, M.R.; Kwak, E.L.; Digumarthy, S.; Muzikansky, A.; et al. Isolation of Rare Circulating Tumour Cells in Cancer Patients by Microchip Technology. Nature 2007, 450, 1235–1239. [Google Scholar] [CrossRef]
- Tomasetti, C.; Marchionni, L.; Nowak, M.A.; Parmigiani, G.; Vogelstein, B. Only Three Driver Gene Mutations Are Required for the Development of Lung and Colorectal Cancers. Proc. Natl. Acad. Sci. USA 2015, 112, 118–123. [Google Scholar] [CrossRef]
- Hanahan, D. Rethinking the War on Cancer. Lancet 2014, 383, 558–563. [Google Scholar] [CrossRef]
- Block, K.I.; Gyllenhaal, C.; Lowe, L.; Amedei, A.; Amin, A.R.M.R.; Amin, A.; Aquilano, K.; Arbiser, J.; Arreola, A.; Arzumanyan, A.; et al. Designing a Broad-Spectrum Integrative Approach for Cancer Prevention and Treatment. Semin. Cancer Biol. 2015, 35, S276–S304. [Google Scholar] [CrossRef]
- Song, M.; Vogelstein, B.; Giovannucci, E.L.; Willett, W.C.; Tomasetti, C. Cancer Prevention: Molecular and Epidemiologic Consensus. Science 2018, 361, 1317–1318. [Google Scholar] [CrossRef]
- Tomasetti, C.; Li, L.; Vogelstein, B. Stem Cell Divisions, Somatic Mutations, Cancer Etiology, and Cancer Prevention. Science 2017, 355, 1330–1334. [Google Scholar] [CrossRef]
- Kirtonia, A.; Sethi, G.; Garg, M. The Multifaceted Role of Reactive Oxygen Species in Tumorigenesis. Cell. Mol. Life Sci. 2020, 77, 4459–4483. [Google Scholar] [CrossRef]
- Chen, C.; Kong, A.-N.T. Dietary Cancer-Chemopreventive Compounds: From Signaling and Gene Expression to Pharmacological Effects. Trends Pharm. Sci. 2005, 26, 318–326. [Google Scholar] [CrossRef]
- Rather, R.A.; Bhagat, M. Cancer Chemoprevention and Piperine: Molecular Mechanisms and Therapeutic Opportunities. Front. Cell. Dev. Biol. 2018, 6, 10. [Google Scholar] [CrossRef]
- Workman, P.; Draetta, G.F.; Schellens, J.H.M.; Bernards, R. How Much Longer Will We Put Up with $100,000 Cancer Drugs? Cell 2017, 168, 579–583. [Google Scholar] [CrossRef]
- Finicelli, M.; Di Salle, A.; Galderisi, U.; Peluso, G. The Mediterranean Diet: An Update of the Clinical Trials. Nutrients 2022, 14, 2956. [Google Scholar] [CrossRef]
- Erb, M.; Kliebenstein, D.J. Plant Secondary Metabolites as Defenses, Regulators, and Primary Metabolites: The Blurred Functional Trichotomy. Plant Physiol. 2020, 184, 39–52. [Google Scholar] [CrossRef]
- Valdés-Jiménez, A.; Peña-Varas, C.; Borrego-Muñoz, P.; Arrue, L.; Alegría-Arcos, M.; Nour-Eldin, H.; Dreyer, I.; Nuñez-Vivanco, G.; Ramírez, D. PSC-Db: A Structured and Searchable 3D-Database for Plant Secondary Compounds. Molecules 2021, 26, 1124. [Google Scholar] [CrossRef]
- War, A.R.; Paulraj, M.G.; Ahmad, T.; Buhroo, A.A.; Hussain, B.; Ignacimuthu, S.; Sharma, H.C. Mechanisms of Plant Defense against Insect Herbivores. Plant Signal. Behav. 2012, 7, 1306–1320. [Google Scholar] [CrossRef]
- Cheynier, V.; Comte, G.; Davies, K.M.; Lattanzio, V.; Martens, S. Plant Phenolics: Recent Advances on Their Biosynthesis, Genetics, and Ecophysiology. Plant Physiol. Biochem. 2013, 72, 1–20. [Google Scholar] [CrossRef]
- El Gharras, H. Polyphenols: Food Sources, Properties and Applications-a Review: Nutraceutical Polyphenols. Int. J. Food Sci. Technol. 2009, 44, 2512–2518. [Google Scholar] [CrossRef]
- De la Rosa, L.A.; Alvarez-Parrilla, E.; Gonzlez-Aguilar, G.A. (Eds.) Fruit and Vegetable Phytochemicals; Wiley-Blackwell: Oxford, UK, 2009; ISBN 978-0-8138-0939-7. [Google Scholar]
- Khoddami, A.; Wilkes, M.; Roberts, T. Techniques for Analysis of Plant Phenolic Compounds. Molecules 2013, 18, 2328–2375. [Google Scholar] [CrossRef]
- Singla, R.K.; Dubey, A.K.; Garg, A.; Sharma, R.K.; Fiorino, M.; Ameen, S.M.; Haddad, M.A.; Al-Hiary, M. Natural Polyphenols: Chemical Classification, Definition of Classes, Subcategories, and Structures. J. AOAC Int. 2019, 102, 1397–1400. [Google Scholar] [CrossRef]
- Soto-Vaca, A.; Gutierrez, A.; Losso, J.N.; Xu, Z.; Finley, J.W. Evolution of Phenolic Compounds from Color and Flavor Problems to Health Benefits. J. Agric. Food Chem. 2012, 60, 6658–6677. [Google Scholar] [CrossRef]
- Manach, C.; Scalbert, A.; Morand, C.; Rémésy, C.; Jiménez, L. Polyphenols: Food Sources and Bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar] [CrossRef]
- Corcoran, M.P.; McKay, D.L.; Blumberg, J.B. Flavonoid Basics: Chemistry, Sources, Mechanisms of Action, and Safety. J. Nutr. Gerontol. Geriatr. 2012, 31, 176–189. [Google Scholar] [CrossRef]
- Pérez-Jiménez, J.; Neveu, V.; Vos, F.; Scalbert, A. Identification of the 100 Richest Dietary Sources of Polyphenols: An Application of the Phenol-Explorer Database. Eur. J. Clin. Nutr. 2010, 64, S112–S120. [Google Scholar] [CrossRef]
- Lambert, J.D.; Lee, M.-J.; Lu, H.; Meng, X.; Hong, J.J.J.; Seril, D.N.; Sturgill, M.G.; Yang, C.S. Epigallocatechin-3-Gallate Is Absorbed but Extensively Glucuronidated Following Oral Administration to Mice. J. Nutr. 2003, 133, 4172–4177. [Google Scholar] [CrossRef]
- Rotches-Ribalta, M.; Andres-Lacueva, C.; Estruch, R.; Escribano, E.; Urpi-Sarda, M. Pharmacokinetics of Resveratrol Metabolic Profile in Healthy Humans after Moderate Consumption of Red Wine and Grape Extract Tablets. Pharmacol. Res. 2012, 66, 375–382. [Google Scholar] [CrossRef]
- Servili, M.; Sordini, B.; Esposto, S.; Urbani, S.; Veneziani, G.; Di Maio, I.; Selvaggini, R.; Taticchi, A. Biological Activities of Phenolic Compounds of Extra Virgin Olive Oil. Antioxidants 2013, 3, 1–23. [Google Scholar] [CrossRef]
- Esatbeyoglu, T.; Huebbe, P.; Ernst, I.M.A.; Chin, D.; Wagner, A.E.; Rimbach, G. Curcumin-From Molecule to Biological Function. Angew. Chem. Int. Ed. 2012, 51, 5308–5332. [Google Scholar] [CrossRef]
- Van Wyk, B.-E.; Wink, M. Medicinal Plants of the World: An Illustrated Scientific Guide to Important Medicinal Plants and Their Uses, 1st ed.; Timber Press: Portland, OR, USA, 2004; ISBN 978-0-88192-602-6. [Google Scholar]
- Kim, S.H.; Kim, M.S.; Lee, M.S.; Park, Y.S.; Lee, H.J.; Kang, S.; Lee, H.S.; Lee, K.-E.; Yang, H.J.; Kim, M.J.; et al. Korean Diet: Characteristics and Historical Background. J. Ethn. Foods 2016, 3, 26–31. [Google Scholar] [CrossRef]
- Pallauf, K.; Giller, K.; Huebbe, P.; Rimbach, G. Nutrition and Healthy Ageing: Calorie Restriction or Polyphenol-Rich “MediterrAsian” Diet? Oxidative Med. Cell. Longev. 2013, 2013, 707421. [Google Scholar] [CrossRef]
- Amiot, M.J.; Riva, C.; Vinet, A. Effects of Dietary Polyphenols on Metabolic Syndrome Features in Humans: A Systematic Review. Obes. Rev. 2016, 17, 573–586. [Google Scholar] [CrossRef]
- Rienks, J.; Barbaresko, J.; Oluwagbemigun, K.; Schmid, M.; Nöthlings, U. Polyphenol Exposure and Risk of Type 2 Diabetes: Dose-Response Meta-Analyses and Systematic Review of Prospective Cohort Studies. Am. J. Clin. Nutr. 2018, 108, 49–61. [Google Scholar] [CrossRef]
- Stefani, M.; Rigacci, S. Beneficial Properties of Natural Phenols: Highlight on Protection against Pathological Conditions Associated with Amyloid Aggregation. Biofactors 2014, 40, 482–493. [Google Scholar] [CrossRef]
- Feinleib, M. Seven Countries: A Multivariate Analysis of Death and Coronary Heart Disease. JAMA 1981, 245, 511. [Google Scholar] [CrossRef]
- Grosso, G.; Godos, J.; Lamuela-Raventos, R.; Ray, S.; Micek, A.; Pajak, A.; Sciacca, S.; D’Orazio, N.; Del Rio, D.; Galvano, F. A Comprehensive Meta-Analysis on Dietary Flavonoid and Lignan Intake and Cancer Risk: Level of Evidence and Limitations. Mol. Nutr. Food Res. 2017, 61, 1600930. [Google Scholar] [CrossRef]
- Patra, S.; Pradhan, B.; Nayak, R.; Behera, C.; Das, S.; Patra, S.K.; Efferth, T.; Jena, M.; Bhutia, S.K. Dietary Polyphenols in Chemoprevention and Synergistic Effect in Cancer: Clinical Evidences and Molecular Mechanisms of Action. Phytomedicine 2021, 90, 153554. [Google Scholar] [CrossRef]
- Kirsanov, K.I.; Vlasova, O.A.; Fetisov, T.I.; Zenkov, R.G.; Lesovaya, E.A.; Belitsky, G.A.; Gurova, K.; Yakubovskaya, M.G. Influence of DNA-Binding Compounds with Cancer Preventive Activity on the Mechanisms of Gene Expression Regulation. Adv. Mol. Onkol. 2019, 5, 41–63. [Google Scholar] [CrossRef]
- Hazafa, A.; Rehman, K.-U.; Jahan, N.; Jabeen, Z. The Role of Polyphenol (Flavonoids) Compounds in the Treatment of Cancer Cells. Nutr. Cancer 2020, 72, 386–397. [Google Scholar] [CrossRef]
- Chou, C.-C.; Yang, J.-S.; Lu, H.-F.; Ip, S.-W.; Lo, C.; Wu, C.-C.; Lin, J.-P.; Tang, N.-Y.; Chung, J.-G.; Chou, M.-J.; et al. Quercetin-Mediated Cell Cycle Arrest and Apoptosis Involving Activation of a Caspase Cascade through the Mitochondrial Pathway in Human Breast Cancer MCF-7 Cells. Arch. Pharm. Res. 2010, 33, 1181–1191. [Google Scholar] [CrossRef]
- Duo, J.; Ying, G.-G.; Wang, G.-W.; Zhang, L. Quercetin Inhibits Human Breast Cancer Cell Proliferation and Induces Apoptosis via Bcl-2 and Bax Regulation. Mol. Med. Rep. 2012, 5, 1453–1456. [Google Scholar] [CrossRef]
- Adhami, V.M.; Malik, A.; Zaman, N.; Sarfaraz, S.; Siddiqui, I.A.; Syed, D.N.; Afaq, F.; Pasha, F.S.; Saleem, M.; Mukhtar, H. Combined Inhibitory Effects of Green Tea Polyphenols and Selective Cyclooxygenase-2 Inhibitors on the Growth of Human Prostate Cancer Cells Both in Vitro and in Vivo. Clin. Cancer Res. 2007, 13, 1611–1619. [Google Scholar] [CrossRef]
- Chua, C.C.; Hamdy, R.C.; Chua, B.H. Mechanism of Transforming Growth Factor-Beta1-Induced Expression of Vascular Endothelial Growth Factor in Murine Osteoblastic MC3T3-E1 Cells. Biochim. Biophys. Acta 2000, 1497, 69–76. [Google Scholar] [CrossRef]
- Chadalapaka, G.; Jutooru, I.; Chintharlapalli, S.; Papineni, S.; Smith, R.; Li, X.; Safe, S. Curcumin Decreases Specificity Protein Expression in Bladder Cancer Cells. Cancer Res. 2008, 68, 5345–5354. [Google Scholar] [CrossRef]
- Namasivayam, N. Chemoprevention in Experimental Animals: Chemoprevention in Experimental Animals. Ann. N. Y. Acad. Sci. 2011, 1215, 60–71. [Google Scholar] [CrossRef]
- Zhou, Y.; Zheng, J.; Li, Y.; Xu, D.-P.; Li, S.; Chen, Y.-M.; Li, H.-B. Natural Polyphenols for Prevention and Treatment of Cancer. Nutrients 2016, 8, 515. [Google Scholar] [CrossRef]
- Kuroiwa, Y.; Nishikawa, A.; Kitamura, Y.; Kanki, K.; Ishii, Y.; Umemura, T.; Hirose, M. Protective Effects of Benzyl Isothiocyanate and Sulforaphane but Not Resveratrol against Initiation of Pancreatic Carcinogenesis in Hamsters. Cancer Lett. 2006, 241, 275–280. [Google Scholar] [CrossRef]
- Sengottuvelan, M.; Senthilkumar, R.; Nalini, N. Modulatory Influence of Dietary Resveratrol during Different Phases of 1,2-Dimethylhydrazine Induced Mucosal Lipid-Peroxidation, Antioxidant Status and Aberrant Crypt Foci Development in Rat Colon Carcinogenesis. Biochim. Biophys. Acta 2006, 1760, 1175–1183. [Google Scholar] [CrossRef]
- Mazué, F.; Delmas, D.; Murillo, G.; Saleiro, D.; Limagne, E.; Latruffe, N. Differential Protective Effects of Red Wine Polyphenol Extracts (RWEs) on Colon Carcinogenesis. Food Funct. 2014, 5, 663–670. [Google Scholar] [CrossRef]
- Fan, P.; Fan, S.; Wang, H.; Mao, J.; Shi, Y.; Ibrahim, M.M.; Ma, W.; Yu, X.; Hou, Z.; Wang, B.; et al. Genistein Decreases the Breast Cancer Stem-like Cell Population through Hedgehog Pathway. Stem Cell. Res. Ther. 2013, 4, 146. [Google Scholar] [CrossRef]
- Kim, H.-S.; Wannatung, T.; Lee, S.; Yang, W.K.; Chung, S.H.; Lim, J.-S.; Choe, W.; Kang, I.; Kim, S.-S.; Ha, J. Quercetin Enhances Hypoxia-Mediated Apoptosis via Direct Inhibition of AMPK Activity in HCT116 Colon Cancer. Apoptosis 2012, 17, 938–949. [Google Scholar] [CrossRef]
- Thomasset, S.C.; Berry, D.P.; Garcea, G.; Marczylo, T.; Steward, W.P.; Gescher, A.J. Dietary Polyphenolic Phytochemicals--Promising Cancer Chemopreventive Agents in Humans? A Review of Their Clinical Properties. Int. J. Cancer 2007, 120, 451–458. [Google Scholar] [CrossRef]
- Choudhari, A.S.; Mandave, P.C.; Deshpande, M.; Ranjekar, P.; Prakash, O. Phytochemicals in Cancer Treatment: From Preclinical Studies to Clinical Practice. Front. Pharm. 2019, 10, 1614. [Google Scholar] [CrossRef]
- Lazarevic, B.; Boezelijn, G.; Diep, L.M.; Kvernrod, K.; Ogren, O.; Ramberg, H.; Moen, A.; Wessel, N.; Berg, R.E.; Egge-Jacobsen, W.; et al. Efficacy and Safety of Short-Term Genistein Intervention in Patients with Localized Prostate Cancer Prior to Radical Prostatectomy: A Randomized, Placebo-Controlled, Double-Blind Phase 2 Clinical Trial. Nutr. Cancer 2011, 63, 889–898. [Google Scholar] [CrossRef]
- Carroll, R.E.; Benya, R.V.; Turgeon, D.K.; Vareed, S.; Neuman, M.; Rodriguez, L.; Kakarala, M.; Carpenter, P.M.; McLaren, C.; Meyskens, F.L.; et al. Phase IIa Clinical Trial of Curcumin for the Prevention of Colorectal Neoplasia. Cancer Prev. Res. 2011, 4, 354–364. [Google Scholar] [CrossRef]
- Kiselev, V.I.; Ashrafyan, L.A.; Muyzhnek, E.L.; Gerfanova, E.V.; Antonova, I.B.; Aleshikova, O.I.; Sarkar, F.H. A New Promising Way of Maintenance Therapy in Advanced Ovarian Cancer: A Comparative Clinical Study. BMC Cancer 2018, 18, 904. [Google Scholar] [CrossRef]
- Bayet-Robert, M.; Kwiatkowski, F.; Leheurteur, M.; Gachon, F.; Planchat, E.; Abrial, C.; Mouret-Reynier, M.-A.; Durando, X.; Barthomeuf, C.; Chollet, P. Phase I Dose Escalation Trial of Docetaxel plus Curcumin in Patients with Advanced and Metastatic Breast Cancer. Cancer Biol. Ther. 2010, 9, 8–14. [Google Scholar] [CrossRef]
- Trudel, D.; Labbé, D.P.; Araya-Farias, M.; Doyen, A.; Bazinet, L.; Duchesne, T.; Plante, M.; Grégoire, J.; Renaud, M.-C.; Bachvarov, D.; et al. A Two-Stage, Single-Arm, Phase II Study of EGCG-Enriched Green Tea Drink as a Maintenance Therapy in Women with Advanced Stage Ovarian Cancer. Gynecol. Oncol. 2013, 131, 357–361. [Google Scholar] [CrossRef]
- Nguyen, M.M.; Ahmann, F.R.; Nagle, R.B.; Hsu, C.-H.; Tangrea, J.A.; Parnes, H.L.; Sokoloff, M.H.; Gretzer, M.B.; Chow, H.-H.S. Randomized, Double-Blind, Placebo-Controlled Trial of Polyphenon E in Prostate Cancer Patients before Prostatectomy: Evaluation of Potential Chemopreventive Activities. Cancer Prev. Res. 2012, 5, 290–298. [Google Scholar] [CrossRef]
- Gontero, P.; Marra, G.; Soria, F.; Oderda, M.; Zitella, A.; Baratta, F.; Chiorino, G.; Gregnanin, I.; Daniele, L.; Cattel, L.; et al. A Randomized Double-Blind Placebo Controlled Phase I-II Study on Clinical and Molecular Effects of Dietary Supplements in Men with Precancerous Prostatic Lesions. Chemoprevention or “Chemopromotion”? Prostate 2015, 75, 1177–1186. [Google Scholar] [CrossRef]
- Wink, M. Modes of Action of Herbal Medicines and Plant Secondary Metabolites. Medicines 2015, 2, 251–286. [Google Scholar] [CrossRef]
- Sah, J.F.; Balasubramanian, S.; Eckert, R.L.; Rorke, E.A. Epigallocatechin-3-Gallate Inhibits Epidermal Growth Factor Receptor Signaling Pathway. J. Biol. Chem. 2004, 279, 12755–12762. [Google Scholar] [CrossRef]
- Nam, S.; Smith, D.M.; Dou, Q.P. Ester Bond-Containing Tea Polyphenols Potently Inhibit Proteasome Activity in Vitro and in Vivo. J. Biol. Chem. 2001, 276, 13322–13330. [Google Scholar] [CrossRef]
- Leone, M.; Zhai, D.; Sareth, S.; Kitada, S.; Reed, J.C.; Pellecchia, M. Cancer Prevention by Tea Polyphenols Is Linked to Their Direct Inhibition of Antiapoptotic Bcl-2-Family Proteins. Cancer Res. 2003, 63, 8118–8121. [Google Scholar]
- Tachibana, H.; Koga, K.; Fujimura, Y.; Yamada, K. A Receptor for Green Tea Polyphenol EGCG. Nat. Struct. Mol. Biol. 2004, 11, 380–381. [Google Scholar] [CrossRef]
- Shimizu, M.; Deguchi, A.; Hara, Y.; Moriwaki, H.; Weinstein, I.B. EGCG Inhibits Activation of the Insulin-like Growth Factor-1 Receptor in Human Colon Cancer Cells. Biochem. Biophys. Res. Commun. 2005, 334, 947–953. [Google Scholar] [CrossRef]
- Kuzuhara, T.; Suganuma, M.; Fujiki, H. Green Tea Catechin as a Chemical Chaperone in Cancer Prevention. Cancer Lett. 2008, 261, 12–20. [Google Scholar] [CrossRef]
- Fujimura, Y.; Tachibana, H.; Yamada, K. Lipid Raft-Associated Catechin Suppresses the FcϵRI Expression by Inhibiting Phosphorylation of the Extracellular Signal-Regulated Kinase1/2. FEBS Lett. 2004, 556, 204–210. [Google Scholar] [CrossRef]
- Tu, B.; Liu, Z.-J.; Chen, Z.-F.; Ouyang, Y.; Hu, Y.-J. Understanding the Structure–Activity Relationship between Quercetin and Naringenin: In Vitro. RSC Adv. 2015, 5, 106171–106181. [Google Scholar] [CrossRef]
- Nair, M.S.; Shukla, A. Molecular Modeling, Simulation and Principal Component Analysis of Binding of Resveratrol and Its Analogues with DNA. J. Biomol. Struct. Dyn. 2020, 38, 3087–3097. [Google Scholar] [CrossRef]
- N’soukpoé-Kossi, C.N.; Bourassa, P.; Mandeville, J.S.; Bekale, L.; Bariyanga, J.; Tajmir-Riahi, H.A. Locating the Binding Sites of Antioxidants Resveratrol, Genistein and Curcumin with TRNA. Int. J. Biol. Macromol. 2015, 80, 41–47. [Google Scholar] [CrossRef]
- Platella, C.; Raucci, U.; Rega, N.; D’Atri, S.; Levati, L.; Roviello, G.N.; Fuggetta, M.P.; Musumeci, D.; Montesarchio, D. Shedding Light on the Interaction of Polydatin and Resveratrol with G-Quadruplex and Duplex DNA: A Biophysical, Computational and Biological Approach. Int. J. Biol. Macromol. 2020, 151, 1163–1172. [Google Scholar] [CrossRef]
- Luch, A.; Baird, W.M. Carcinogenic Polycyclic Aromatic Hydrocarbons. In Comprehensive Toxicology; Elsevier: Amsterdam, The Netherlands, 2010; pp. 85–123. ISBN 978-0-08-046884-6. [Google Scholar]
- Gibis, M. Heterocyclic Aromatic Amines in Cooked Meat Products: Causes, Formation, Occurrence, and Risk Assessment: Heterocyclic Amines in Cooked Meat Products…. Compr. Rev. Food Sci. Food Saf. 2016, 15, 269–302. [Google Scholar] [CrossRef]
- Attaluri, S.; Bonala, R.R.; Yang, I.-Y.; Lukin, M.A.; Wen, Y.; Grollman, A.P.; Moriya, M.; Iden, C.R.; Johnson, F. DNA Adducts of Aristolochic Acid II: Total Synthesis and Site-Specific Mutagenesis Studies in Mammalian Cells. Nucleic Acids Res. 2010, 38, 339–352. [Google Scholar] [CrossRef]
- Wu, H.-C.; Santella, R. The Role of Aflatoxins in Hepatocellular Carcinoma. Hepat. Mon. 2012, 12. [Google Scholar] [CrossRef]
- Omiecinski, C.J.; Vanden Heuvel, J.P.; Perdew, G.H.; Peters, J.M. Xenobiotic Metabolism, Disposition, and Regulation by Receptors: From Biochemical Phenomenon to Predictors of Major Toxicities. Toxicol. Sci. 2011, 120, S49–S75. [Google Scholar] [CrossRef]
- Beischlag, T.V.; Luis Morales, J.; Hollingshead, B.D.; Perdew, G.H. The Aryl Hydrocarbon Receptor Complex and the Control of Gene Expression. Crit. Rev. Eukaryot. Gene Expr. 2008, 18, 207–250. [Google Scholar] [CrossRef]
- Denison, M.S.; Soshilov, A.A.; He, G.; DeGroot, D.E.; Zhao, B. Exactly the Same but Different: Promiscuity and Diversity in the Molecular Mechanisms of Action of the Aryl Hydrocarbon (Dioxin) Receptor. Toxicol. Sci. 2011, 124, 1–22. [Google Scholar] [CrossRef]
- Lindsey, S.; Papoutsakis, E.T. The Evolving Role of the Aryl Hydrocarbon Receptor (AHR) in the Normophysiology of Hematopoiesis. Stem Cell. Rev. Rep. 2012, 8, 1223–1235. [Google Scholar] [CrossRef]
- Xue, Z.; Li, D.; Yu, W.; Zhang, Q.; Hou, X.; He, Y.; Kou, X. Mechanisms and Therapeutic Prospects of Polyphenols as Modulators of the Aryl Hydrocarbon Receptor. Food Funct. 2017, 8, 1414–1437. [Google Scholar] [CrossRef]
- Ciolino, H.P.; Daschner, P.J.; Wang, T.T.Y.; Yeh, G.C. Effect of Curcumin on the Aryl Hydrocarbon Receptor and Cytochrome P450 1A1 in MCF-7 Human Breast Carcinoma Cells. Biochem. Pharmacol. 1998, 56, 197–206. [Google Scholar] [CrossRef]
- Ciolino, H.P.; Daschner, P.J.; Yeh, G.C. Dietary Flavonols Quercetin and Kaempferol Are Ligands of the Aryl Hydrocarbon Receptor That Affect CYP1A1 Transcription Differentially. Biochem. J. 1999, 340 Pt 3, 715–722. [Google Scholar] [CrossRef]
- Perdew, G.H.; Hollingshead, B.D.; Dinatale, B.C.; Morales, J.L.; Labrecque, M.P.; Takhar, M.K.; Tam, K.J.; Beischlag, T.V. Estrogen Receptor Expression Is Required for Low-Dose Resveratrol-Mediated Repression of Aryl Hydrocarbon Receptor Activity. J. Pharm. Exp. Ther. 2010, 335, 273–283. [Google Scholar] [CrossRef]
- Fukuda, I.; Mukai, R.; Kawase, M.; Yoshida, K.; Ashida, H. Interaction between the Aryl Hydrocarbon Receptor and Its Antagonists, Flavonoids. Biochem. Biophys. Res. Commun. 2007, 359, 822–827. [Google Scholar] [CrossRef]
- Jin, U.-H.; Park, H.; Li, X.; Davidson, L.A.; Allred, C.; Patil, B.; Jayaprakasha, G.; Orr, A.A.; Mao, L.; Chapkin, R.S.; et al. Structure-Dependent Modulation of Aryl Hydrocarbon Receptor-Mediated Activities by Flavonoids. Toxicol. Sci. 2018, 164, 205–217. [Google Scholar] [CrossRef]
- Kaur, M.; Badhan, R.K.S. Phytochemical Mediated-Modulation of the Expression and Transporter Function of Breast Cancer Resistance Protein at the Blood-Brain Barrier: An in-Vitro Study. Brain Res. 2017, 1654, 9–23. [Google Scholar] [CrossRef]
- Goya-Jorge, E.; Giner, R.M.; Sylla-Iyarreta Veitía, M.; Gozalbes, R.; Barigye, S.J. Predictive Modeling of Aryl Hydrocarbon Receptor (AhR) Agonism. Chemosphere 2020, 256, 127068. [Google Scholar] [CrossRef]
- Goya-Jorge, E.; Jorge Rodríguez, M.E.; Veitía, M.S.-I.; Giner, R.M. Plant Occurring Flavonoids as Modulators of the Aryl Hydrocarbon Receptor. Molecules 2021, 26, 2315. [Google Scholar] [CrossRef]
- Mukai, R.; Shirai, Y.; Saito, N.; Fukuda, I.; Nishiumi, S.; Yoshida, K.-I.; Ashida, H. Suppression Mechanisms of Flavonoids on Aryl Hydrocarbon Receptor-Mediated Signal Transduction. Arch. Biochem. Biophys. 2010, 501, 134–141. [Google Scholar] [CrossRef]
- Nishiumi, S.; Yoshida, K.-I.; Ashida, H. Curcumin Suppresses the Transformation of an Aryl Hydrocarbon Receptor through Its Phosphorylation. Arch. Biochem. Biophys. 2007, 466, 267–273. [Google Scholar] [CrossRef]
- Quadri, S.A.; Qadri, A.N.; Hahn, M.E.; Mann, K.K.; Sherr, D.H. The Bioflavonoid Galangin Blocks Aryl Hydrocarbon Receptor Activation and Polycyclic Aromatic Hydrocarbon-Induced Pre-B Cell Apoptosis. Mol. Pharm. 2000, 58, 515–525. [Google Scholar] [CrossRef]
- Palermo, C.M.; Westlake, C.A.; Gasiewicz, T.A. Epigallocatechin Gallate Inhibits Aryl Hydrocarbon Receptor Gene Transcription through an Indirect Mechanism Involving Binding to a 90 KDa Heat Shock Protein. Biochemistry 2005, 44, 5041–5052. [Google Scholar] [CrossRef]
- Ciolino, H.P.; Daschner, P.J.; Yeh, G.C. Resveratrol Inhibits Transcription of CYP1A1 in Vitro by Preventing Activation of the Aryl Hydrocarbon Receptor. Cancer Res. 1998, 58, 5707–5712. [Google Scholar]
- Froyen, E.B.; Steinberg, F.M. Genistein Decreases Basal Hepatic Cytochrome P450 1A1 Protein Expression and Activity in Swiss Webster Mice. Nutr. Res. 2016, 36, 430–439. [Google Scholar] [CrossRef]
- Macpherson, L.; Matthews, J. Inhibition of Aryl Hydrocarbon Receptor-Dependent Transcription by Resveratrol or Kaempferol Is Independent of Estrogen Receptor α Expression in Human Breast Cancer Cells. Cancer Lett. 2010, 299, 119–129. [Google Scholar] [CrossRef]
- Manikandan, P.; Nagini, S. Cytochrome P450 Structure, Function and Clinical Significance: A Review. Curr. Drug Targets 2018, 19, 38–54. [Google Scholar] [CrossRef]
- Wahlang, B.; Falkner, K.C.; Cave, M.C.; Prough, R.A. Role of Cytochrome P450 Monooxygenase in Carcinogen and Chemotherapeutic Drug Metabolism. In Advances in Pharmacology; Elsevier: Amsterdam, The Netherlands, 2015; Volume 74, pp. 1–33. ISBN 978-0-12-803119-3. [Google Scholar]
- Shimada, T.; Tanaka, K.; Takenaka, S.; Murayama, N.; Martin, M.V.; Foroozesh, M.K.; Yamazaki, H.; Guengerich, F.P.; Komori, M. Structure−Function Relationships of Inhibition of Human Cytochromes P450 1A1, 1A2, 1B1, 2C9, and 3A4 by 33 Flavonoid Derivatives. Chem. Res. Toxicol. 2010, 23, 1921–1935. [Google Scholar] [CrossRef]
- Kimura, Y.; Ito, H.; Ohnishi, R.; Hatano, T. Inhibitory Effects of Polyphenols on Human Cytochrome P450 3A4 and 2C9 Activity. Food Chem. Toxicol. 2010, 48, 429–435. [Google Scholar] [CrossRef]
- Matsuno, Y.; Atsumi, Y.; Alauddin, M.; Rana, M.M.; Fujimori, H.; Hyodo, M.; Shimizu, A.; Ikuta, T.; Tani, H.; Torigoe, H.; et al. Resveratrol and Its Related Polyphenols Contribute to the Maintenance of Genome Stability. Sci. Rep. 2020, 10, 5388. [Google Scholar] [CrossRef]
- Yan, H.; Jiang, J.; Du, A.; Gao, J.; Zhang, D.; Song, L. Genistein Enhances Radiosensitivity of Human Hepatocellular Carcinoma Cells by Inducing G2/M Arrest and Apoptosis. Radiat. Res. 2020, 193, 286. [Google Scholar] [CrossRef]
- Majidinia, M.; Bishayee, A.; Yousefi, B. Polyphenols: Major Regulators of Key Components of DNA Damage Response in Cancer. DNA Repair. 2019, 82, 102679. [Google Scholar] [CrossRef]
- Zhou, Z.; Liu, Y.; Jiang, X.; Zheng, C.; Luo, W.; Xiang, X.; Qi, X.; Shen, J. Metformin Modified Chitosan as a Multi-Functional Adjuvant to Enhance Cisplatin-Based Tumor Chemotherapy Efficacy. Int. J. Biol. Macromol. 2023, 224, 797–809. [Google Scholar] [CrossRef]
- Kryston, T.B.; Georgiev, A.B.; Pissis, P.; Georgakilas, A.G. Role of Oxidative Stress and DNA Damage in Human Carcinogenesis. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2011, 711, 193–201. [Google Scholar] [CrossRef]
- Gates, K.S. An Overview of Chemical Processes That Damage Cellular DNA: Spontaneous Hydrolysis, Alkylation, and Reactions with Radicals. Chem. Res. Toxicol. 2009, 22, 1747–1760. [Google Scholar] [CrossRef]
- Pereira, C.; Grácio, D.; Teixeira, J.P.; Magro, F. Oxidative Stress and DNA Damage: Implications in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2015, 21, 2403–2417. [Google Scholar] [CrossRef]
- Jomova, K.; Valko, M. Advances in Metal-Induced Oxidative Stress and Human Disease. Toxicology 2011, 283, 65–87. [Google Scholar] [CrossRef]
- Gebicki, J.M. Oxidative Stress, Free Radicals and Protein Peroxides. Arch. Biochem. Biophys. 2016, 595, 33–39. [Google Scholar] [CrossRef]
- Paulsen, C.E.; Carroll, K.S. Cysteine-Mediated Redox Signaling: Chemistry, Biology, and Tools for Discovery. Chem. Rev. 2013, 113, 4633–4679. [Google Scholar] [CrossRef]
- Cos, P.; Ying, L.; Calomme, M.; Hu, J.P.; Cimanga, K.; Van Poel, B.; Pieters, L.; Vlietinck, A.J.; Vanden Berghe, D. Structure-Activity Relationship and Classification of Flavonoids as Inhibitors of Xanthine Oxidase and Superoxide Scavengers. J. Nat. Prod. 1998, 61, 71–76. [Google Scholar] [CrossRef]
- Chen, J.-W.; Zhu, Z.-Q.; Hu, T.-X.; Zhu, D.-Y. Structure-Activity Relationship of Natural Flavonoids in Hydroxyl Radical-Scavenging Effects. Acta Pharm. Sin. 2002, 23, 667–672. [Google Scholar]
- Olszowy, M. What Is Responsible for Antioxidant Properties of Polyphenolic Compounds from Plants? Plant Physiol. Biochem. 2019, 144, 135–143. [Google Scholar] [CrossRef]
- Guo, Q.; Zhao, B.; Shen, S.; Hou, J.; Hu, J.; Xin, W. ESR Study on the Structure–Antioxidant Activity Relationship of Tea Catechins and Their Epimers. Biochim. Biophys. Acta (BBA) Gen. Subj. 1999, 1427, 13–23. [Google Scholar] [CrossRef]
- Nakagawa, T.; Yokozawa, T. Direct Scavenging of Nitric Oxide and Superoxide by Green Tea. Food Chem. Toxicol. 2002, 40, 1745–1750. [Google Scholar] [CrossRef]
- Shanmugam, T.; Selvaraj, M.; Poomalai, S. Epigallocatechin Gallate Potentially Abrogates Fluoride Induced Lung Oxidative Stress, Inflammation via Nrf2/Keap1 Signaling Pathway in Rats: An in-Vivo and in-Silico Study. Int. Immunopharmacol. 2016, 39, 128–139. [Google Scholar] [CrossRef]
- Giftson, J.S.; Jayanthi, S.; Nalini, N. Chemopreventive Efficacy of Gallic Acid, an Antioxidant and Anticarcinogenic Polyphenol, against 1,2-Dimethyl Hydrazine Induced Rat Colon Carcinogenesis. Investig. New Drugs 2010, 28, 251–259. [Google Scholar] [CrossRef]
- Sharmila, G.; Athirai, T.; Kiruthiga, B.; Senthilkumar, K.; Elumalai, P.; Arunkumar, R.; Arunakaran, J. Chemopreventive Effect of Quercetin in MNU and Testosterone Induced Prostate Cancer of Sprague-Dawley Rats. Nutr. Cancer 2014, 66, 38–46. [Google Scholar] [CrossRef]
- Henning, S.M.; Niu, Y.; Lee, N.H.; Thames, G.D.; Minutti, R.R.; Wang, H.; Go, V.L.W.; Heber, D. Bioavailability and Antioxidant Activity of Tea Flavanols after Consumption of Green Tea, Black Tea, or a Green Tea Extract Supplement. Am. J. Clin. Nutr. 2004, 80, 1558–1564. [Google Scholar] [CrossRef] [PubMed]
- Young, J.F.; Dragsted, L.O.; Haraldsdóttir, J.; Daneshvar, B.; Kall, M.A.; Loft, S.; Nilsson, L.; Nielsen, S.E.; Mayer, B.; Skibsted, L.H.; et al. Green Tea Extract Only Affects Markers of Oxidative Status Postprandially: Lasting Antioxidant Effect of Flavonoid-Free Diet. Br. J. Nutr. 2002, 87, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Fallah, A.A.; Sarmast, E.; Jafari, T. Effect of Dietary Anthocyanins on Biomarkers of Oxidative Stress and Antioxidative Capacity: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. J. Funct. Foods 2020, 68, 103912. [Google Scholar] [CrossRef]
- Mira, L.; Tereza Fernandez, M.; Santos, M.; Rocha, R.; Helena Florêncio, M.; Jennings, K.R. Interactions of Flavonoids with Iron and Copper Ions: A Mechanism for Their Antioxidant Activity. Free Radic. Res. 2002, 36, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Adjimani, J.P.; Asare, P. Antioxidant and Free Radical Scavenging Activity of Iron Chelators. Toxicol. Rep. 2015, 2, 721–728. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Lertpiriyapong, K.; Steelman, L.S.; Abrams, S.L.; Yang, L.V.; Murata, R.M.; Rosalen, P.L.; Scalisi, A.; Neri, L.M.; Cocco, L.; et al. Effects of Resveratrol, Curcumin, Berberine and Other Nutraceuticals on Aging, Cancer Development, Cancer Stem Cells and MicroRNAs. Aging 2017, 9, 1477–1536. [Google Scholar] [CrossRef]
- León-González, A.J.; Auger, C.; Schini-Kerth, V.B. Pro-Oxidant Activity of Polyphenols and Its Implication on Cancer Chemoprevention and Chemotherapy. Biochem. Pharmacol. 2015, 98, 371–380. [Google Scholar] [CrossRef]
- Kim, H.-S.; Quon, M.J.; Kim, J. New Insights into the Mechanisms of Polyphenols beyond Antioxidant Properties; Lessons from the Green Tea Polyphenol, Epigallocatechin 3-Gallate. Redox Biol. 2014, 2, 187–195. [Google Scholar] [CrossRef]
- He, F.; Antonucci, L.; Karin, M. NRF2 as a Regulator of Cell Metabolism and Inflammation in Cancer. Carcinogenesis 2020, 41, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Tanigawa, S.; Fujii, M.; Hou, D. Action of Nrf2 and Keap1 in ARE-Mediated NQO1 Expression by Quercetin. Free Radic. Biol. Med. 2007, 42, 1690–1703. [Google Scholar] [CrossRef] [PubMed]
- Feng, R.-B.; Wang, Y.; He, C.; Yang, Y.; Wan, J.-B. Gallic Acid, a Natural Polyphenol, Protects against Tert-Butyl Hydroperoxide- Induced Hepatotoxicity by Activating ERK-Nrf2-Keap1-Mediated Antioxidative Response. Food Chem. Toxicol. 2018, 119, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Jiang, Z.; Li, C.; Zhu, Y.; Li, Z.; Tang, Y.; Ni, C. Apigenin Prevents Metabolic Syndrome in High-Fructose Diet-Fed Mice by Keap1-Nrf2 Pathway. Biomed. Pharmacother. 2018, 105, 1283–1290. [Google Scholar] [CrossRef]
- Lin, Y.-L.; Tsai, S.-H.; Lin-Shiau, S.-Y.; Ho, C.-T.; Lin, J.-K. Theaflavin-3,3′-Digallate from Black Tea Blocks the Nitric Oxide Synthase by down-Regulating the Activation of NF-ΚB in Macrophages. Eur. J. Pharmacol. 1999, 367, 379–388. [Google Scholar] [CrossRef]
- Lin, Y.-L.; Lin, J.-K. (−)-Epigallocatechin-3-Gallate Blocks the Induction of Nitric Oxide Synthase by Down-Regulating Lipopolysaccharide-Induced Activity of Transcription Factor Nuclear Factor-ΚB. Mol. Pharmacol. 1997, 52, 465–472. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M.; Forman, H.J. Redox Homeostasis: The Golden Mean of Healthy Living. Redox Biol. 2016, 8, 205–215. [Google Scholar] [CrossRef]
- Forman, H.J.; Davies, K.J.A.; Ursini, F. How Do Nutritional Antioxidants Really Work: Nucleophilic Tone and Para-Hormesis versus Free Radical Scavenging in Vivo. Free Radic. Biol. Med. 2014, 66, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Watson, J. Oxidants, Antioxidants and the Current Incurability of Metastatic Cancers. Open Biol. 2013, 3, 120144. [Google Scholar] [CrossRef]
- Wang, J.; Yi, J. Cancer Cell Killing via ROS: To Increase or Decrease, That Is the Question. Cancer Biol. Ther. 2008, 7, 1875–1884. [Google Scholar] [CrossRef]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting Cancer Cells by ROS-Mediated Mechanisms: A Radical Therapeutic Approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Russo, G.L.; Tedesco, I.; Spagnuolo, C.; Russo, M. Antioxidant Polyphenols in Cancer Treatment: Friend, Foe or Foil? Semin. Cancer Biol. 2017, 46, 1–13. [Google Scholar] [CrossRef]
- Peixoto, P.; Cartron, P.-F.; Serandour, A.A.; Hervouet, E. From 1957 to Nowadays: A Brief History of Epigenetics. IJMS 2020, 21, 7571. [Google Scholar] [CrossRef]
- Nam, A.S.; Chaligne, R.; Landau, D.A. Integrating Genetic and Non-Genetic Determinants of Cancer Evolution by Single-Cell Multi-Omics. Nat. Rev. Genet. 2021, 22, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Darwiche, N. Epigenetic Mechanisms and the Hallmarks of Cancer: An Intimate Affair. Am. J. Cancer Res. 2020, 10, 1954–1978. [Google Scholar] [PubMed]
- Nishiyama, A.; Nakanishi, M. Navigating the DNA Methylation Landscape of Cancer. Trends Genet. 2021, 37, 1012–1027. [Google Scholar] [CrossRef] [PubMed]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef] [PubMed]
- Martire, S.; Banaszynski, L.A. The Roles of Histone Variants in Fine-Tuning Chromatin Organization and Function. Nat. Rev. Mol. Cell. Biol. 2020, 21, 522–541. [Google Scholar] [CrossRef]
- Vardabasso, C.; Hasson, D.; Ratnakumar, K.; Chung, C.-Y.; Duarte, L.F.; Bernstein, E. Histone Variants: Emerging Players in Cancer Biology. Cell. Mol. Life Sci. 2014, 71, 379–404. [Google Scholar] [CrossRef]
- Scaffidi, P. Histone H1 Alterations in Cancer. Biochim. Biophys. Acta 2016, 1859, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Lyubitelev, A.V.; Kirpichnikov, M.P.; Studitsky, V.M. The Role of Linker Histones in Carcinogenesis. Russ. J. Bioorg. Chem. 2021, 47, 278–287. [Google Scholar] [CrossRef]
- Di Leva, G.; Garofalo, M.; Croce, C.M. MicroRNAs in Cancer. Annu. Rev. Pathol. 2014, 9, 287–314. [Google Scholar] [CrossRef] [PubMed]
- Zyner, K.G.; Simeone, A.; Flynn, S.M.; Doyle, C.; Marsico, G.; Adhikari, S.; Portella, G.; Tannahill, D.; Balasubramanian, S. G-Quadruplex DNA Structures in Human Stem Cells and Differentiation. Nat. Commun. 2022, 13, 142. [Google Scholar] [CrossRef]
- Nandakumar, V.; Vaid, M.; Katiyar, S.K. (-)-Epigallocatechin-3-Gallate Reactivates Silenced Tumor Suppressor Genes, Cip1/P21 and P16INK4a, by Reducing DNA Methylation and Increasing Histones Acetylation in Human Skin Cancer Cells. Carcinogenesis 2011, 32, 537–544. [Google Scholar] [CrossRef]
- Henning, S.M.; Wang, P.; Said, J.; Magyar, C.; Castor, B.; Doan, N.; Tosity, C.; Moro, A.; Gao, K.; Li, L.; et al. Polyphenols in Brewed Green Tea Inhibit Prostate Tumor Xenograft Growth by Localizing to the Tumor and Decreasing Oxidative Stress and Angiogenesis. J. Nutr. Biochem. 2012, 23, 1537–1542. [Google Scholar] [CrossRef]
- Khan, M.A.; Hussain, A.; Sundaram, M.K.; Alalami, U.; Gunasekera, D.; Ramesh, L.; Hamza, A.; Quraishi, U. (−)-Epigallocatechin-3-Gallate Reverses the Expression of Various Tumor-Suppressor Genes by Inhibiting DNA Methyltransferases and Histone Deacetylases in Human Cervical Cancer Cells. Oncol. Rep. 2015, 33, 1976–1984. [Google Scholar] [CrossRef]
- Fang, M.Z.; Wang, Y.; Ai, N.; Hou, Z.; Sun, Y.; Lu, H.; Welsh, W.; Yang, C.S. Tea Polyphenol (-)-Epigallocatechin-3-Gallate Inhibits DNA Methyltransferase and Reactivates Methylation-Silenced Genes in Cancer Cell Lines. Cancer Res. 2003, 63, 7563–7570. [Google Scholar]
- Jiang, A.; Wang, X.; Shan, X.; Li, Y.; Wang, P.; Jiang, P.; Feng, Q. Curcumin Reactivates Silenced Tumor Suppressor Gene RARβ by Reducing DNA Methylation. Phytother. Res. 2015, 29, 1237–1245. [Google Scholar] [CrossRef]
- Du, L.; Xie, Z.; Wu, L.; Chiu, M.; Lin, J.; Chan, K.K.; Liu, S.; Liu, Z. Reactivation of RASSF1A in Breast Cancer Cells by Curcumin. Nutr. Cancer 2012, 64, 1228–1235. [Google Scholar] [CrossRef]
- Kumar, U.; Sharma, U.; Rathi, G. Reversal of Hypermethylation and Reactivation of Glutathione S-Transferase Pi 1 Gene by Curcumin in Breast Cancer Cell Line. Tumour Biol. 2017, 39, 1010428317692258. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Kumar, L.; Mohanty, S.K.; Maikhuri, J.P.; Rajender, S.; Gupta, G. Sensitization of Androgen Refractory Prostate Cancer Cells to Anti-Androgens through Re-Expression of Epigenetically Repressed Androgen Receptor-Synergistic Action of Quercetin and Curcumin. Mol. Cell. Endocrinol. 2016, 431, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Kala, R.; Shah, H.N.; Martin, S.L.; Tollefsbol, T.O. Epigenetic-Based Combinatorial Resveratrol and Pterostilbene Alters DNA Damage Response by Affecting SIRT1 and DNMT Enzyme Expression, Including SIRT1-Dependent γ-H2AX and Telomerase Regulation in Triple-Negative Breast Cancer. BMC Cancer 2015, 15, 672. [Google Scholar] [CrossRef] [PubMed]
- Medina-Aguilar, R.; Pérez-Plasencia, C.; Marchat, L.A.; Gariglio, P.; García Mena, J.; Rodríguez Cuevas, S.; Ruíz-García, E.; Astudillo-de la Vega, H.; Hernández Juárez, J.; Flores-Pérez, A.; et al. Methylation Landscape of Human Breast Cancer Cells in Response to Dietary Compound Resveratrol. PLoS ONE 2016, 11, e0157866. [Google Scholar] [CrossRef]
- Zhu, W.; Qin, W.; Zhang, K.; Rottinghaus, G.E.; Chen, Y.-C.; Kliethermes, B.; Sauter, E.R. Trans-Resveratrol Alters Mammary Promoter Hypermethylation in Women at Increased Risk for Breast Cancer. Nutr. Cancer 2012, 64, 393–400. [Google Scholar] [CrossRef]
- Majid, S.; Dar, A.A.; Ahmad, A.E.; Hirata, H.; Kawakami, K.; Shahryari, V.; Saini, S.; Tanaka, Y.; Dahiya, A.V.; Khatri, G.; et al. BTG3 Tumor Suppressor Gene Promoter Demethylation, Histone Modification and Cell Cycle Arrest by Genistein in Renal Cancer. Carcinogenesis 2009, 30, 662–670. [Google Scholar] [CrossRef]
- Majid, S.; Dar, A.A.; Shahryari, V.; Hirata, H.; Ahmad, A.; Saini, S.; Tanaka, Y.; Dahiya, A.V.; Dahiya, R. Genistein Reverses Hypermethylation and Induces Active Histone Modifications in Tumor Suppressor Gene B-Cell Translocation Gene 3 in Prostate Cancer. Cancer 2010, 116, 66–76. [Google Scholar] [CrossRef]
- King-Batoon, A.; Leszczynska, J.M.; Klein, C.B. Modulation of Gene Methylation by Genistein or Lycopene in Breast Cancer Cells. Environ. Mol. Mutagen. 2008, 49, 36–45. [Google Scholar] [CrossRef]
- Pandey, M.; Shukla, S.; Gupta, S. Promoter Demethylation and Chromatin Remodeling by Green Tea Polyphenols Leads to Re-Expression of GSTP1 in Human Prostate Cancer Cells. Int. J. Cancer 2010, 126, 2520–2533. [Google Scholar] [CrossRef]
- Saldanha, S.N.; Kala, R.; Tollefsbol, T.O. Molecular Mechanisms for Inhibition of Colon Cancer Cells by Combined Epigenetic-Modulating Epigallocatechin Gallate and Sodium Butyrate. Exp. Cell. Res. 2014, 324, 40–53. [Google Scholar] [CrossRef]
- Choi, K.-C.; Jung, M.G.; Lee, Y.-H.; Yoon, J.C.; Kwon, S.H.; Kang, H.-B.; Kim, M.-J.; Cha, J.-H.; Kim, Y.J.; Jun, W.J.; et al. Epigallocatechin-3-Gallate, a Histone Acetyltransferase Inhibitor, Inhibits EBV-Induced B Lymphocyte Transformation via Suppression of RelA Acetylation. Cancer Res. 2009, 69, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Krauthauser, C.; Maduskuie, V.; Fawcett, P.T.; Olson, J.M.; Rajasekaran, S.A. Curcumin-Induced HDAC Inhibition and Attenuation of Medulloblastoma Growth in Vitro and in Vivo. BMC Cancer 2011, 11, 144. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Shu, W.; Chen, W.; Wu, Q.; Liu, H.; Cui, G. Curcumin, Both Histone Deacetylase and P300/CBP-Specific Inhibitor, Represses the Activity of Nuclear Factor Kappa B and Notch 1 in Raji Cells. Basic Clin. Pharm. Toxicol. 2007, 101, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-J.; Chen, Y.-R.; Tseng, T.-H. Quercetin Induces FasL-Related Apoptosis, in Part, through Promotion of Histone H3 Acetylation in Human Leukemia HL-60 Cells. Oncol. Rep. 2011, 25, 583–591. [Google Scholar] [CrossRef]
- Xiao, X.; Shi, D.; Liu, L.; Wang, J.; Xie, X.; Kang, T.; Deng, W. Quercetin Suppresses Cyclooxygenase-2 Expression and Angiogenesis through Inactivation of P300 Signaling. PLoS ONE 2011, 6, e22934. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.-H.; Zheng, Y.; Kim, H.-S.; Xu, X.; Cao, L.; Luhasen, T.; Lee, M.-H.; Xiao, C.; Vassilopoulos, A.; Chen, W.; et al. Interplay among BRCA1, SIRT1, and Survivin during BRCA1-Associated Tumorigenesis. Mol. Cell 2008, 32, 11–20. [Google Scholar] [CrossRef]
- Dhar, S.; Kumar, A.; Li, K.; Tzivion, G.; Levenson, A.S. Resveratrol Regulates PTEN/Akt Pathway through Inhibition of MTA1/HDAC Unit of the NuRD Complex in Prostate Cancer. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2015, 1853, 265–275. [Google Scholar] [CrossRef]
- Kai, L.; Samuel, S.K.; Levenson, A.S. Resveratrol Enhances P53 Acetylation and Apoptosis in Prostate Cancer by Inhibiting MTA1/NuRD Complex. Int. J. Cancer 2010, 126, 1538–1548. [Google Scholar] [CrossRef]
- Majid, S.; Kikuno, N.; Nelles, J.; Noonan, E.; Tanaka, Y.; Kawamoto, K.; Hirata, H.; Li, L.C.; Zhao, H.; Okino, S.T.; et al. Genistein Induces the P21WAF1/CIP1 and P16INK4a Tumor Suppressor Genes in Prostate Cancer Cells by Epigenetic Mechanisms Involving Active Chromatin Modification. Cancer Res. 2008, 68, 2736–2744. [Google Scholar] [CrossRef]
- Li, Y.; Chen, H.; Hardy, T.M.; Tollefsbol, T.O. Epigenetic Regulation of Multiple Tumor-Related Genes Leads to Suppression of Breast Tumorigenesis by Dietary Genistein. PLoS ONE 2013, 8, e54369. [Google Scholar] [CrossRef]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [PubMed]
- Hombach, S.; Kretz, M. Non-Coding RNAs: Classification, Biology and Functioning. Adv. Exp. Med. Biol. 2016, 937, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Bu, P. Non-Coding RNA in Cancer. Essays Biochem. 2021, 65, 625–639. [Google Scholar] [CrossRef]
- Slack, F.J.; Chinnaiyan, A.M. The Role of Non-Coding RNAs in Oncology. Cell 2019, 179, 1033–1055. [Google Scholar] [CrossRef]
- Tsang, W.P.; Kwok, T.T. Epigallocatechin Gallate Up-Regulation of MiR-16 and Induction of Apoptosis in Human Cancer Cells. J. Nutr. Biochem. 2010, 21, 140–146. [Google Scholar] [CrossRef]
- Chakrabarti, M.; Khandkar, M.; Banik, N.L.; Ray, S.K. Alterations in Expression of Specific MicroRNAs by Combination of 4-HPR and EGCG Inhibited Growth of Human Malignant Neuroblastoma Cells. Brain Res. 2012, 1454, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Bian, S.; Yang, C.S. Green Tea Polyphenol EGCG Suppresses Lung Cancer Cell Growth through Upregulating MiR-210 Expression Caused by Stabilizing HIF-1α. Carcinogenesis 2011, 32, 1881–1889. [Google Scholar] [CrossRef] [PubMed]
- Zan, L.; Chen, Q.; Zhang, L.; Li, X. Epigallocatechin Gallate (EGCG) Suppresses Growth and Tumorigenicity in Breast Cancer Cells by Downregulation of MiR-25. Bioengineered 2019, 10, 374–382. [Google Scholar] [CrossRef]
- Zhong, Z.; Dong, Z.; Yang, L.; Chen, X.; Gong, Z. Inhibition of Proliferation of Human Lung Cancer Cells by Green Tea Catechins Is Mediated by Upregulation of Let-7. Exp. Ther. Med. 2012, 4, 267–272. [Google Scholar] [CrossRef]
- Appari, M.; Babu, K.R.; Kaczorowski, A.; Gross, W.; Herr, I. Sulforaphane, Quercetin and Catechins Complement Each Other in Elimination of Advanced Pancreatic Cancer by MiR-Let-7 Induction and K-Ras Inhibition. Int. J. Oncol. 2014, 45, 1391–1400. [Google Scholar] [CrossRef]
- Nirgude, S.; Desai, S.; Choudhary, B. Curcumin Alters Distinct Molecular Pathways in Breast Cancer Subtypes Revealed by Integrated miRNA/mRNA Expression Analysis. Cancer Rep. 2022, 5, e1596. [Google Scholar] [CrossRef]
- Wang, X.; Hang, Y.; Liu, J.; Hou, Y.; Wang, N.; Wang, M. Anticancer Effect of Curcumin Inhibits Cell Growth through MiR-21/PTEN/Akt Pathway in Breast Cancer Cell. Oncol. Lett. 2017, 13, 4825–4831. [Google Scholar] [CrossRef]
- Liu, W.; Huang, M.; Zou, Q.; Lin, W. Curcumin Suppresses Gastric Cancer Biological Activity by Regulation of MiRNA-21: An in Vitro Study. Int. J. Clin. Exp. Pathol. 2018, 11, 5820–5829. [Google Scholar]
- Zhang, W.; Bai, W.; Zhang, W. MiR-21 Suppresses the Anticancer Activities of Curcumin by Targeting PTEN Gene in Human Non-Small Cell Lung Cancer A549 Cells. Clin. Transl. Oncol. 2014, 16, 708–713. [Google Scholar] [CrossRef]
- Mudduluru, G.; George-William, J.N.; Muppala, S.; Asangani, I.A.; Kumarswamy, R.; Nelson, L.D.; Allgayer, H. Curcumin Regulates MiR-21 Expression and Inhibits Invasion and Metastasis in Colorectal Cancer. Biosci. Rep. 2011, 31, 185–197. [Google Scholar] [CrossRef]
- Roy, S.; Yu, Y.; Padhye, S.B.; Sarkar, F.H.; Majumdar, A.P.N. Difluorinated-Curcumin (CDF) Restores PTEN Expression in Colon Cancer Cells by down-Regulating MiR-21. PLoS ONE 2013, 8, e68543. [Google Scholar] [CrossRef]
- Yang, C.H.; Yue, J.; Sims, M.; Pfeffer, L.M. The Curcumin Analog EF24 Targets NF-ΚB and MiRNA-21, and Has Potent Anticancer Activity in Vitro and in Vivo. PLoS ONE 2013, 8, e71130. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xie, W.; Xie, C.; Huang, C.; Zhu, J.; Liang, Z.; Deng, F.; Zhu, M.; Zhu, W.; Wu, R.; et al. Curcumin Modulates MiR-19/PTEN/AKT/P53 Axis to Suppress Bisphenol A-Induced MCF-7 Breast Cancer Cell Proliferation: CURCUMIN MODULATES BPA-DYSREGULATED MIR-19/PTEN/AKT/P53 AXIS. Phytother. Res. 2014, 28, 1553–1560. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Estrov, Z.; Ji, Y.; Coombes, K.R.; Harris, D.H.; Kurzrock, R. Curcumin (Diferuloylmethane) Alters the Expression Profiles of MicroRNAs in Human Pancreatic Cancer Cells. Mol. Cancer Ther. 2008, 7, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, T.; Ti, X.; Shi, J.; Wu, C.; Ren, X.; Yin, H. Curcumin Promotes Apoptosis in A549/DDP Multidrug-Resistant Human Lung Adenocarcinoma Cells through an MiRNA Signaling Pathway. Biochem. Biophys. Res. Commun. 2010, 399, 1–6. [Google Scholar] [CrossRef]
- Zhu, M.; Zheng, Z.; Huang, J.; Ma, X.; Huang, C.; Wu, R.; Li, X.; Liang, Z.; Deng, F.; Wu, J.; et al. Modulation of MiR-34a in Curcumin-Induced Antiproliferation of Prostate Cancer Cells. J. Cell. Biochem. 2019, 120, 15616–15624. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.-L.; Chang, J.-M.; Chong, I.-W.; Hung, Y.-L.; Chen, Y.-H.; Huang, W.-T.; Kuo, H.-F.; Hsieh, C.-C.; Liu, P.-L. Curcumin Inhibits LIN-28A through the Activation of MiRNA-98 in the Lung Cancer Cell Line A549. Molecules 2017, 22, 929. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Fang, Z.; Zha, Z.; Sun, Q.; Wang, H.; Sun, M.; Qiao, B. Quercetin Inhibits Cell Viability, Migration and Invasion by Regulating MiR-16/HOXA10 Axis in Oral Cancer. Eur. J. Pharmacol. 2019, 847, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Sonoki, H.; Sato, T.; Endo, S.; Matsunaga, T.; Yamaguchi, M.; Yamazaki, Y.; Sugatani, J.; Ikari, A. Quercetin Decreases Claudin-2 Expression Mediated by Up-Regulation of MicroRNA MiR-16 in Lung Adenocarcinoma A549 Cells. Nutrients 2015, 7, 4578–4592. [Google Scholar] [CrossRef]
- Lou, G.; Liu, Y.; Wu, S.; Xue, J.; Yang, F.; Fu, H.; Zheng, M.; Chen, Z. The P53/MiR-34a/SIRT1 Positive Feedback Loop in Quercetin-Induced Apoptosis. Cell. Physiol. Biochem. 2015, 35, 2192–2202. [Google Scholar] [CrossRef]
- Tao, S.; He, H.; Chen, Q. Quercetin Inhibits Proliferation and Invasion Acts by Up-Regulating MiR-146a in Human Breast Cancer Cells. Mol. Cell. Biochem. 2015, 402, 93–100. [Google Scholar] [CrossRef]
- Pratheeshkumar, P.; Son, Y.-O.; Divya, S.P.; Wang, L.; Turcios, L.; Roy, R.V.; Hitron, J.A.; Kim, D.; Dai, J.; Asha, P.; et al. Quercetin Inhibits Cr(VI)-Induced Malignant Cell Transformation by Targeting MiR-21-PDCD4 Signaling Pathway. Oncotarget 2017, 8, 52118–52131. [Google Scholar] [CrossRef]
- Nwaeburu, C.C.; Bauer, N.; Zhao, Z.; Abukiwan, A.; Gladkich, J.; Benner, A.; Herr, I. Up-Regulation of MicroRNA Let-7c by Quercetin Inhibits Pancreatic Cancer Progression by Activation of Numbl. Oncotarget 2016, 7, 58367–58380. [Google Scholar] [CrossRef]
- Del Follo-Martinez, A.; Banerjee, N.; Li, X.; Safe, S.; Mertens-Talcott, S. Resveratrol and Quercetin in Combination Have Anticancer Activity in Colon Cancer Cells and Repress Oncogenic MicroRNA-27a. Nutr. Cancer 2013, 65, 494–504. [Google Scholar] [CrossRef]
- Wang, G.; Dai, F.; Yu, K.; Jia, Z.; Zhang, A.; Huang, Q.; Kang, C.; Jiang, H.; Pu, P. Resveratrol Inhibits Glioma Cell Growth via Targeting Oncogenic MicroRNAs and Multiple Signaling Pathways. Int. J. Oncol. 2015, 46, 1739–1747. [Google Scholar] [CrossRef]
- Li, H.; Jia, Z.; Li, A.; Jenkins, G.; Yang, X.; Hu, J.; Guo, W. Resveratrol Repressed Viability of U251 Cells by MiR-21 Inhibiting of NF-ΚB Pathway. Mol. Cell. Biochem. 2013, 382, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Ding, J.; Wu, Y. Resveratrol Induces Apoptosis of Bladder Cancer Cells via MiR-21 Regulation of the Akt/Bcl-2 Signaling Pathway. Mol. Med. Rep. 2014, 9, 1467–1473. [Google Scholar] [CrossRef] [PubMed]
- Sheth, S.; Jajoo, S.; Kaur, T.; Mukherjea, D.; Sheehan, K.; Rybak, L.P.; Ramkumar, V. Resveratrol Reduces Prostate Cancer Growth and Metastasis by Inhibiting the Akt/MicroRNA-21 Pathway. PLoS ONE 2012, 7, e51655. [Google Scholar] [CrossRef]
- Wang, H.; Feng, H.; Zhang, Y. Resveratrol Inhibits Hypoxia-Induced Glioma Cell Migration and Invasion by the p-STAT3/MiR-34a Axis. Neoplasma 2016, 63, 532–539. [Google Scholar] [CrossRef]
- Yang, S.; Li, W.; Sun, H.; Wu, B.; Ji, F.; Sun, T.; Chang, H.; Shen, P.; Wang, Y.; Zhou, D. Resveratrol Elicits Anti-Colorectal Cancer Effect by Activating MiR-34c-KITLG in Vitro and in Vivo. BMC Cancer 2015, 15, 969. [Google Scholar] [CrossRef]
- Venkatadri, R.; Muni, T.; Iyer, A.K.V.; Yakisich, J.S.; Azad, N. Role of Apoptosis-Related MiRNAs in Resveratrol-Induced Breast Cancer Cell Death. Cell Death Dis. 2016, 7, e2104. [Google Scholar] [CrossRef]
- Sun, Q.; Cong, R.; Yan, H.; Gu, H.; Zeng, Y.; Liu, N.; Chen, J.; Wang, B. Genistein Inhibits Growth of Human Uveal Melanoma Cells and Affects MicroRNA-27a and Target Gene Expression. Oncol. Rep. 2009, 22, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Xiang, J.; Shen, J.; Zou, X.; Zhai, S.; Yin, Y.; Li, P.; Wang, X.; Sun, Q. Oncogenic MicroRNA-27a Is a Target for Genistein in Ovarian Cancer Cells. Anticancer Agents Med. Chem. 2013, 13, 1126–1132. [Google Scholar] [CrossRef]
- Yang, Y.; Zang, A.; Jia, Y.; Shang, Y.; Zhang, Z.; Ge, K.; Zhang, J.; Fan, W.; Wang, B. Genistein Inhibits A549 Human Lung Cancer Cell Proliferation via MiR-27a and MET Signaling. Oncol. Lett. 2016, 12, 2189–2193. [Google Scholar] [CrossRef]
- Avci, C.B.; Susluer, S.Y.; Caglar, H.O.; Balci, T.; Aygunes, D.; Dodurga, Y.; Gunduz, C. Genistein-Induced Mir-23b Expression Inhibits the Growth of Breast Cancer Cells. Contemp. Oncol. 2015, 19, 32–35. [Google Scholar] [CrossRef]
- Chiyomaru, T.; Yamamura, S.; Zaman, M.S.; Majid, S.; Deng, G.; Shahryari, V.; Saini, S.; Hirata, H.; Ueno, K.; Chang, I.; et al. Genistein Suppresses Prostate Cancer Growth through Inhibition of Oncogenic MicroRNA-151. PLoS ONE 2012, 7, e43812. [Google Scholar] [CrossRef]
- De la Parra, C.; Castillo-Pichardo, L.; Cruz-Collazo, A.; Cubano, L.; Redis, R.; Calin, G.A.; Dharmawardhane, S. Soy Isoflavone Genistein-Mediated Downregulation of MiR-155 Contributes to the Anticancer Effects of Genistein. Nutr. Cancer 2016, 68, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Duan, Q.; Ahmad, A.; Bao, B.; Banerjee, S.; Shi, Y.; Ma, J.; Geng, J.; Chen, Z.; Rahman, K.M.W.; et al. Genistein Inhibits Cell Growth and Induces Apoptosis through Up-Regulation of MiR-34a in Pancreatic Cancer Cells. Curr. Drug Targets 2012, 13, 1750–1756. [Google Scholar] [CrossRef] [PubMed]
- Chiyomaru, T.; Yamamura, S.; Fukuhara, S.; Yoshino, H.; Kinoshita, T.; Majid, S.; Saini, S.; Chang, I.; Tanaka, Y.; Enokida, H.; et al. Genistein Inhibits Prostate Cancer Cell Growth by Targeting MiR-34a and Oncogenic HOTAIR. PLoS ONE 2013, 8, e70372. [Google Scholar] [CrossRef]
- Mishra, S.; Verma, S.S.; Rai, V.; Awasthee, N.; Chava, S.; Hui, K.M.; Kumar, A.P.; Challagundla, K.B.; Sethi, G.; Gupta, S.C. Long Non-Coding RNAs Are Emerging Targets of Phytochemicals for Cancer and Other Chronic Diseases. Cell. Mol. Life Sci. 2019, 76, 1947–1966. [Google Scholar] [CrossRef]
- Bouyahya, A.; Mechchate, H.; Oumeslakht, L.; Zeouk, I.; Aboulaghras, S.; Balahbib, A.; Zengin, G.; Kamal, M.A.; Gallo, M.; Montesano, D.; et al. The Role of Epigenetic Modifications in Human Cancers and the Use of Natural Compounds as Epidrugs: Mechanistic Pathways and Pharmacodynamic Actions. Biomolecules 2022, 12, 367. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Origin and Physiological Roles of Inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Mitchell, J.P.; Carmody, R.J. NF-ΚB and the Transcriptional Control of Inflammation. In International Review of Cell and Molecular Biology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 335, pp. 41–84. ISBN 978-0-12-812339-3. [Google Scholar]
- Liu, F.; Xia, Y.; Parker, A.S.; Verma, I.M. IKK Biology. Immunol. Rev. 2012, 246, 239–253. [Google Scholar] [CrossRef]
- Sun, S.-C. Non-Canonical NF-ΚB Signaling Pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NFκB System. WIREs Mech. Dis. 2016, 8, 227–241. [Google Scholar] [CrossRef]
- Disis, M.L. Immune Regulation of Cancer. J. Clin. Oncol. 2010, 28, 4531–4538. [Google Scholar] [CrossRef] [PubMed]
- Laveti, D.; Kumar, M.; Hemalatha, R.; Sistla, R.; Naidu, V.; Talla, V.; Verma, V.; Kaur, N.; Nagpal, R. Anti-Inflammatory Treatments for Chronic Diseases: A Review. Inflamm. Allergy Drug Targets 2013, 12, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.-X.; Xia, Z.; Zhang, N.; Gong, W.; Huang, S. Constitutive NF-KappaB Activity Regulates the Expression of VEGF and IL-8 and Tumor Angiogenesis of Human Glioblastoma. Oncol. Rep. 2010, 23, 725–732. [Google Scholar] [PubMed]
- Kumar, S.; Mehta, K. Tissue Transglutaminase Constitutively Activates HIF-1α Promoter and Nuclear Factor-ΚB via a Non-Canonical Pathway. PLoS ONE 2012, 7, e49321. [Google Scholar] [CrossRef]
- Peng, G.; Dixon, D.A.; Muga, S.J.; Smith, T.J.; Wargovich, M.J. Green Tea Polyphenol (−)-Epigallocatechin-3-Gallate Inhibits Cyclooxygenase-2 Expression in Colon Carcinogenesis. Mol. Carcinog. 2006, 45, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.C.; Wheeler, D.S.; Malhotra, V.; Odoms, K.; Denenberg, A.G.; Wong, H.R. A Green Tea-Derived Polyphenol, Epigallocatechin-3-Gallate, Inhibits IkappaB Kinase Activation and IL-8 Gene Expression in Respiratory Epithelium. Inflammation 2002, 26, 233–241. [Google Scholar] [CrossRef]
- Wheeler, D.S.; Catravas, J.D.; Odoms, K.; Denenberg, A.; Malhotra, V.; Wong, H.R. Epigallocatechin-3-Gallate, a Green Tea–Derived Polyphenol, Inhibits IL-1β-Dependent Proinflammatory Signal Transduction in Cultured Respiratory Epithelial Cells. J. Nutr. 2004, 134, 1039–1044. [Google Scholar] [CrossRef]
- Yang, F.; Oz, H.S.; Barve, S.; de Villiers, W.J.; McClain, C.J.; Varilek, G.W. The Green Tea Polyphenol (-)-Epigallocatechin-3-Gallate Blocks Nuclear Factor-Kappa B Activation by Inhibiting I Kappa B Kinase Activity in the Intestinal Epithelial Cell Line IEC-6. Mol. Pharmacol. 2001, 60, 528–533. [Google Scholar]
- Fechtner, S.; Singh, A.; Chourasia, M.; Ahmed, S. Molecular Insights into the Differences in Anti-Inflammatory Activities of Green Tea Catechins on IL-1β Signaling in Rheumatoid Arthritis Synovial Fibroblasts. Toxicol. Appl. Pharmacol. 2017, 329, 112–120. [Google Scholar] [CrossRef]
- Singh, A.K.; Umar, S.; Riegsecker, S.; Chourasia, M.; Ahmed, S. Regulation of Transforming Growth Factor β-Activated Kinase Activation by Epigallocatechin-3-Gallate in Rheumatoid Arthritis Synovial Fibroblasts: Suppression of K63 -Linked Autoubiquitination of Tumor Necrosis Factor Receptor-Associated Factor 6: TAK1 INHIBITION BY EGCG IN RA. Arthritis Rheumatol. 2016, 68, 347–358. [Google Scholar] [CrossRef]
- Varthya, S.B.; Sarma, P.; Bhatia, A.; Shekhar, N.; Prajapat, M.; Kaur, H.; Thangaraju, P.; Kumar, S.; Singh, R.; Siingh, A.; et al. Efficacy of Green Tea, Its Polyphenols and Nanoformulation in Experimental Colitis and the Role of Non-Canonical and Canonical Nuclear Factor Kappa Beta (NF-KB) Pathway: A Preclinical in-Vivo and in-Silico Exploratory Study. J. Biomol. Struct. Dyn. 2021, 39, 5314–5326. [Google Scholar] [CrossRef]
- Lakshmi, S.P.; Reddy, A.T.; Kodidhela, L.D.; Varadacharyulu, N.C. The Tea Catechin Epigallocatechin Gallate Inhibits NF-ΚB-Mediated Transcriptional Activation by Covalent Modification. Arch. Biochem. Biophys. 2020, 695, 108620. [Google Scholar] [CrossRef]
- Bachmeier, B.E.; Mohrenz, I.V.; Mirisola, V.; Schleicher, E.; Romeo, F.; Höhneke, C.; Jochum, M.; Nerlich, A.G.; Pfeffer, U. Curcumin Downregulates the Inflammatory Cytokines CXCL1 and -2 in Breast Cancer Cells via NFκB. Carcinogenesis 2008, 29, 779–789. [Google Scholar] [CrossRef]
- Olivera, A.; Moore, T.W.; Hu, F.; Brown, A.P.; Sun, A.; Liotta, D.C.; Snyder, J.P.; Yoon, Y.; Shim, H.; Marcus, A.I.; et al. Inhibition of the NF-ΚB Signaling Pathway by the Curcumin Analog, 3,5-Bis(2-Pyridinylmethylidene)-4-Piperidone (EF31): Anti-Inflammatory and Anti-Cancer Properties. Int. Immunopharmacol. 2012, 12, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Hakeem, M.A.; Mongy, S.; Hassan, B.; Tantawi, O.I.; Badawy, I. Curcumin Loaded Chitosan-Protamine Nanoparticles Revealed Antitumor Activity via Suppression of NF-ΚB, Proinflammatory Cytokines and Bcl-2 Gene Expression in the Breast Cancer Cells. J. Pharm. Sci. 2021, 110, 3298–3305. [Google Scholar] [CrossRef]
- Coker-Gurkan, A.; Bulut, D.; Genc, R.; Arisan, E.-D.; Obakan-Yerlikaya, P.; Palavan-Unsal, N. Curcumin Prevented Human Autocrine Growth Hormone (GH) Signaling Mediated NF-ΚB Activation and MiR-183-96-182 Cluster Stimulated Epithelial Mesenchymal Transition in T47D Breast Cancer Cells. Mol. Biol. Rep. 2019, 46, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, F.; Shafiee, M.; Banikazemi, Z.; Pourhanifeh, M.H.; Khanbabaei, H.; Shamshirian, A.; Amiri Moghadam, S.; ArefNezhad, R.; Sahebkar, A.; Avan, A.; et al. Curcumin Inhibits NF-KB and Wnt/β-Catenin Pathways in Cervical Cancer Cells. Pathol.-Res. Pract. 2019, 215, 152556. [Google Scholar] [CrossRef]
- Tian, F.; Zhang, C.; Tian, W.; Jiang, Y.; Zhang, X. Comparison of the Effect of P65 SiRNA and Curcumin in Promoting Apoptosis in Esophageal Squamous Cell Carcinoma Cells and in Nude Mice. Oncol. Rep. 2012, 28, 232–240. [Google Scholar] [CrossRef]
- Mu, Y.-T.; Feng, H.-H.; Yu, J.-Q.; Liu, Z.-K.; Wang, Y.; Shao, J.; Li, R.-H.; Li, D.-K. Curcumin Suppressed Proliferation and Migration of Human Retinoblastoma Cells through Modulating NF-ΚB Pathway. Int. Ophthalmol. 2020, 40, 2435–2440. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Song, Y.; Zhang, X. Quercetin Suppresses the Migration and Invasion in Human Colon Cancer Caco-2 Cells Through Regulating Toll-like Receptor 4/Nuclear Factor-Kappa B Pathway. Pharmacogn. Mag. 2016, 12, S237–S244. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-A.; Zhang, S.; Yin, Q.; Zhang, J. Quercetin Induces Human Colon Cancer Cells Apoptosis by Inhibiting the Nuclear Factor-Kappa B Pathway. Pharmacogn. Mag. 2015, 11, 404–409. [Google Scholar] [CrossRef]
- Youn, H.; Jeong, J.-C.; Jeong, Y.S.; Kim, E.-J.; Um, S.-J. Quercetin Potentiates Apoptosis by Inhibiting Nuclear Factor-KappaB Signaling in H460 Lung Cancer Cells. Biol. Pharm. Bull. 2013, 36, 944–951. [Google Scholar] [CrossRef]
- Lai, W.-W.; Hsu, S.-C.; Chueh, F.-S.; Chen, Y.-Y.; Yang, J.-S.; Lin, J.-P.; Lien, J.-C.; Tsai, C.-H.; Chung, J.-G. Quercetin Inhibits Migration and Invasion of SAS Human Oral Cancer Cells through Inhibition of NF-ΚB and Matrix Metalloproteinase-2/-9 Signaling Pathways. Anticancer. Res. 2013, 33, 1941–1950. [Google Scholar]
- Zhang, W.; Yin, G.; Dai, J.; Sun, Y.; Hoffman, R.M.; Yang, Z.; Fan, Y. Chemoprevention by Quercetin of Oral Squamous Cell Carcinoma by Suppression of the NF-ĸB Signaling Pathway in DMBA-Treated Hamsters. Anticancer. Res. 2017, 37, 4041–4049. [Google Scholar] [CrossRef] [PubMed]
- Shree, A.; Islam, J.; Sultana, S. Quercetin Ameliorates Reactive Oxygen Species Generation, Inflammation, Mucus Depletion, Goblet Disintegration, and Tumor Multiplicity in Colon Cancer: Probable Role of Adenomatous Polyposis Coli, β-catenin. Phytother. Res. 2021, 35, 2171–2184. [Google Scholar] [CrossRef] [PubMed]
- García-Zepeda, S.P.; García-Villa, E.; Díaz-Chávez, J.; Hernández-Pando, R.; Gariglio, P. Resveratrol Induces Cell Death in Cervical Cancer Cells through Apoptosis and Autophagy. Eur. J. Cancer Prev. 2013, 22, 577–584. [Google Scholar] [CrossRef]
- Dariya, B.; Behera, S.K.; Srivani, G.; Farran, B.; Alam, A.; Nagaraju, G.P. Computational Analysis of Nuclear Factor-ΚB and Resveratrol in Colorectal Cancer. J. Biomol. Struct. Dyn. 2021, 39, 2914–2922. [Google Scholar] [CrossRef]
- Wu, F.; Cui, L. Resveratrol Suppresses Melanoma by Inhibiting NF-ΚB/MiR-221 and Inducing TFG Expression. Arch. Derm. Res. 2017, 309, 823–831. [Google Scholar] [CrossRef]
- Qian, W.; Xiao, Q.; Wang, L.; Qin, T.; Xiao, Y.; Li, J.; Yue, Y.; Zhou, C.; Duan, W.; Ma, Q.; et al. Resveratrol Slows the Tumourigenesis of Pancreatic Cancer by Inhibiting NFκB Activation. Biomed. Pharmacother. 2020, 127, 110116. [Google Scholar] [CrossRef]
- Rawat, D.; Chhonker, S.K.; Naik, R.A.; Koiri, R.K. Modulation of Antioxidant Enzymes, SIRT1 and NF-κB by Resveratrol and Nicotinamide in Alcohol-aflatoxin B1-induced Hepatocellular Carcinoma. J. Biochem. Mol. Toxicol. 2021, 35, e22625. [Google Scholar] [CrossRef]
- Tino, A.B.; Chitcholtan, K.; Sykes, P.H.; Garrill, A. Resveratrol and Acetyl-Resveratrol Modulate Activity of VEGF and IL-8 in Ovarian Cancer Cell Aggregates via Attenuation of the NF-ΚB Protein. J. Ovarian Res. 2016, 9, 84. [Google Scholar] [CrossRef] [PubMed]
- Fukui, M.; Yamabe, N.; Kang, K.S.; Zhu, B.T. Growth-Stimulatory Effect of Resveratrol in Human Cancer Cells. Mol. Carcinog. 2010, 49, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Wang, J.; Zhu, B. Genistein Inhibits the Proliferation of Human Multiple Myeloma Cells through Suppression of Nuclear Factor-ΚB and Upregulation of MicroRNA-29b. Mol. Med. Rep. 2016, 13, 1627–1632. [Google Scholar] [CrossRef] [PubMed]
- Ozturk, S.; Alp, E.; Saglam, A.Y.; Konac, E.; Menevse, E. The Effects of Thymoquinone and Genistein Treatment on Telomerase Activity, Apoptosis, Angiogenesis, and Survival in Thyroid Cancer Cell Lines. J. Can. Res. Ther. 2017, 14, 328–334. [Google Scholar] [CrossRef]
- Zhang, J.; Su, H.; Li, Q.; Li, J.; Zhao, Q. Genistein Decreases A549 Cell Viability via Inhibition of the PI3K/AKT/HIF-1α/VEGF and NF-ΚB/COX-2 Signaling Pathways. Mol. Med. Rep. 2017, 15, 2296–2302. [Google Scholar] [CrossRef]
- Zhou, P.; Wang, C.; Hu, Z.; Chen, W.; Qi, W.; Li, A. Genistein Induces Apoptosis of Colon Cancer Cells by Reversal of Epithelial-to-Mesenchymal via a Notch1/NF-ΚB/Slug/E-Cadherin Pathway. BMC Cancer 2017, 17, 813. [Google Scholar] [CrossRef]
- Pan, H.; Zhou, W.; He, W.; Liu, X.; Ding, Q.; Ling, L.; Zha, X.; Wang, S. Genistein Inhibits MDA-MB-231 Triple-Negative Breast Cancer Cell Growth by Inhibiting NF-ΚB Activity via the Notch-1 Pathway. Int. J. Mol. Med. 2012, 30, 337–343. [Google Scholar] [CrossRef]
- Luo, Y.; Wang, S.; Zhou, Z.; Wang, Z.; Zhang, Y.; Zhang, Y.; Zhao, P. Apoptotic Effect of Genistein on Human Colon Cancer Cells via Inhibiting the Nuclear Factor-Kappa B (NF-ΚB) Pathway. Tumor Biol. 2014, 35, 11483–11488. [Google Scholar] [CrossRef]
- Vilela, F.M.P.; Syed, D.N.; Chamcheu, J.C.; Calvo-Castro, L.A.; Fortes, V.S.; Fonseca, M.J.V.; Mukhtar, H. Biotransformed Soybean Extract (BSE) Inhibits Melanoma Cell Growth and Viability in Vitro: Involvement of Nuclear Factor-Kappa B Signaling. PLoS ONE 2014, 9, e103248. [Google Scholar] [CrossRef]
- Alorda-Clara, M.; Torrens-Mas, M.; Morla-Barcelo, P.M.; Roca, P.; Sastre-Serra, J.; Pons, D.G.; Oliver, J. High Concentrations of Genistein Decrease Cell Viability Depending on Oxidative Stress and Inflammation in Colon Cancer Cell Lines. IJMS 2022, 23, 7526. [Google Scholar] [CrossRef] [PubMed]
- Al-Khayri, J.M.; Sahana, G.R.; Nagella, P.; Joseph, B.V.; Alessa, F.M.; Al-Mssallem, M.Q. Flavonoids as Potential Anti-Inflammatory Molecules: A Review. Molecules 2022, 27, 2901. [Google Scholar] [CrossRef]
- Ambati, G.G.; Jachak, S.M. Natural Product Inhibitors of Cyclooxygenase (COX) Enzyme: A Review on Current Status and Future Perspectives. Curr. Med. Chem. 2021, 28, 1877–1905. [Google Scholar] [CrossRef]
- Yahfoufi, N.; Alsadi, N.; Jambi, M.; Matar, C. The Immunomodulatory and Anti-Inflammatory Role of Polyphenols. Nutrients 2018, 10, 1618. [Google Scholar] [CrossRef]
- Barrett, K.E.; Wu, G.D. Influence of the Microbiota on Host Physiology-Moving beyond the Gut: Editorial. J. Physiol. 2017, 595, 433–435. [Google Scholar] [CrossRef] [PubMed]
- Young, V.B. The Role of the Microbiome in Human Health and Disease: An Introduction for Clinicians. BMJ 2017, 356, j831. [Google Scholar] [CrossRef] [PubMed]
- LaCourse, K.D.; Johnston, C.D.; Bullman, S. The Relationship between Gastrointestinal Cancers and the Microbiota. Lancet Gastroenterol. Hepatol. 2021, 6, 498–509. [Google Scholar] [CrossRef]
- Wilson, M.R.; Jiang, Y.; Villalta, P.W.; Stornetta, A.; Boudreau, P.D.; Carrá, A.; Brennan, C.A.; Chun, E.; Ngo, L.; Samson, L.D.; et al. The Human Gut Bacterial Genotoxin Colibactin Alkylates DNA. Science 2019, 363, eaar7785. [Google Scholar] [CrossRef]
- He, Z.; Gharaibeh, R.Z.; Newsome, R.C.; Pope, J.L.; Dougherty, M.W.; Tomkovich, S.; Pons, B.; Mirey, G.; Vignard, J.; Hendrixson, D.R.; et al. Campylobacter jejuni Promotes Colorectal Tumorigenesis through the Action of Cytolethal Distending Toxin. Gut 2019, 68, 289–300. [Google Scholar] [CrossRef]
- Hashemi Goradel, N.; Heidarzadeh, S.; Jahangiri, S.; Farhood, B.; Mortezaee, K.; Khanlarkhani, N.; Negahdari, B. Fusobacterium nucleatum and Colorectal Cancer: A Mechanistic Overview. J. Cell. Physiol. 2019, 234, 2337–2344. [Google Scholar] [CrossRef]
- Baj, J.; Forma, A.; Sitarz, M.; Portincasa, P.; Garruti, G.; Krasowska, D.; Maciejewski, R. Helicobacter pylori Virulence Factors—Mechanisms of Bacterial Pathogenicity in the Gastric Microenvironment. Cells 2020, 10, 27. [Google Scholar] [CrossRef]
- Flukes, S.L.; IARC Working Group on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer. Schistosomes, Liver Flukes and Helicobacter pylori: This Publication Represents the Views and Expert Opinions of an IARC Working Group on the Evaluation of Carcinogenic Risks to Humans, Which Met in Lyon, 7–14 June 1994; International Agency for Research on Cancer, Ed.; IARC monographs on the evaluation of carcinogenic risks to humans; IARC: Lyon, France, 1994; ISBN 978-92-832-1261-4. [Google Scholar]
- Fang, Y.; Yan, C.; Zhao, Q.; Xu, J.; Liu, Z.; Gao, J.; Zhu, H.; Dai, Z.; Wang, D.; Tang, D. The Roles of Microbial Products in the Development of Colorectal Cancer: A Review. Bioengineered 2021, 12, 720–735. [Google Scholar] [CrossRef] [PubMed]
- Wirbel, J.; Pyl, P.T.; Kartal, E.; Zych, K.; Kashani, A.; Milanese, A.; Fleck, J.S.; Voigt, A.Y.; Palleja, A.; Ponnudurai, R.; et al. Meta-Analysis of Fecal Metagenomes Reveals Global Microbial Signatures That Are Specific for Colorectal Cancer. Nat. Med. 2019, 25, 679–689. [Google Scholar] [CrossRef]
- Ternes, D.; Karta, J.; Tsenkova, M.; Wilmes, P.; Haan, S.; Letellier, E. Microbiome in Colorectal Cancer: How to Get from Meta-Omics to Mechanism? Trends Microbiol. 2020, 28, 401–423. [Google Scholar] [CrossRef]
- Daglia, M. Polyphenols as Antimicrobial Agents. Curr. Opin. Biotechnol. 2012, 23, 174–181. [Google Scholar] [CrossRef]
- Bittencourt, M.L.F.; Rodrigues, R.P.; Kitagawa, R.R.; Gonçalves, R.D.C.R. The Gastroprotective Potential of Silibinin against Helicobacter pylori Infection and Gastric Tumor Cells. Life Sci. 2020, 256, 117977. [Google Scholar] [CrossRef]
- Escandón, R.A.; del Campo, M.; López-Solis, R.; Obreque-Slier, E.; Toledo, H. Antibacterial Effect of Kaempferol and (−)-Epicatechin on Helicobacter pylori. Eur. Food Res. Technol. 2016, 242, 1495–1502. [Google Scholar] [CrossRef]
- Martini, S.; D’Addario, C.; Colacevich, A.; Focardi, S.; Borghini, F.; Santucci, A.; Figura, N.; Rossi, C. Antimicrobial Activity against Helicobacter pylori Strains and Antioxidant Properties of Blackberry Leaves (Rubus ulmifolius) and Isolated Compounds. Int. J. Antimicrob. Agents 2009, 34, 50–59. [Google Scholar] [CrossRef]
- Chen, M.; Su, C.; Yang, J.; Lu, C.; Hou, Y.; Wu, J.; Hsu, Y. Baicalin, Baicalein, and Lactobacillus Rhamnosus JB3 Alleviated Helicobacter pylori Infections in Vitro and in Vivo. J. Food Sci. 2018, 83, 3118–3125. [Google Scholar] [CrossRef] [PubMed]
- Mandalari, G.; Bisignano, C.; Cirmi, S.; Navarra, M. Effectiveness of Citrus Fruits on Helicobacter pylori. Evid.-Based Complement Altern. Med. 2017, 2017, 8379262. [Google Scholar] [CrossRef] [PubMed]
- Park, J.U.; Cho, J.S.; Kim, J.S.; Kim, H.K.; Jo, Y.H.; Rahman, M.A.A.; Lee, Y.I. Synergistic Effect of Rubus crataegifolius and Ulmus macrocarpa against Helicobacter pylori Clinical Isolates and Gastritis. Front. Pharmacol. 2020, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- Ranilla, L.G.; Apostolidis, E.; Shetty, K. Antimicrobial Activity of an Amazon Medicinal Plant (Chancapiedra) (Phyllanthus niruri L.) against Helicobacter pylori and Lactic Acid Bacteria: Phyllanthus niruri L. and antimicrobial activity. Phytother. Res. 2012, 26, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Kareem, S.M.; Mahmood, S.S.; Hindi, N.K. Effects of Curcumin and Silymarin on the Shigella dysenteriae and Campylobacter jejuni In Vitro. J. Gastrointest. Cancer 2020, 51, 824–828. [Google Scholar] [CrossRef]
- Castillo, S.; Heredia, N.; García, S. 2(5H)-Furanone, Epigallocatechin Gallate, and a Citric-Based Disinfectant Disturb Quorum-Sensing Activity and Reduce Motility and Biofilm Formation of Campylobacter jejuni. Folia Microbiol. 2015, 60, 89–95. [Google Scholar] [CrossRef]
- Lobo de Sá, F.D.; Heimesaat, M.M.; Bereswill, S.; Nattramilarasu, P.K.; Schulzke, J.D.; Bücker, R. Resveratrol Prevents Campylobacter jejuni-Induced Leaky Gut by Restoring Occludin and Claudin-5 in the Paracellular Leak Pathway. Front. Pharmacol. 2021, 12, 640572. [Google Scholar] [CrossRef]
- Heimesaat, M.M.; Mousavi, S.; Escher, U.; Lobo de Sá, F.D.; Peh, E.; Schulzke, J.-D.; Kittler, S.; Bücker, R.; Bereswill, S. Resveratrol Alleviates Acute Campylobacter jejuni Induced Enterocolitis in a Preclinical Murine Intervention Study. Microorganisms 2020, 8, 1858. [Google Scholar] [CrossRef]
- Ben Lagha, A.; Haas, B.; Grenier, D. Tea Polyphenols Inhibit the Growth and Virulence Properties of Fusobacterium nucleatum. Sci. Rep. 2017, 7, 44815. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.R.I.; Preshaw, P.M.; Lim, L.P.; Ong, M.M.A.; Lin, H.-S.; Tan, K.S. Pterostilbene Complexed with Cyclodextrin Exerts Antimicrobial and Anti-Inflammatory Effects. Sci. Rep. 2020, 10, 9072. [Google Scholar] [CrossRef]
- Andrade, F.D.O.; Liu, F.; Zhang, X.; Rosim, M.P.; Dani, C.; Cruz, I.; Wang, T.T.Y.; Helferich, W.; Li, R.W.; Hilakivi-Clarke, L. Genistein Reduces the Risk of Local Mammary Cancer Recurrence and Ameliorates Alterations in the Gut Microbiota in the Offspring of Obese Dams. Nutrients 2021, 13, 201. [Google Scholar] [CrossRef] [PubMed]
- Fernández, J.; García, L.; Monte, J.; Villar, C.; Lombó, F. Functional Anthocyanin-Rich Sausages Diminish Colorectal Cancer in an Animal Model and Reduce Pro-Inflammatory Bacteria in the Intestinal Microbiota. Genes 2018, 9, 133. [Google Scholar] [CrossRef]
- McFadden, R.-M.T.; Larmonier, C.B.; Shehab, K.W.; Midura-Kiela, M.; Ramalingam, R.; Harrison, C.A.; Besselsen, D.G.; Chase, J.H.; Caporaso, J.G.; Jobin, C.; et al. The Role of Curcumin in Modulating Colonic Microbiota during Colitis and Colon Cancer Prevention. Inflamm. Bowel Dis. 2015, 21, 2483–2494. [Google Scholar] [CrossRef]
- Chen, L.; Jiang, B.; Zhong, C.; Guo, J.; Zhang, L.; Mu, T.; Zhang, Q.; Bi, X. Chemoprevention of Colorectal Cancer by Black Raspberry Anthocyanins Involved the Modulation of Gut Microbiota and SFRP2 Demethylation. Carcinogenesis 2018, 39, 471–481. [Google Scholar] [CrossRef]
- Wu, J.-C.; Tsai, M.-L.; Lai, C.-S.; Lo, C.-Y.; Ho, C.-T.; Wang, Y.-J.; Pan, M.-H. Polymethoxyflavones Prevent Benzo[a]Pyrene/Dextran Sodium Sulfate-Induced Colorectal Carcinogenesis through Modulating Xenobiotic Metabolism and Ameliorate Autophagic Defect in ICR Mice: PMFs Prevent B a P/DSS-Induced Colorectal Carcinogenesis. Int. J. Cancer 2018, 142, 1689–1701. [Google Scholar] [CrossRef]
- Wu, M.; Wu, Y.; Deng, B.; Li, J.; Cao, H.; Qu, Y.; Qian, X.; Zhong, G. Isoliquiritigenin Decreases the Incidence of Colitis-Associated Colorectal Cancer by Modulating the Intestinal Microbiota. Oncotarget 2016, 7, 85318–85331. [Google Scholar] [CrossRef]
- Gong, Y.; Dong, R.; Gao, X.; Li, J.; Jiang, L.; Zheng, J.; Cui, S.; Ying, M.; Yang, B.; Cao, J.; et al. Neohesperidin Prevents Colorectal Tumorigenesis by Altering the Gut Microbiota. Pharmacol. Res. 2019, 148, 104460. [Google Scholar] [CrossRef]
- Flemer, B.; Lynch, D.B.; Brown, J.M.R.; Jeffery, I.B.; Ryan, F.J.; Claesson, M.J.; O’Riordain, M.; Shanahan, F.; O’Toole, P.W. Tumour-Associated and Non-Tumour-Associated Microbiota in Colorectal Cancer. Gut 2017, 66, 633–643. [Google Scholar] [CrossRef]
- Wang, X.; Ye, T.; Chen, W.-J.; Lv, Y.; Hao, Z.; Chen, J.; Zhao, J.-Y.; Wang, H.-P.; Cai, Y.-K. Structural shift of gut microbiota during chemo-preventive effects of epigallocatechin gallate on colorectal carcinogenesis in mice. World J. Gastroenterol. 2017, 23, 8128–8139. [Google Scholar] [CrossRef]
- Messaoudene, M.; Pidgeon, R.; Richard, C.; Ponce, M.; Diop, K.; Benlaifaoui, M.; Nolin-Lapalme, A.; Cauchois, F.; Malo, J.; Belkaid, W.; et al. A Natural Polyphenol Exerts Antitumor Activity and Circumvents Anti–PD-1 Resistance through Effects on the Gut Microbiota. Cancer Discov. 2022, 12, 1070–1087. [Google Scholar] [CrossRef]
- Jain, N. The Early Life Education of the Immune System: Moms, Microbes and (Missed) Opportunities. Gut Microbes 2020, 12, 1824564. [Google Scholar] [CrossRef]
- Minarrieta, L.; Ghorbani, P.; Sparwasser, T.; Berod, L. Metabolites: Deciphering the Molecular Language between DCs and Their Environment. Semin. Immunopathol. 2017, 39, 177–198. [Google Scholar] [CrossRef]
- Suzuki, H.; Jeong, K.I.; Itoh, K.; Doi, K. Regional Variations in the Distributions of Small Intestinal Intraepithelial Lymphocytes in Germ-Free and Specific Pathogen-Free Mice. Exp. Mol. Pathol. 2002, 72, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Sefik, E.; Geva-Zatorsky, N.; Oh, S.; Konnikova, L.; Zemmour, D.; McGuire, A.M.; Burzyn, D.; Ortiz-Lopez, A.; Lobera, M.; Yang, J.; et al. Individual Intestinal Symbionts Induce a Distinct Population of RORγ+ Regulatory T Cells. Science 2015, 349, 993–997. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, D.; Sefik, E.; Galván-Peña, S.; Wu, M.; Yang, L.; Yang, Z.; Kostic, A.; Golovkina, T.V.; Kasper, D.L.; Mathis, D.; et al. An Immunologic Mode of Multigenerational Transmission Governs a Gut Treg Setpoint. Cell 2020, 181, 1276–1290.e13. [Google Scholar] [CrossRef] [PubMed]
- Mandaliya, D.K.; Patel, S.; Seshadri, S. The Combinatorial Effect of Acetate and Propionate on High-Fat Diet Induced Diabetic Inflammation or Metaflammation and T Cell Polarization. Inflammation 2021, 44, 68–79. [Google Scholar] [CrossRef]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly-Y, M.; Glickman, J.N.; Garrett, W.S. The Microbial Metabolites, Short-Chain Fatty Acids, Regulate Colonic Treg Cell Homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef]
- Hang, S.; Paik, D.; Yao, L.; Kim, E.; Trinath, J.; Lu, J.; Ha, S.; Nelson, B.N.; Kelly, S.P.; Wu, L.; et al. Bile Acid Metabolites Control TH17 and Treg Cell Differentiation. Nature 2019, 576, 143–148. [Google Scholar] [CrossRef]
- Song, X.; Sun, X.; Oh, S.F.; Wu, M.; Zhang, Y.; Zheng, W.; Geva-Zatorsky, N.; Jupp, R.; Mathis, D.; Benoist, C.; et al. Microbial Bile Acid Metabolites Modulate Gut RORγ+ Regulatory T Cell Homeostasis. Nature 2020, 577, 410–415. [Google Scholar] [CrossRef]
- Son, B.; Lee, S.; Youn, H.; Kim, E.; Kim, W.; Youn, B. The Role of Tumor Microenvironment in Therapeutic Resistance. Oncotarget 2017, 8, 3933–3945. [Google Scholar] [CrossRef]
- Prager, I.; Watzl, C. Mechanisms of Natural Killer Cell-Mediated Cellular Cytotoxicity. J. Leukoc. Biol. 2019, 105, 1319–1329. [Google Scholar] [CrossRef]
- Martínez-Lostao, L.; Anel, A.; Pardo, J. How Do Cytotoxic Lymphocytes Kill Cancer Cells? Clin. Cancer Res. 2015, 21, 5047–5056. [Google Scholar] [CrossRef]
- Kroemer, G.; Zitvogel, L. The Breakthrough of the Microbiota. Nat. Rev. Immunol. 2018, 18, 87–88. [Google Scholar] [CrossRef] [PubMed]
- Focaccetti, C.; Izzi, V.; Benvenuto, M.; Fazi, S.; Ciuffa, S.; Giganti, M.G.; Potenza, V.; Manzari, V.; Modesti, A.; Bei, R. Polyphenols as Immunomodulatory Compounds in the Tumor Microenvironment: Friends or Foes? IJMS 2019, 20, 1714. [Google Scholar] [CrossRef]
- Liu, D.; You, M.; Xu, Y.; Li, F.; Zhang, D.; Li, X.; Hou, Y. Inhibition of Curcumin on Myeloid-Derived Suppressor Cells Is Requisite for Controlling Lung Cancer. Int. Immunopharmacol. 2016, 39, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Halder, R.C.; Almasi, A.; Sagong, B.; Leung, J.; Jewett, A.; Fiala, M. Curcuminoids and ω-3 Fatty Acids with Anti-Oxidants Potentiate Cytotoxicity of Natural Killer Cells against Pancreatic Ductal Adenocarcinoma Cells and Inhibit Interferon γ Production. Front. Physiol. 2015, 6, 129. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Shin, H.; Kim, J. In Vivo Anti-Cancer Effects of Resveratrol Mediated by NK Cell Activation. J. Innate Immun. 2021, 13, 94–106. [Google Scholar] [CrossRef]
- Zhang, X.; Cook, K.L.; Warri, A.; Cruz, I.M.; Rosim, M.; Riskin, J.; Helferich, W.; Doerge, D.; Clarke, R.; Hilakivi-Clarke, L. Lifetime Genistein Intake Increases the Response of Mammary Tumors to Tamoxifen in Rats. Clin. Cancer Res. 2017, 23, 814–824. [Google Scholar] [CrossRef]
- Li, B.; Chan, H.L.; Chen, P. Immune Checkpoint Inhibitors: Basics and Challenges. Curr. Med. Chem. 2019, 26, 3009–3025. [Google Scholar] [CrossRef]
- Zhou, Z.; Liu, Y.; Song, W.; Jiang, X.; Deng, Z.; Xiong, W.; Shen, J. Metabolic Reprogramming Mediated PD-L1 Depression and Hypoxia Reversion to Reactivate Tumor Therapy. J. Control. Release 2022, 352, 793–812. [Google Scholar] [CrossRef]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef]
- Verdura, S.; Cuyàs, E.; Cortada, E.; Brunet, J.; Lopez-Bonet, E.; Martin-Castillo, B.; Bosch-Barrera, J.; Encinar, J.A.; Menendez, J.A. Resveratrol Targets PD-L1 Glycosylation and Dimerization to Enhance Antitumor T-Cell Immunity. Aging 2020, 12, 8–34. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, Y.; Tian, K.; Chen, X.; Zhang, R.; Mu, X.; Wu, Y.; Wang, D.; Wang, S.; Liu, F.; et al. Apigenin Suppresses PD-L1 Expression in Melanoma and Host Dendritic Cells to Elicit Synergistic Therapeutic Effects. J. Exp. Clin. Cancer Res. 2018, 37, 261. [Google Scholar] [CrossRef]
- Jiang, Z.-B.; Wang, W.-J.; Xu, C.; Xie, Y.-J.; Wang, X.-R.; Zhang, Y.-Z.; Huang, J.-M.; Huang, M.; Xie, C.; Liu, P.; et al. Luteolin and Its Derivative Apigenin Suppress the Inducible PD-L1 Expression to Improve Anti-Tumor Immunity in KRAS-Mutant Lung Cancer. Cancer Lett. 2021, 515, 36–48. [Google Scholar] [CrossRef]
- Liao, F.; Liu, L.; Luo, E.; Hu, J. Curcumin Enhances Anti-Tumor Immune Response in Tongue Squamous Cell Carcinoma. Arch. Oral Biol. 2018, 92, 32–37. [Google Scholar] [CrossRef]
- Guo, L.; Li, H.; Fan, T.; Ma, Y.; Wang, L. Synergistic Efficacy of Curcumin and Anti-Programmed Cell Death-1 in Hepatocellular Carcinoma. Life Sci. 2021, 279, 119359. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Sun, Q.; Liu, Q.; Li, H.; Zhang, W.; Sun, C. Focus on Immune Checkpoint PD-1/PD-L1 Pathway: New Advances of Polyphenol Phytochemicals in Tumor Immunotherapy. Biomed. Pharmacother. 2022, 154, 113618. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Li, Z.; Tao, J.; Hu, H.; Li, Z.; Zhang, Z.; Cheng, F.; Sun, Y.; Zhang, Y.; Yang, J.; et al. Resveratrol Induces PD-L1 Expression through Snail-Driven Activation of Wnt Pathway in Lung Cancer Cells. J. Cancer Res. Clin. Oncol. 2021, 147, 1101–1113. [Google Scholar] [CrossRef]
- Kawabata, K.; Yoshioka, Y.; Terao, J. Role of Intestinal Microbiota in the Bioavailability and Physiological Functions of Dietary Polyphenols. Molecules 2019, 24, 370. [Google Scholar] [CrossRef]
- Mohos, V.; Fliszár-Nyúl, E.; Lemli, B.; Zsidó, B.Z.; Hetényi, C.; Mladěnka, P.; Horký, P.; Pour, M.; Poór, M. Testing the Pharmacokinetic Interactions of 24 Colonic Flavonoid Metabolites with Human Serum Albumin and Cytochrome P450 Enzymes. Biomolecules 2020, 10, 409. [Google Scholar] [CrossRef] [PubMed]
- Fatima, A.; Khan, M.S.; Ahmad, M.W. Therapeutic Potential of Equol: A Comprehensive Review. Curr. Pharm. Des. 2020, 26, 5837–5843. [Google Scholar] [CrossRef]
- Tuli, H.S.; Kumar, A.; Sak, K.; Aggarwal, D.; Gupta, D.S.; Kaur, G.; Vashishth, K.; Dhama, K.; Kaur, J.; Saini, A.K.; et al. Gut Microbiota-Assisted Synthesis, Cellular Interactions and Synergistic Perspectives of Equol as a Potent Anticancer Isoflavone. Pharmaceuticals 2022, 15, 1418. [Google Scholar] [CrossRef]
- Rogovskii, V.S. The Therapeutic Potential of Urolithin A for Cancer Treatment AndPrevention. Curr. Cancer Drug Targets 2022, 22, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Al-Harbi, S.A.; Abdulrahman, A.O.; Zamzami, M.A.; Khan, M.I. Urolithins: The Gut Based Polyphenol Metabolites of Ellagitannins in Cancer Prevention, a Review. Front. Nutr. 2021, 8, 647582. [Google Scholar] [CrossRef]
- Zhang, J.; Ren, L.; Yu, M.; Liu, X.; Ma, W.; Huang, L.; Li, X.; Ye, X. S-Equol Inhibits Proliferation and Promotes Apoptosis of Human Breast Cancer MCF-7 Cells via Regulating MiR-10a-5p and PI3K/AKT Pathway. Arch. Biochem. Biophys. 2019, 672, 108064. [Google Scholar] [CrossRef]
- Zou, Y.; Wang, Y.; Cai, Y.; Ma, D. Effects of equol on proliferation of colorectal cancer HCT-15 cell. Wei Sheng Yan Jiu 2019, 48, 803–806. [Google Scholar] [PubMed]
- Magee, P.J.; Allsopp, P.; Samaletdin, A.; Rowland, I.R. Daidzein, R-(+)Equol and S-(-)Equol Inhibit the Invasion of MDA-MB-231 Breast Cancer Cells Potentially via the down-Regulation of Matrix Metalloproteinase-2. Eur. J. Nutr. 2014, 53, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Bao, C.; Namgung, H.; Lee, J.; Park, H.-C.; Ko, J.; Moon, H.; Ko, H.W.; Lee, H.J. Daidzein Suppresses Tumor Necrosis Factor-α Induced Migration and Invasion by Inhibiting Hedgehog/Gli1 Signaling in Human Breast Cancer Cells. J. Agric. Food Chem. 2014, 62, 3759–3767. [Google Scholar] [CrossRef]
- Brown, N.M.; Belles, C.A.; Lindley, S.L.; Zimmer-Nechemias, L.D.; Zhao, X.; Witte, D.P.; Kim, M.O.; Setchell, K.D.R. The Chemopreventive Action of Equol Enantiomers in a Chemically Induced Animal Model of Breast Cancer. Carcinogenesis 2010, 31, 886–893. [Google Scholar] [CrossRef]
- Yu, X.; Zou, Y.Q.; Wang, Y.; Chen, Z.K.; Ma, D.F. Equol and its enantiomers inhibited urethane-induced lung cancer in mice. Beijing Da Xue Xue Bao Yi Xue Ban 2022, 54, 244–248. [Google Scholar]
- González-Sarrías, A.; Núñez-Sánchez, M.Á.; García-Villalba, R.; Tomás-Barberán, F.A.; Espín, J.C. Antiproliferative Activity of the Ellagic Acid-Derived Gut Microbiota Isourolithin A and Comparison with Its Urolithin A Isomer: The Role of Cell Metabolism. Eur. J. Nutr. 2017, 56, 831–841. [Google Scholar] [CrossRef]
- González-Sarrías, A.; Núñez-Sánchez, M.Á.; Tomé-Carneiro, J.; Tomás-Barberán, F.A.; García-Conesa, M.T.; Espín, J.C. Comprehensive Characterization of the Effects of Ellagic Acid and Urolithins on Colorectal Cancer and Key-Associated Molecular Hallmarks: MicroRNA Cell Specific Induction of CDKN1A (P21) as a Common Mechanism Involved. Mol. Nutr. Food Res. 2016, 60, 701–716. [Google Scholar] [CrossRef]
- Mohammed Saleem, Y.I.; Albassam, H.; Selim, M. Urolithin A Induces Prostate Cancer Cell Death in P53-Dependent and in P53-Independent Manner. Eur. J. Nutr. 2020, 59, 1607–1618. [Google Scholar] [CrossRef] [PubMed]
- El-Wetidy, M.S.; Ahmad, R.; Rady, I.; Helal, H.; Rady, M.I.; Vaali-Mohammed, M.-A.; Al-Khayal, K.; Traiki, T.B.; Abdulla, M.-H. Urolithin A Induces Cell Cycle Arrest and Apoptosis by Inhibiting Bcl-2, Increasing P53-P21 Proteins and Reactive Oxygen Species Production in Colorectal Cancer Cells. Cell Stress Chaperones 2021, 26, 473–493. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-F.; Li, X.-L.; Zhang, Z.-L.; Qiu, L.; Ding, S.-X.; Xue, J.-X.; Zhao, G.-P.; Li, J. Antiaging Effects of Urolithin A on Replicative Senescent Human Skin Fibroblasts. Rejuvenation Res. 2019, 22, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Djedjibegovic, J.; Marjanovic, A.; Panieri, E.; Saso, L. Ellagic Acid-Derived Urolithins as Modulators of Oxidative Stress. Oxid. Med. Cell. Longev. 2020, 2020, 5194508. [Google Scholar] [CrossRef]
- Si, W.; Zhang, Y.; Li, X.; Du, Y.; Xu, Q. Understanding the Functional Activity of Polyphenols Using Omics-Based Approaches. Nutrients 2021, 13, 3953. [Google Scholar] [CrossRef] [PubMed]
- Di Lorenzo, C.; Colombo, F.; Biella, S.; Stockley, C.; Restani, P. Polyphenols and Human Health: The Role of Bioavailability. Nutrients 2021, 13, 273. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Dong, Y.; Wang, F.; Zhang, Y. Nanoformulations to Enhance the Bioavailability and Physiological Functions of Polyphenols. Molecules 2020, 25, 4613. [Google Scholar] [CrossRef]




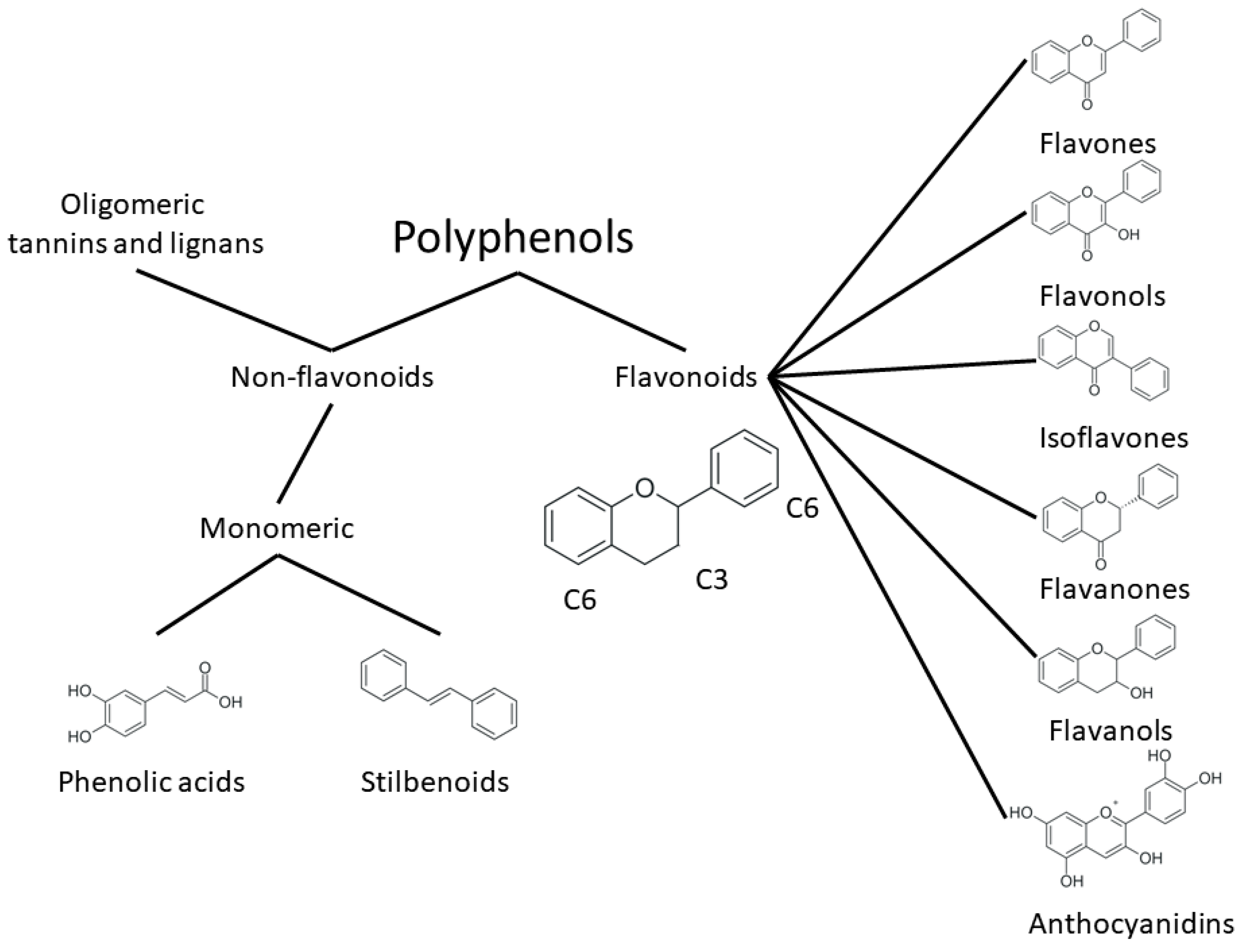{kind=link}
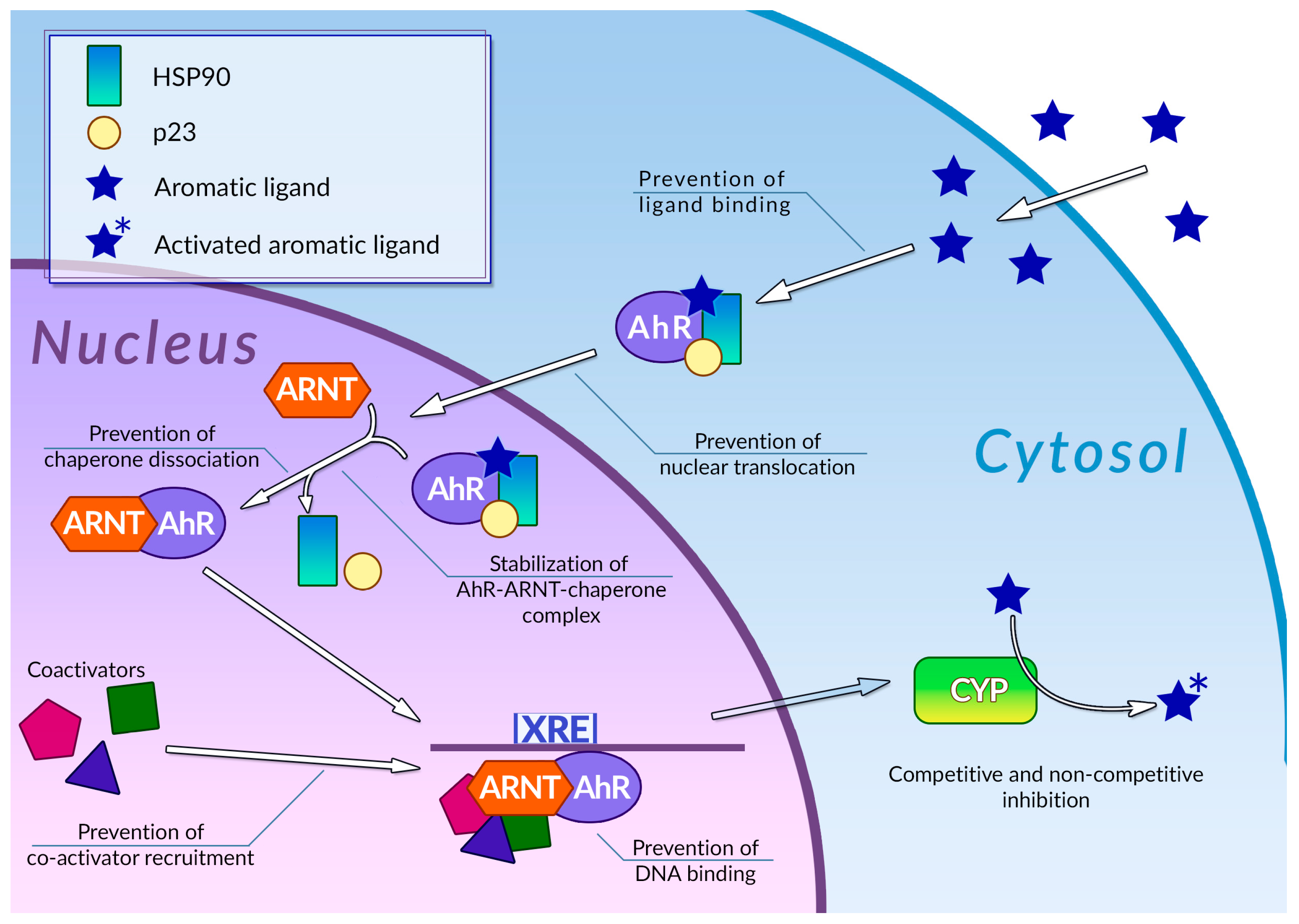{kind=link}
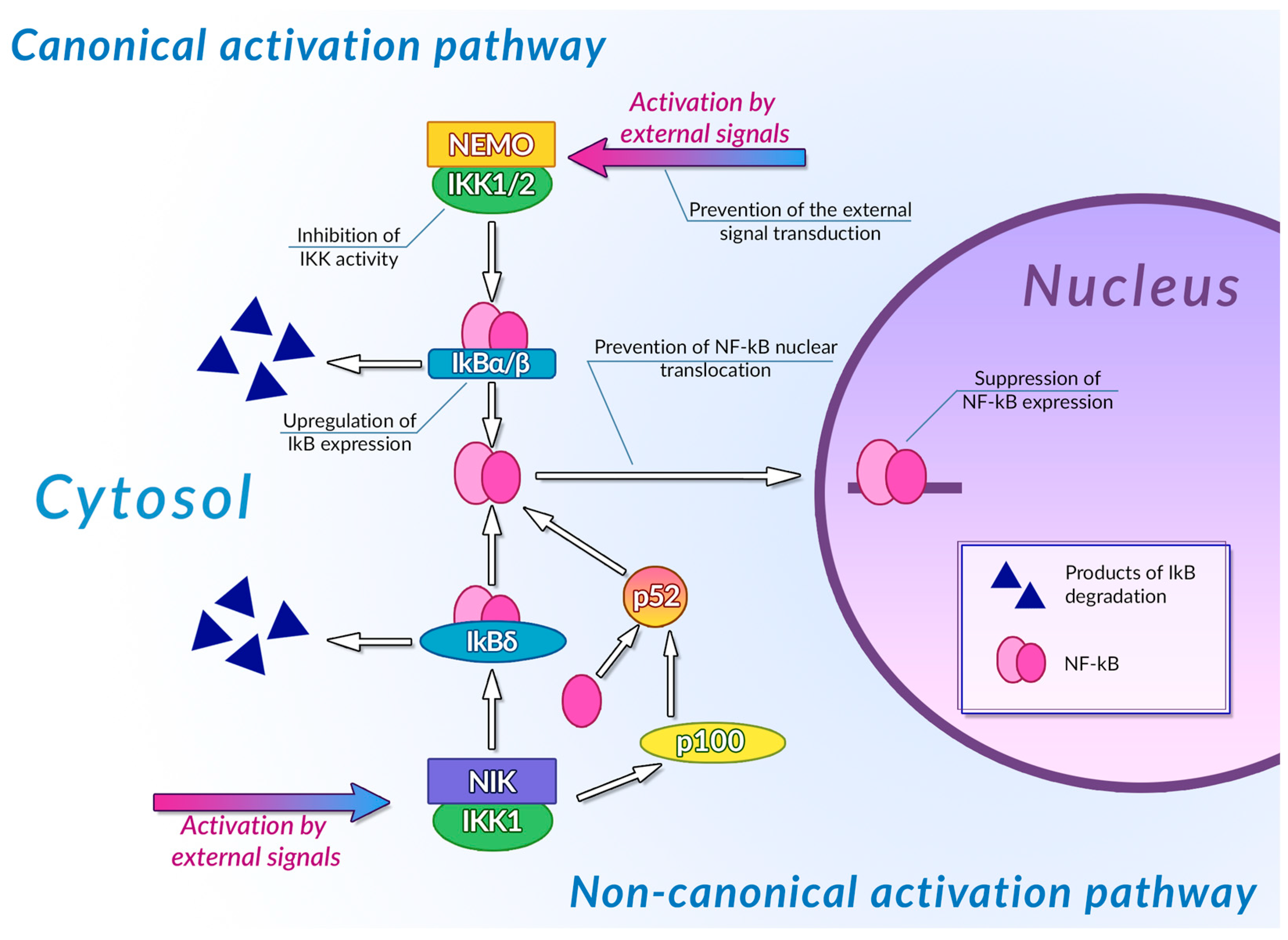{kind=link}
| Compound | Structure | Mechanism | Reference |
|---|---|---|---|
| Curcumin | Phenolic acid homodimer | Direct competitive inhibition of AhR in cell-free extracts | [92] |
| Quercetin | Flavonol | [93] | |
| Resveratrol | Stilbenoid | [94] | |
| Flavone | Flavone | [95] | |
| Luteolin | Flavone | ||
| Apigenin | Flavone | ||
| Galangin | Flavonol | ||
| Kaempferol | Flavonol | Inhibition of AhR in Caco2 cell culture | [96] |
| Luteolin | Flavone | ||
| Apigenin | Flavone | ||
| Fisetin | Flavonol | ||
| Quercetin | Flavonol | Agonism of AhR in Caco2 cell culture | |
| Robinetin | Flavone | ||
| Taxifolin | Flavonol | ||
| Morin | Flavone | ||
| Quercetin | Flavonol | Stimulation of AhR-regulated luciferase gene expression in H1L6.1c2 reporter cell culture | [97] |
| Naringin | Glycosylated flavanone | ||
| Hesperidin | Glycosylated flavanone | ||
| Hesperitin | Flavanone | ||
| Galangin | Flavonol | Prevention of nuclear translocation of AhR in MCF-7 and Hepa-1c1c7 cell cultures | [100] |
| Apigenin | Flavone | ||
| Luteolin | Flavone | ||
| Flavone | Flavone | ||
| Kaempferol | Flavonol | ||
| Curcumin | Phenolic acid homodimer | [101] | |
| Resveratrol | Stilbenoid | [94] | |
| Naringenin | Flavanone | Prevention of active AhR-Arnt heterodimer formation in cell cultures | [100] |
| Apigenin | Flavone | ||
| Kaempferol | Flavonol | ||
| Galangin | Flavonol | [102] | |
| EGCG | Flavanol and phenolic acid heterodimer | [100,103] | |
| Curcumin | Phenolic acid homodimer | Prevention of XRE binding by AhR-Arnt dimer in tumor cell cultures | [101] |
| Galangin | Flavonol | [102] | |
| Resveratrol | Stilbenoid | [104] | |
| Quercetin | Flavonol | [93] | |
| Kaempferol | Flavonol | ||
| Resveratrol | Stilbenoid | Prevention of AhR-Arnt coactivator recruitment | [94] |
| Genistein | Isoflavone | [105] | |
| Kaempferol | Flavonol | [106] |
| Compound | Structure | Mechanism | Reference |
|---|---|---|---|
| Curcumin |  | Downregulation of DNMT DNA methyltransferases expression | [164,165] |
| Downregulation of histone deacetylase expression | [177,178] | ||
| Modulation of various miRNAs expression | [196] | ||
| Downregulation of miR-21 expression | [197,198,199,200,201,202] | ||
| Downregulation of miR-19a and -19b | [203] | ||
| Modulation of various miRNAs expression | [204] | ||
| Downregulation of miR-136 and -186* | [205] | ||
| Upregulation of miR-34 | [206] | ||
| Upregulation of miR-98 | [207] | ||
| EGCG |  | Downregulation of DNMT DNA methyltransferases expression | [160] |
| Inhibition of DNMT catalytic activity | [161] | ||
| Inhibition of HDAC 1-3 histone deacetylase expression | [174,175] | ||
| Inhibition of histone acetyltransferase catalytic activity | [176] | ||
| Modulation of various miRNAs expression, including miR-16 | [190] | ||
| Downregulation of miR-92, -93 and -106b, upregulation of miR-7-1, -34a and -99a | [191] | ||
| Upregulation of miR-210 expression | [192] | ||
| Downregulation of miR-25 expression | [193] | ||
| Upregulation of let-7 family miRNAs expression | [194] | ||
| Upregulation of miR-let7a (in combination with quercetin) | [195] | ||
| Quercetin |  | Inhibition of DNMT catalytic activity | [167] |
| Inhibition of histone deacetylase and increase in histone acetyltransferase activity | [179] | ||
| Inhibition of p300 acetyltransferase activity | [180] | ||
| Upregulation of miR-16 | [208] | ||
| Downregulation of miR-16 | [209] | ||
| Upregulation of miR-34 | [210] | ||
| Upregulation of miR-146a | [211] | ||
| Downregulation of miR-21 | [212] | ||
| Upregulation of let-7c | [213] | ||
| Downregulation of miR-27a (in combination with resveratrol) | [214] | ||
| Resveratrol |  | Inhibition of DNA methyltransferase recruitment by SIRT1 | [168] |
| Selective reversion of hypermethylation of tumor suppressor genes (in combination with pterostilbene) | [169,170] | ||
| Recruitment of DNMT3b DNA methyltransferase to oncogenes | [169] | ||
| Enhancement of SIRT1 histone deacetylase activity | [181] | ||
| Modulation of NuRD histone deacetylase complex activity | [182,183] | ||
| Downregulation of miR-19, -21 and -30-a5p | [215] | ||
| Downregulation of miR-21 | [216,217,218] | ||
| Upregulation of miR-34a and -34c | [219,220] | ||
| Modulation of various miRNAs expression, including miR-20a-5p, -140-5p, -125b-1-3p, -199a-5p, -122-5p and -542-3p | [221] | ||
| Genistein |  | Inhibition of DNMT catalytic activity | [171,172,173] |
| Promotion of histone acetylation at regulatory regions of tumor suppressor genes | [171,172,184,185] | ||
| Downregulation of miR-27a | [222,223] | ||
| Upregulation of miR-27a | [224] | ||
| Upregulation of miR-23b | [225] | ||
| Downregulation of miR-151 | [226] | ||
| Downregulation of miR-155 | [227] | ||
| Upregulation of miR-34a | [228,229] |
| Compound | Structure | Mechanism | Reference |
|---|---|---|---|
| Curcumin |  | Prevention of IκB phosphorylation | [250] |
| Reduction in NF-κB DNA binding efficiency | [251] | ||
| Reduction in NF-κB level | [252] | ||
| Downregulation of NF-κB and IKK expression | [253] | ||
| Downregulation of NF-κB expression | [254] | ||
| Downregulation of NF-κB and suppression of IκB phosphorylation | [255] | ||
| Prevention of NF-κB activation and nuclear translocation | [256] | ||
| EGCG |  | General decrease in NF-κB activity | [242] |
| Inhibition of IKK activity | [243,245] | ||
| Prevention of IRAK receptor-asociated kinase degradation | [244] | ||
| Inhibition of TAK-1 TNF-β-dependent kinase | [246,247] | ||
| Inhibition of NF-κB activity by binding to IKK inhibitor binding site | [248] | ||
| Prevention of NF-κB interaction with DNA | [249] | ||
| Quercetin |  | Downregulation of NF-κB p65 expression | [257] |
| Upregulation of IκBα expression | [258] | ||
| Downregulation of NF-κB expression, combined with upregulation of IκB expression | [259] | ||
| Downregulation of NF-κB and IκB expression, reduction in IKK1/2 phosphorylation | [260] | ||
| Downregulation of NF-κB p65 and p50 expression | [261] | ||
| Prevention of upregulation of NF-κB expression | [262] | ||
| Resveratrol |  | Decrease in NF-κB p65 expression | [263] |
| Prevention ov NF-κB dimerization and nuclear translocation | [264] | ||
| Downregulation of NF-κB expression | [265] | ||
| Suppression of NF-κB activity | [266] | ||
| Promotion of NF-κB-inhibiting activity of SIRT1 | [267] | ||
| Inhibition of NF-κB activation, combined with an increased secretion of IL-8 | [268] | ||
| Promotion of NF-κB expression and nuclear translocation, stimulation of IκB phosphorylation | [269] | ||
| Genistein |  | Suppression of NF-κB expression by upregulation of miR-29 | [270] |
| Downregulation of NF-κB expression | [271,272] | ||
| Downregulation of NF-κB expression by suppression of Notch-1 activity | [273,274] | ||
| Inhibition of IκB and NF-κB phosphorylation, prevention of NF-κB nuclear translocation | [275] | ||
| Activation of IKK, increase in NF-κB phosphorylation (in combination with daidzein) | [276] | ||
| Facilitation of NF-κB activation and nuclear translocation | [277] |
| Compound | Structure | Mechanism | Reference |
|---|---|---|---|
| Silibinin | Flavonol and phenolic acid heterodimer | Suppression of H. pylori growth | [293] |
| Kaempferol | Flavonol | [294] | |
| (-)-epicatechin | Flavanol | ||
| Ellagic acid | Tannin | [295] | |
| Gallic acid | Phenolic acid | ||
| Quercitin | Favonol | ||
| Curcumin | Phenolic acid homodimer | Suppression of C. jejuni growth and prevention of C. jejuni-induced lesions | [300] |
| EGCG | Flavanol and phenolic acid heterodimer | [301] | |
| Resveratrol | Stilbene | [302,303] | |
| EGCG | Flavanol and phenolic acid heterodimer | Suppression of biofilm formation by F. nucleatum, combined with alleviation of inflammation induced by this microorganism | [304] |
| Theaflavines | Condensated flavanols | ||
| Resveratrol | Stilbene | [305] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lyubitelev, A.; Studitsky, V. Inhibition of Cancer Development by Natural Plant Polyphenols: Molecular Mechanisms. Int. J. Mol. Sci. 2023, 24, 10663. https://doi.org/10.3390/ijms241310663
Lyubitelev A, Studitsky V. Inhibition of Cancer Development by Natural Plant Polyphenols: Molecular Mechanisms. International Journal of Molecular Sciences. 2023; 24(13):10663. https://doi.org/10.3390/ijms241310663
Chicago/Turabian StyleLyubitelev, Alexander, and Vasily Studitsky. 2023. "Inhibition of Cancer Development by Natural Plant Polyphenols: Molecular Mechanisms" International Journal of Molecular Sciences 24, no. 13: 10663. https://doi.org/10.3390/ijms241310663
APA StyleLyubitelev, A., & Studitsky, V. (2023). Inhibition of Cancer Development by Natural Plant Polyphenols: Molecular Mechanisms. International Journal of Molecular Sciences, 24(13), 10663. https://doi.org/10.3390/ijms241310663