
Central and Peripheral Inflammation in Mild Cognitive Impairment in the Context of Alzheimer’s Disease



Abstract
1. Introduction
2. Results
2.1. Comparison of Alzheimer’s Disease Biomarkers: Amyloid and Tau Proteins
2.2. Neuroinflammation, Astrogliosis, and Microglia Activation Biomarkers in CSF and Serum
2.3. Systemic Inflammation Biomarkers
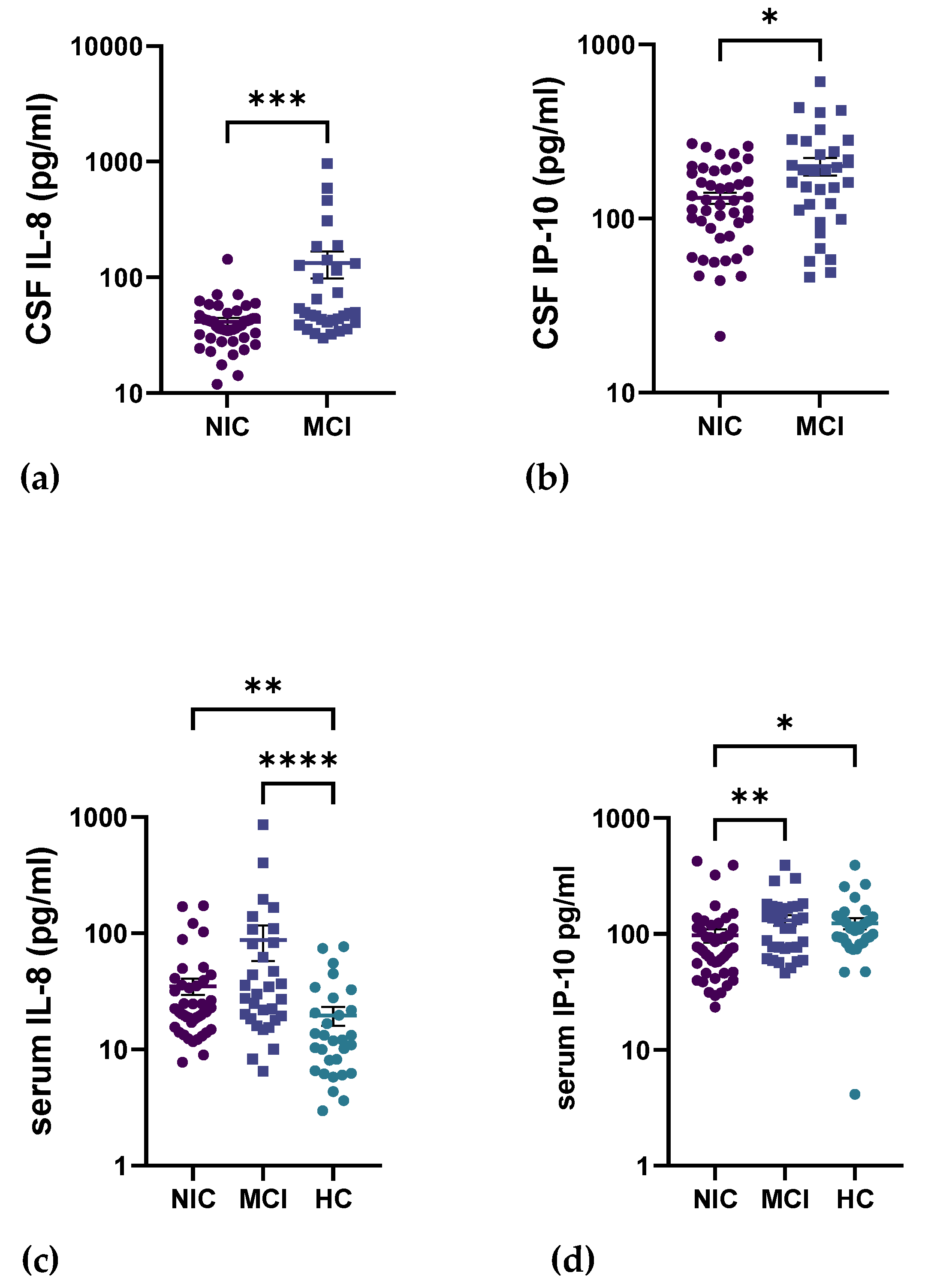
2.4. Circulating Inflammatory Cytokines in the CNS
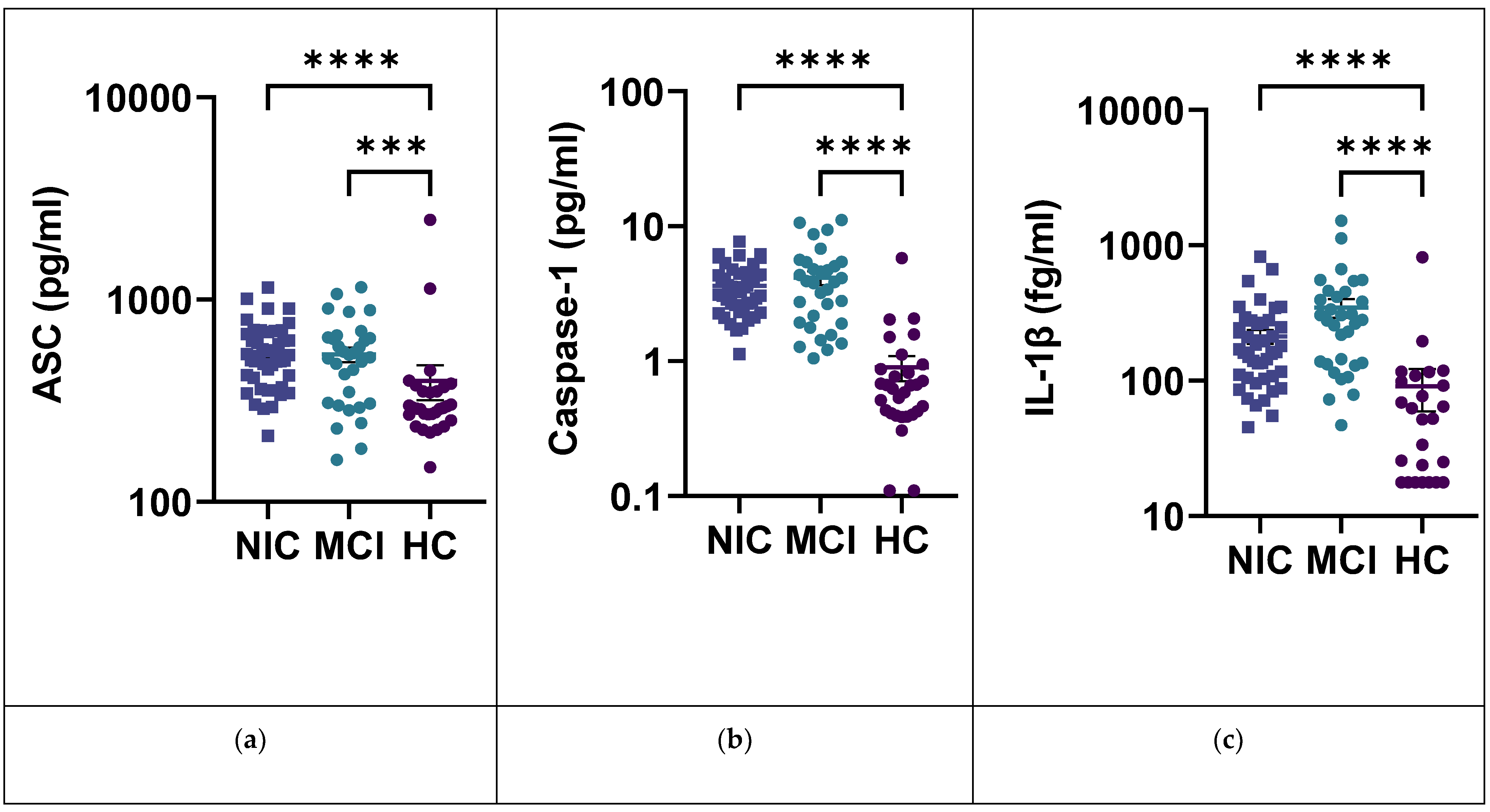
2.5. IL-1β and the Inflammasome Pathway
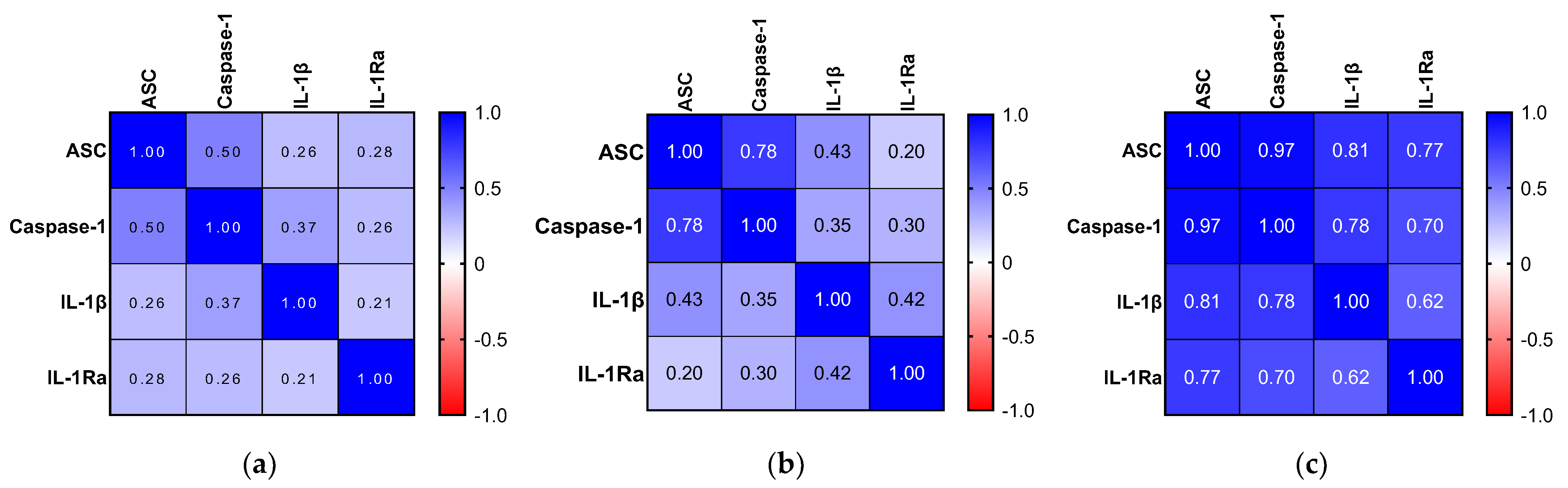
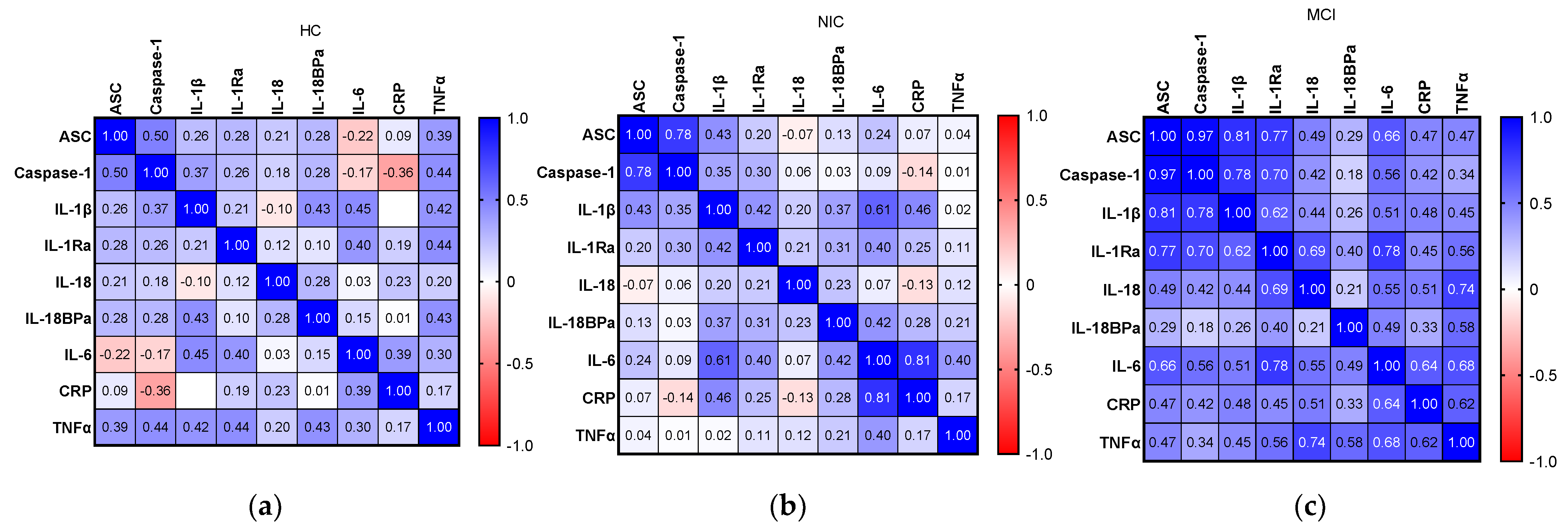
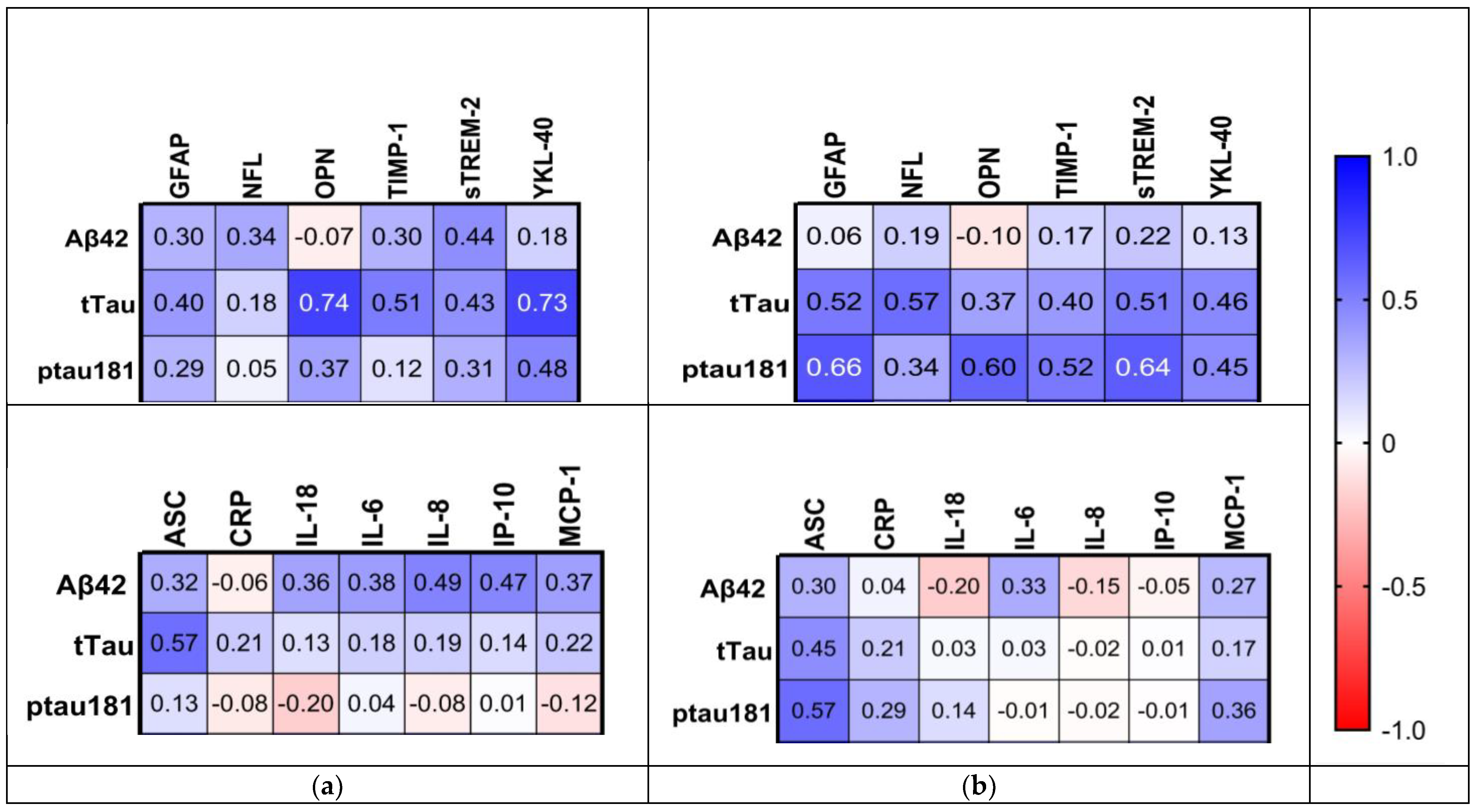
2.6. Relationship between Inflammatory Biomarkers and AD Hallmarks
3. Discussion
4. Materials and Methods
4.1. Participants
4.2. Ethical Consent
4.3. Sample Collection
4.4. CSF and Serum Analyses
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vega, J.N.; Newhouse, P.A. Mild Cognitive Impairment: Diagnosis, Longitudinal Course, and Emerging Treatments. Curr. Psychiatry Rep. 2014, 16, 490. [Google Scholar] [CrossRef]
- Mitchell, A.J.; Shiri-Feshki, M. Rate of Progression of Mild Cognitive Impairment to Dementia—Meta-Analysis of 41 Robust Inception Cohort Studies. Acta Psychiatr. Scand. 2009, 119, 252–265. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.Y.; Cui, K.; Liu, L.; Qin, Y.; Cui, J.; Han, H.J.; Luo, Y.H.; Yu, H.M. Screening and Predicting Progression from High-Risk Mild Cognitive Impairment to Alzheimer’s Disease. Sci. Rep. 2021, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- World Alzheimer Report 2021: Journey through the Diagnosis of Dementia; Alzheimer’s Disease International: Chicago, IL, USA, 2021.
- Winblad, B.; Amouyel, P.; Andrieu, S.; Ballard, C.; Brayne, C.; Brodaty, H.; Cedazo-Minguez, A.; Dubois, B.; Edvardsson, D.; Feldman, H.; et al. Defeating Alzheimer’s Disease and Other Dementias: A Priority for European Science and Society. Lancet Neurol. 2016, 15, 455–532. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2018, 14, 367–429. [Google Scholar] [CrossRef]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef]
- Cummings, J.L.; Tong, G.; Ballard, C. Treatment Combinations for Alzheimer’s Disease: Current and Future Pharmacotherapy Options. J. Alzheimer’s Dis. 2019, 67, 779. [Google Scholar] [CrossRef]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s Disease Drug-Development Pipeline: Few Candidates, Frequent Failures. Alzheimers Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef]
- Mullane, K.; Williams, M. Alzheimer’s Disease (AD) Therapeutics—1: Repeated Clinical Failures Continue to Question the Amyloid Hypothesis of AD and the Current Understanding of AD Causality. Biochem. Pharmacol. 2018, 158, 359–375. [Google Scholar] [CrossRef]
- Heneka, M.T.; O’Banion, M.K.; Terwel, D.; Kummer, M.P. Neuroinflammatory Processes in Alzheimer’s Disease. J. Neural Transm. 2010, 117, 919–947. [Google Scholar] [CrossRef]
- Heneka, M.T.; O’Banion, M.K. Inflammatory Processes in Alzheimer’s Disease. J. Neuroimmunol. 2007, 184, 69–91. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Cui, G.; Zhu, M.; Kang, X.; Guo, H. Neuroinflammation in Alzheimer’s Disease: Chemokines Produced by Astrocytes and Chemokine Receptors. Int. J. Clin. Exp. Pathol. 2014, 7, 8342. [Google Scholar]
- Zaheer, S.; Thangavel, R.; Wu, Y.; Khan, M.M.; Kempuraj, D.; Zaheer, A. Enhanced Expression of Glia Maturation Factor Correlates with Glial Activation in the Brain of Triple Transgenic Alzheimer’s Disease Mice. Neurochem. Res. 2013, 38, 218. [Google Scholar] [CrossRef]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s Disease. Neurobiol. Aging 2000, 21, 383. [Google Scholar] [CrossRef] [PubMed]
- Hansson, O. Biomarkers for Neurodegenerative Diseases. Nat. Med. 2021, 27, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Aisen, P.S.; Davis, K.L. Inflammatory Mechanisms in Alzheimer’s Disease: Implications for Therapy. Am. J. Psychiatry 2006, 151, 1105–1113. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic Reactive Astrocytes Are Induced by Activated Microglia. Nature 2017, 541, 481. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Jack, C.R.; Albert, M.S.; Knopman, D.S.; McKhann, G.M.; Sperling, R.A.; Carrillo, M.C.; Thies, B.; Phelps, C.H. Introduction to the Recommendations from the National Institute on Aging-Alzheimer’s Association Workgroups on Diagnostic Guidelines for Alzheimer’s Disease. Alzheimer’s Dement. 2011, 7, 257–262. [Google Scholar] [CrossRef]
- Varesi, A.; Carrara, A.; Pires, V.G.; Floris, V.; Pierella, E.; Savioli, G.; Prasad, S.; Esposito, C.; Ricevuti, G.; Chirumbolo, S.; et al. Blood-Based Biomarkers for Alzheimer’s Disease Diagnosis and Progression: An Overview. Cells 2022, 11, 1367. [Google Scholar] [CrossRef] [PubMed]
- Olsson, B.; Lautner, R.; Andreasson, U.; Öhrfelt, A.; Portelius, E.; Bjerke, M.; Hölttä, M.; Rosén, C.; Olsson, C.; Strobel, G.; et al. CSF and Blood Biomarkers for the Diagnosis of Alzheimer’s Disease: A Systematic Review and Meta-Analysis. Lancet Neurol. 2016, 15, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Mattsson, N.; Cullen, N.C.; Andreasson, U.; Zetterberg, H.; Blennow, K. Association Between Longitudinal Plasma Neurofilament Light and Neurodegeneration in Patients With Alzheimer Disease. JAMA Neurol. 2019, 76, 791. [Google Scholar] [CrossRef]
- Weston, P.S.J.; Poole, T.; Ryan, N.S.; Nair, A.; Liang, Y.; Macpherson, K.; Druyeh, R.; Malone, I.B.; Ahsan, R.L.; Pemberton, H.; et al. Serum Neurofilament Light in Familial Alzheimer Disease: A Marker of Early Neurodegeneration. Neurology 2017, 89, 2167. [Google Scholar] [CrossRef] [PubMed]
- Preische, O.; Schultz, S.A.; Apel, A.; Kuhle, J.; Kaeser, S.A.; Barro, C.; Gräber, S.; Kuder-Buletta, E.; LaFougere, C.; Laske, C.; et al. Serum Neurofilament Dynamics Predicts Neurodegeneration and Clinical Progression in Presymptomatic Alzheimer’s Disease. Nat. Med. 2019, 25, 277. [Google Scholar] [CrossRef] [PubMed]
- Milà-Alomà, M.; Salvadó, G.; Gispert, J.D.; Vilor-Tejedor, N.; Grau-Rivera, O.; Sala-Vila, A.; Sánchez-Benavides, G.; Arenaza-Urquijo, E.M.; Crous-Bou, M.; González-de-Echávarri, J.M.; et al. Amyloid Beta, Tau, Synaptic, Neurodegeneration, and Glial Biomarkers in the Preclinical Stage of the Alzheimer’s Continuum. Alzheimer’s Dement. 2020, 16, 1358. [Google Scholar] [CrossRef] [PubMed]
- Ng, A.; Tam, W.W.; Zhang, M.W.; Ho, C.S.; Husain, S.F.; McIntyre, R.S.; Ho, R.C. IL-1β, IL-6, TNF- α and CRP in Elderly Patients with Depression or Alzheimer’s Disease: Systematic Review and Meta-Analysis. Sci. Rep. 2018, 8, 12050. [Google Scholar] [CrossRef]
- Van Hulle, C.; Jonaitis, E.M.; Betthauser, T.J.; Batrla, R.; Wild, N.; Kollmorgen, G.; Andreasson, U.; Okonkwo, O.; Bendlin, B.B.; Asthana, S.; et al. An Examination of a Novel Multipanel of CSF Biomarkers in the Alzheimer’s Disease Clinical and Pathological Continuum. Alzheimer’s Dement. 2021, 17, 431. [Google Scholar] [CrossRef]
- Lee, W.J.; Liao, Y.C.; Wang, Y.F.; Lin, I.F.; Wang, S.J.; Fuh, J.L. Plasma MCP-1 and Cognitive Decline in Patients with Alzheimer’s Disease and Mild Cognitive Impairment: A Two-Year Follow-up Study. Sci. Rep. 2018, 8, 1–8. [Google Scholar] [CrossRef]
- Hu, W.T.; Howell, J.C.; Ozturk, T.; Gangishetti, U.; Kollhoff, A.L.; Hatcher-Martin, J.M.; Anderson, A.M.; Tyor, W.R. CSF Cytokines in Aging, Multiple Sclerosis, and Dementia. Front. Immunol. 2019, 10, 480. [Google Scholar] [CrossRef]
- Bettcher, B.M.; Johnson, S.C.; Fitch, R.; Casaletto, K.B.; Heffernan, K.S.; Asthana, S.; Zetterberg, H.; Blennow, K.; Carlsson, C.M.; Neuhaus, J.; et al. CSF and Plasma Levels of Inflammation Differentially Relate to CNS Markers of Alzheimer’s Disease Pathology and Neuronal Damage. J. Alzheimers Dis. 2018, 62, 385. [Google Scholar] [CrossRef]
- Dinarello, C.A. IL-1: Discoveries, Controversies and Future Directions. Eur. J. Immunol. 2010, 40, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Testa, G.; Staurenghi, E.; Zerbinati, C.; Gargiulo, S.; Iuliano, L.; Giaccone, G.; Fantò, F.; Poli, G.; Leonarduzzi, G.; Gamba, P. Changes in Brain Oxysterols at Different Stages of Alzheimer’s Disease: Their Involvement in Neuroinflammation. Redox. Biol. 2016, 10, 24. [Google Scholar] [CrossRef] [PubMed]
- Forlenza, O.V.; Diniz, B.S.; Talib, L.L.; Mendonça, V.A.; Ojopi, E.B.; Gattaz, W.F.; Teixeira, A.L. Increased Serum IL-1β Level in Alzheimer’s Disease and Mild Cognitive Impairment. Dement. Geriatr. Cogn. Disord. 2009, 28, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Leung, R.; Proitsi, P.; Simmons, A.; Lunnon, K.; Güntert, A.; Kronenberg, D.; Pritchard, M.; Tsolaki, M.; Mecocci, P.; Kloszewska, I.; et al. Inflammatory Proteins in Plasma Are Associated with Severity of Alzheimer’s Disease. PLoS ONE 2013, 8, 64971. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.Y. The NLRP3 Inflammasome: Molecular Activation and Regulation to Therapeutics. Nat. Rev. Immunol. 2019, 19, 477. [Google Scholar] [CrossRef]
- Liu, L.; Chan, C. The Role of Inflammasome in Alzheimer’s Disease. Ageing Res. Rev. 2014, 15, 6. [Google Scholar] [CrossRef]
- Hanslik, K.L.; Ulland, T.K. The Role of Microglia and the Nlrp3 Inflammasome in Alzheimer’s Disease. Front. Neurol. 2020, 11, 570711. [Google Scholar] [CrossRef]
- Hulstaert, F.; Blennow, K.; Ivanoiu, A.; Schoonderwaldt, H.C.; Riemenschneider, M.; de Deyn, P.P.; Bancher, C.; Cras, P.; Wiltfang, J.; Mehta, P.D.; et al. Improved Discrimination of AD Patients Using β-Amyloid(1-42) and Tau Levels in CSF. Neurology 1999, 52, 1555. [Google Scholar] [CrossRef]
- van Harten, A.C.; Wiste, H.J.; Weigand, S.D.; Mielke, M.M.; Kremers, W.K.; Eichenlaub, U.; Dyer, R.B.; Algeciras-Schimnich, A.; Knopman, D.S.; Jack, C.R.; et al. Detection of Alzheimer’s Disease Amyloid Beta 1-42, p-Tau, and t-Tau Assays. Alzheimer’s Dement. 2022, 18, 635–644. [Google Scholar] [CrossRef]
- Gulisano, W.; Maugeri, D.; Baltrons, M.A.; Fà, M.; Amato, A.; Palmeri, A.; D’Adamio, L.; Grassi, C.; Devanand, D.P.; Honig, L.S.; et al. Role of Amyloid-β and Tau Proteins in Alzheimer’s Disease: Confuting the Amyloid Cascade. J. Alzheimers Dis. 2018, 64, S611. [Google Scholar] [CrossRef]
- Bertens, D.; Tijms, B.M.; Scheltens, P.; Teunissen, C.E.; Visser, P.J. Unbiased Estimates of Cerebrospinal Fluid β-Amyloid 1-42 Cutoffs in a Large Memory Clinic Population. Alzheimers Res. Ther. 2017, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Palmqvist, S.; Zetterberg, H.; Blennow, K.; Vestberg, S.; Andreasson, U.; Brooks, D.J.; Owenius, R.; Hägerström, D.; Wollmer, P.; Minthon, L.; et al. Accuracy of Brain Amyloid Detection in Clinical Practice Using Cerebrospinal Fluid β-Amyloid 42: A Cross-Validation Study Against Amyloid Positron Emission Tomography. JAMA Neurol. 2014, 71, 1282–1289. [Google Scholar] [CrossRef]
- de Riva, V.; Galloni, E.; Marcon, M.; di Dionisio, L.; Deluca, C.; Meligrana, L.; Bolner, A.; Perini, F. Analysis of Combined CSF Biomarkers in AD Diagnosis. Clin. Lab. 2014, 60, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Mormino, E.C. The Relevance of Beta-Amyloid on Markers of Alzheimer’s Disease in Clinically Normal Individuals and Factors That Influence These Associations. Neuropsychol. Rev. 2014, 24, 300. [Google Scholar] [CrossRef] [PubMed]
- Perez-Nievas, B.G.; Stein, T.D.; Tai, H.C.; Dols-Icardo, O.; Scotton, T.C.; Barroeta-Espar, I.; Fernandez-Carballo, L.; De Munain, E.L.; Perez, J.; Marquie, M.; et al. Dissecting Phenotypic Traits Linked to Human Resilience to Alzheimer’s Pathology. Brain 2013, 136, 2510. [Google Scholar] [CrossRef]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased Clearance of CNS Amyloid-β in Alzheimer’s Disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef]
- Jack, C.R.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical Model of Dynamic Biomarkers of the Alzheimer’s Pathological Cascade. Lancet Neurol. 2010, 9, 119. [Google Scholar] [CrossRef]
- Huang, L.K.; Chao, S.P.; Hu, C.J. Clinical Trials of New Drugs for Alzheimer Disease. J. Biomed. Sci. 2020, 27, 1–13. [Google Scholar] [CrossRef]
- Carecchio, M.; Chiocchetti, A.; Galimberti, D. Osteopontin Is Increased in the Cerebrospinal Fluid of Patients with Alzheimer’s Disease and Its Levels Correlate with Cognitive Decline. J. Alzheimer’s Dis. 2010, 19, 1143–1148. [Google Scholar] [CrossRef]
- Craig-Schapiro, R.; Perrin, R.J.; Roe, C.M.; Xiong, C.; Carter, D.; Cairns, N.J.; Mintun, M.A.; Peskind, E.R.; Li, G.; Galasko, D.R.; et al. YKL-40: A Novel Prognostic Fluid Biomarker for Preclinical Alzheimer’s Disease. Biol. Psychiatry 2010, 68, 903. [Google Scholar] [CrossRef]
- Risbud, M.V.; Shapiro, I.M. Role of Cytokines in Intervertebral Disc Degeneration: Pain and Disc-Content. Nat. Rev. Rheumatol. 2014, 10, 44. [Google Scholar] [CrossRef] [PubMed]
- Livshits, G.; Zhai, G.; Hart, D.J.; Kato, B.S.; Wang, H.; Williams, F.M.K.; Spector, T.D. Interleukin-6 Is a Significant Predictor of Radiographic Knee Osteoarthritis: The Chingford Study. Arthritis Rheum 2009, 60, 2037. [Google Scholar] [CrossRef] [PubMed]
- Gundogdu, G.; Gundogdu, K. A Novel Biomarker in Patients with Knee Osteoarthritis: Adropin. Clin. Rheumatol. 2018, 37, 2179–2186. [Google Scholar] [CrossRef]
- Bromander, S.; Anckarsäter, R.; Kristiansson, M.; Blennow, K.; Zetterberg, H.; Anckarsäter, H.; Wass, C.E. Changes in Serum and Cerebrospinal Fluid Cytokines in Response to Non-Neurological Surgery: An Observational Study. J. Neuroinflamm. 2012, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Fertleman, M.; Pereira, C.; Dani, M.; Harris, B.H.L.; Di Giovannantonio, M.; Taylor-Robinson, S.D. Cytokine Changes in Cerebrospinal Fluid and Plasma after Emergency Orthopaedic Surgery. Sci. Rep. 2022, 12, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Varatharaj, A.; Galea, I. The Blood-Brain Barrier in Systemic Inflammation. Brain Behav. Immun. 2017, 60, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bettcher, B.M.; Tansey, M.G.; Dorothée, G.; Heneka, M.T. Peripheral and Central Immune System Crosstalk in Alzheimer Disease–A Research Prospectus. Nat. Rev. Neurol. 2021, 17, 689–701. [Google Scholar] [CrossRef]
- Prinz, M.; Priller, J. The Role of Peripheral Immune Cells in the CNS in Steady State and Disease. Nat. Neurosci. 2017, 20, 136–144. [Google Scholar] [CrossRef]
- Shang, D.S.; Yang, Y.M.; Zhang, H.; Tian, L.; Jiang, J.S.; Dong, Y.B.; Zhang, K.; Li, B.; Zhao, W.D.; Fang, W.G.; et al. Intracerebral GM-CSF Contributes to Transendothelial Monocyte Migration in APP/PS1 Alzheimer’s Disease Mice. J. Cereb. Blood Flow Metab. 2016, 36, 1978. [Google Scholar] [CrossRef]
- Kierdorf, K.; Masuda, T.; Jordão, M.J.C.; Prinz, M. Macrophages at CNS Interfaces: Ontogeny and Function in Health and Disease. Nat. Rev. Neurosci. 2019, 20, 547–562. [Google Scholar] [CrossRef]
- Bowman, G.L.; Dayon, L.; Kirkland, R.; Wojcik, J.; Peyratout, G.; Severin, I.C.; Henry, H.; Oikonomidi, A.; Migliavacca, E.; Bacher, M.; et al. Blood-Brain Barrier Breakdown, Neuroinflammation, and Cognitive Decline in Older Adults. Alzheimer’s Dement. 2018, 14, 1640–1650. [Google Scholar] [CrossRef] [PubMed]
- Takata, F.; Nakagawa, S.; Matsumoto, J.; Dohgu, S. Blood-Brain Barrier Dysfunction Amplifies the Development of Neuroinflammation: Understanding of Cellular Events in Brain Microvascular Endothelial Cells for Prevention and Treatment of BBB Dysfunction. Front. Cell Neurosci. 2021, 15, 661838. [Google Scholar] [CrossRef] [PubMed]
- Felger, J.C.; Haroon, E.; Patel, T.A.; Goldsmith, D.R.; Wommack, E.C.; Woolwine, B.J.; Le, N.A.; Feinberg, R.; Tansey, M.G.; Miller, A.H. What Does Plasma CRP Tell Us about Peripheral and Central Inflammation in Depression? Mol. Psychiatry 2020, 25, 1301. [Google Scholar] [CrossRef]
- Hillmer, L.; Erhardt, E.B.; Caprihan, A.; Adair, J.C.; Knoefel, J.E.; Prestopnik, J.; Thompson, J.; Hobson, S.; Rosenberg, G.A. Blood-Brain Barrier Disruption Measured by Albumin Index Correlates with Inflammatory Fluid Biomarkers. J. Cereb. Blood Flow Metab. 2023, 43, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wang, H.; Lou, W.; Ma, L.; Li, Y.; Zhang, N.; Wang, C.; Li, F.; Awais, M.; Cao, S.; et al. IP-10 Promotes Blood–Brain Barrier Damage by Inducing Tumor Necrosis Factor Alpha Production in Japanese Encephalitis. Front. Immunol. 2018, 9, 1148. [Google Scholar] [CrossRef]
- Franciotta, D.; Martino, G.; Zardini, E.; Furlan, R.; Bergamaschi, R.; Andreoni, L.; Cosi, V. Serum and CSF Levels of MCP-1 and IP-10 in Multiple Sclerosis Patients with Acute and Stable Disease and Undergoing Immunomodulatory Therapies. J. Neuroimmunol. 2001, 115, 192–198. [Google Scholar] [CrossRef]
- Gertje, E.C.; Janelidze, S.; van Westen, D.; Cullen, N.; Stomrud, E.; Palmqvist, S.; Hansson, O.; Mattsson-Carlgren, N. Associations Between CSF Markers of Inflammation, White Matter Lesions, and Cognitive Decline in Individuals without Dementia. Neurology 2023, 100, e1812. [Google Scholar] [CrossRef]
- Michlmayr, D.; McKimmie, C.S. Role of CXCL10 in Central Nervous System Inflammation. Int. J. Interferon Cytokine Mediat. Res. 2014, 6, 1–18. [Google Scholar] [CrossRef]
- Scarpini, E.; Galimberti, D.; Baron, P.; Clerici, R.; Ronzoni, M.; Conti, G.; Scarlato, G. IP-10 and MCP-1 Levels in CSF and Serum from Multiple Sclerosis Patients with Different Clinical Subtypes of the Disease. J. Neurol. Sci. 2002, 195, 41–46. [Google Scholar] [CrossRef]
- Jiang, S.; Maphis, N.M.; Binder, J.; Chisholm, D.; Weston, L.; Duran, W.; Peterson, C.; Zimmerman, A.; Mandell, M.A.; Jett, S.D.; et al. Proteopathic Tau Primes and Activates Interleukin-1β via Myeloid-Cell-Specific MyD88- and NLRP3-ASC-Inflammasome Pathway. Cell Rep. 2021, 36, 109720. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total n = 107 | HC n = 30 | NIC n = 45 | MCI n = 32 | p-Value | |
|---|---|---|---|---|---|
| Age, years | 64.7 (7.8) | 59.6 (6.1) | 64.8 (6.5) | 69.1 (8.3) | <0.0001 |
| Sex (female), n (%) | 63 (58.9) | 15 (50) | 28 (62.2) | 20 (62.5) | 0.0655 |
| MMSE (0–30) * | 26.4 (22–30) | - | 30 (30–30)] | 22 (21.75–24) | <0.0001 |
| Comorbidities, n (%) | |||||
| Hypertension | 12 (11.2) | - | 2 (4.4) | 10 (31.3) | 0.014 |
| Ischemic heart disease | 12 (11.2) | - | 0 (0.0) | 12 (37.5) | <0.0001 |
| Diabetes | 2 (1.9) | - | 0 (0.0) | 2 (6.3) | 0.0893 |
| Concomitant disease, n (%) | |||||
| Knee arthritis | 32 (29.9) | - | 32 (71.1) | - | - |
| Disc herniation | 11 (10.3) | - | 11 (24.4) | - | - |
| Analyte | HC | NIC | MCI | NIC vs. MCI |
|---|---|---|---|---|
| n = 30 | n = 45 | n = 32 | p-Value | |
| CSF | ||||
| Aβ42 IT pg/mL | - | 496.3 (235.6) | 735.7 (285.1) | 0.0002 |
| tTau pg/mL | - | 211.6 (235.3) | 198.1 (186.8) | 0.1263 |
| pTau181 pg/mL | - | 15.51 (8.149) | 16.22 (12.95) | 0.3872 |
| Serum | ||||
| tTau pg/mL | 10.01 (12.04) | 17.10 (10.87) | 13.93 (14.70) | 0.0003 * |
| pTau181 pg/mL | - | 1.238 (1.989) | 0.9307 (1.371) | 0.0173 |
| AD cutoffs, n (%) | ||||
| Aβ42 < 556 pg/mL | - | 28 (62%) | 8 (25%) | 0.0023 |
| pTau181/Aβ42 ratio | ||||
| pTau181/Aβ42 † | - | 0.035 | 0.018 | 0.0008 |
| Analyte | HC | NIC | MCI | HC vs. NIC | HC vs. MCI | NIC vs. MCI | Overall † |
|---|---|---|---|---|---|---|---|
| n = 30 | n = 45 | n = 32 | p-Value | p-Value | p-Value | p-Value | |
| CSF | |||||||
| GFAP pg/mL | - | 5737 (3396) | 9189 (11,108) | - | - | 0.1386 | - |
| NFL pg/mL | - | 1164 (1685) | 1639 (2086) | - | - | 0.0723 | - |
| OPN ng/mL | - | 254.8 (119.9) | 512.8 (318.0) | - | - | <0.0001 | - |
| TIMP-1 ng/mL | - | 53.36 (16.96) | 63.90 (40.00) | - | - | 0.5072 | - |
| sTREM-2 ng/mL | - | 14.16 (5.948) | 18.05 (10.03) | - | - | 0.1332 | - |
| YKL-40 ng/mL | - | 172.5 (75.31) | 233.6 (123.6) | - | - | 0.0165 | - |
| Serum | |||||||
| GFAP pg/mL | 94.38 (39.56) | 133.0 (84.99) | 151.8 (132.1) | 0.1209 | 0.0288 | >0.9999 | 0.0272 |
| NFL pg/mL | 14.01 (8.115) | 33.67 (29.70) | 58.10 (106.5) | 0.0001 | 0.2455 | 0.0699 | 0.0002 |
| OPN ng/mL | 41.57 (19.80) | 93.07 (42.22) | 51.53 (25.25) | <0.0001 | 0.3558 | <0.0001 | <0.0001 |
| TIMP-1 ng/mL | 111.1 (39.74) | 230.6 (58.89) | 225.4 (35.75) | <0.0001 | <0.0001 | >0.9999 | <0.0001 |
| sTREM-2 ng/mL | 40.49 (19.42) | 30.90 (12.31) | 28.28 (11.85) | 0.0584 | 0.0088 | >0.9999 | 0.0086 |
| YKL-40 ng/mL | 67.05 (69.11) | 156.2 (274.9) | 66.73 (60.43) | 0.0426 | >0.9999 | 0.3484 | 0.0407 |
| Analyte | HC | NIC | MCI | HC vs. NIC | HC vs. MCI | NIC vs. MCI | Overall |
|---|---|---|---|---|---|---|---|
| n = 30 | n = 45 | n = 32 | p-Value | p-Value | p-Value | p-Value | |
| CRP ng/mL | 3922 (3604) | 12,856 (19798) | 3722 (8060) | 0.0816 | 0.0724 | <0.0001 | <0.0001 |
| IL-10 pg/mL | 2.190 (1.151) | 6.094 (5.195) | 3.456 (2.120) | <0.0001 | 0.0086 | 0.0295 | <0.0001 |
| IL-6 pg/mL | 7.807 (18.71) | 42.76 (71.71) | 3.162 (3.983) | 0.4752 | 0.0066 | <0.0001 | <0.0001 |
| IL-8 pg/mL | 19.67 (19.65) | 35.19 (37.69) | 87.74 (164.6) | 0.0054 | <0.0001 | 0.3771 | <0.0001 |
| IP-10 pg/mL | 123.4 (75.34) | 97.20 (85.05) | 131.7 (79.53) | 0.0238 | >0.9999 | 0.0098 | 0.0037 |
| MCP-1 pg/mL | 207.1 (68.54) | 536.1 (220.5) | 389.6 (155.0) | <0.0001 | <0.0001 | 0.0171 | <0.0001 |
| TNFα fg/mL | 602.7 (249.0) | 1592 (648.5) | 1235 (447.9) | <0.0001 | <0.0001 | 0.2253 | <0.0001 |
| Analyte | NIC | MCI | NIC vs. MCI |
|---|---|---|---|
| n = 45 | n = 32 | p-Value | |
| CRP ng/mL | 7.595 (11.18) | 11.93 (17.76) | 0.8531 |
| IL-6 pg/mL | 3. 541 (3.332) | 11.63 (25.81) | 0.0572 |
| IL-8 pg/mL | 41.36 (20.63) | 132.8 (198.1) | 0.0002 |
| IP-10 pg/mL | 131.4 (65.38) | 199.6 (130.2) | 0.0187 |
| MCP-1 pg/mL | 602.7 (232.4) | 671.0 (324.5) | 0.3126 |
| Analyte | HC | NIC | MCI | HC vs. NIC | HC vs. MCI | NIC vs. MCI | Overall |
|---|---|---|---|---|---|---|---|
| n = 30 | n = 45 | n = 32 | p-Value | p-Value | p-Value | p-Value | |
| CSF | |||||||
| ASC pg/mL | - | 45.20 (15.75) | 48.83 (26.97) | - | - | 0.9243 | - |
| IL-18 pg/mL | - | 2.622 (1.830) | 3.325 (2.997) | - | - | 0.9897 | - |
| Serum | |||||||
| ASC pg/mL | 396.3 (426.2) | 550.0 (199.3) | 535.3 (247.8) | <0.0001 | 0.0002 | >0.9999 | <0.0001 |
| Caspase-1 pg/mL | 0.9008 (1.049) | 3.626 (1.453) | 4.151 (2.716) | <0.0001 | <0.0001 | >0.9999 | <0.0001 |
| IL-1β fg/mL | 90.74 (157.6) | 211.7 (161.5) | 345.2 (306.9) | <0.0001 | <0.0001 | 0.1801 | <0.0001 |
| IL-1Ra pg/mL | 272.3 (164.4) | 833.0 (443.2) | 684.3 (967.3) | 0.0005 | 0.0177 | 0.2925 | 0.0007 |
| IL-18 pg/mL | 222.6 (100.1) | 223.7 (154.5) | 234.0 (114.3) | >0.9999 | >0.9999 | 0.4652 | 0.3580 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmidt-Morgenroth, I.; Michaud, P.; Gasparini, F.; Avrameas, A. Central and Peripheral Inflammation in Mild Cognitive Impairment in the Context of Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 10523. https://doi.org/10.3390/ijms241310523
Schmidt-Morgenroth I, Michaud P, Gasparini F, Avrameas A. Central and Peripheral Inflammation in Mild Cognitive Impairment in the Context of Alzheimer’s Disease. International Journal of Molecular Sciences. 2023; 24(13):10523. https://doi.org/10.3390/ijms241310523
Chicago/Turabian StyleSchmidt-Morgenroth, Inès, Philippe Michaud, Fabrizio Gasparini, and Alexandre Avrameas. 2023. "Central and Peripheral Inflammation in Mild Cognitive Impairment in the Context of Alzheimer’s Disease" International Journal of Molecular Sciences 24, no. 13: 10523. https://doi.org/10.3390/ijms241310523
APA StyleSchmidt-Morgenroth, I., Michaud, P., Gasparini, F., & Avrameas, A. (2023). Central and Peripheral Inflammation in Mild Cognitive Impairment in the Context of Alzheimer’s Disease. International Journal of Molecular Sciences, 24(13), 10523. https://doi.org/10.3390/ijms241310523