
An Update on MYBPC3 Gene Mutation in Hypertrophic Cardiomyopathy

 ,
,  , , ,
, , ,  , ,
, , 

Abstract
1. Introduction
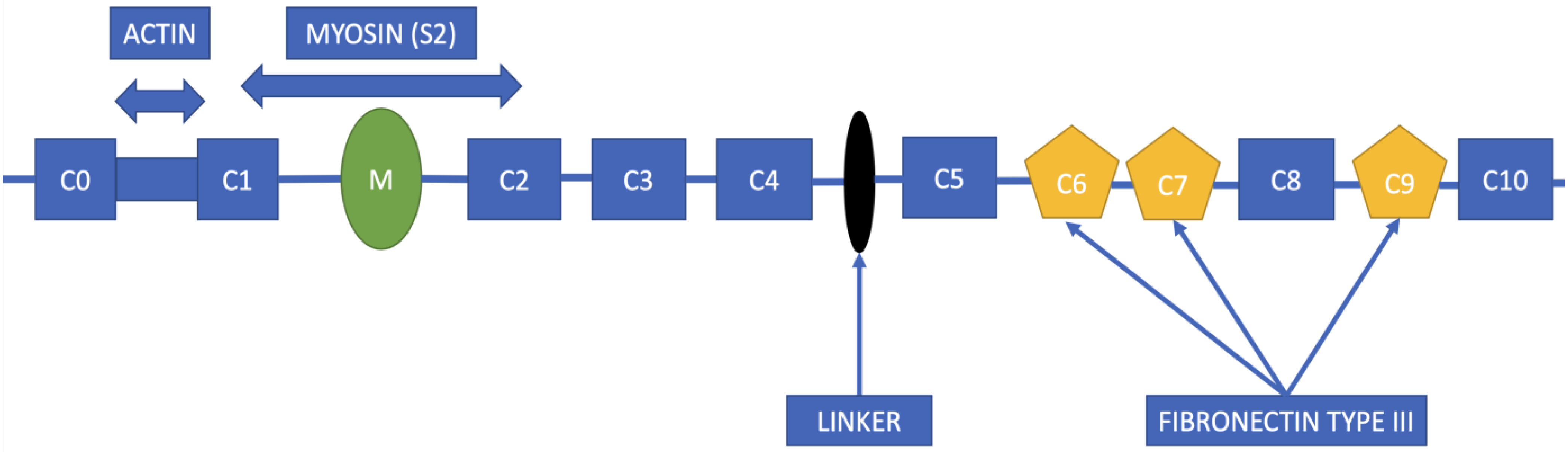
2. Structure of the MYBPC3 Gene
3. MYBPC3 Mutations in HCM Patients
3.1. Truncating Mutations
3.2. Nontruncating Mutations
3.3. Ubiquitin–Proteasome System and Nonsense-Mediated RNA Dysfunctions
3.4. Allelic Imbalance
4. From Genetics to Clinical Implications
4.1. Demography
4.2. Phenotype
4.3. Clinical Implications of Mutation Type
4.4. Major Outcomes
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bai, Y.; Zheng, J.-P.; Lu, F.; Zhang, X.-L.; Sun, C.-P.; Guo, W.-H.; Zou, Y.-X.; Lip, G.Y.H.; Shi, X.-B. Prevalence, incidence and mortality of hypertrophic cardiomyopathy based on a population cohort of 21.9 million in China. Sci. Rep. 2022, 12, 18799. [Google Scholar] [CrossRef] [PubMed]
- Carrier, L. Targeting the population for gene therapy with MYBPC. J. Mol. Cell. Cardiol. 2021, 150, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Seeger, T.; Shrestha, R.; Lam, C.K.; Chen, C.; McKeithan, W.L.; Lau, E.; Wnorowski, A.; McMullen, G.; Greenhaw, M.; Lee, J.; et al. A Premature Termination Codon Mutation in MYBPC3 Causes Hypertrophic Cardio-myopathy via Chronic Activation of Nonsense-Mediated Decay. Circulation 2019, 139, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Alphonse, P.; Virk, S.; Collins, J.; Campbell, T.; Thomas, S.P.; Semsarian, C.; Kumar, S. Prognostic impact of atrial fibrillation in hypertrophic cardiomyopathy: A systematic review. Clin. Res. Cardiol. 2021, 110, 544–554. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Norton, N.; Morales, A.; Li, D.; Siegfried, J.D.; Gonzalez-Quintana, J. Coding sequence rare variants identified in MYBPC3, MYH6, TPM1, TNNC1, and TNNI3 from 312 patients with familial or idiopathic dilated cardiomyopathy. Circ. Cardiovasc. Genet. 2010, 3, 155–161. [Google Scholar] [CrossRef]
- Viswanathan, S.K.; Sanders, H.K.; McNamara, J.W.; Jagadeesan, A.; Jahangir, A.; Tajik, A.J.; Sadayappan, S. Hypertrophic cardiomyopathy clinical phenotype is independent of gene mutation and mutation dosage. PLoS ONE 2017, 12, e0187948. [Google Scholar] [CrossRef]
- Yuan, C.-C.; Kazmierczak, K.; Liang, J.; Kanashiro-Takeuchi, R.; Irving, T.C.; Gomes, A.V.; Wang, Y.; Burghardt, T.P.; Szczesna-Cordary, D. Hypercontractile mutant of ventricular myosin essential light chain leads to disruption of sarcomeric structure and function and results in restrictive cardiomyopathy in mice. Cardiovasc. Res. 2017, 113, 1124–1136. [Google Scholar] [CrossRef]
- Carrier, L.; Bonne, G.; Bährend, E.; Yu, B.; Richard, P.; Niel, F.; Hainque, B.; Cruaud, C.; Gary, F.; Labeit, S.; et al. Organization and sequence of human cardiac myosin binding protein C gene (MYBPC3) and identification of mutations predicted to produce truncated proteins in familial hypertrophic cardiomyopathy. Circ. Res. 1997, 80. [Google Scholar] [CrossRef]
- Flavigny, J.; Robert, P.; Camelin, J.C.; Schwartz, K.; Carrier, L.; Berrebi-Bertrand, I. Biomolecular interactions between human recombinant beta-MyHC and cMyBP-Cs implicated in familial hypertrophic cardiomyopathy. Cardiovasc. Res. 2003, 60, 388–396. [Google Scholar] [CrossRef]
- Shaffer, J.F.; Kensler, R.W.; Harris, S.P. The Myosin-binding Protein C Motif Binds to F-actin in a Phosphorylation-sensitive Manner. J. Biol. Chem. 2009, 284, 12318–12327. [Google Scholar] [CrossRef]
- Fougerousse, F.; Delezoide, A.-L.; Fiszman, M.Y.; Schwartz, K.; Beckmann, J.S.; Carrier, L. Cardiac myosin binding protein C gene is specifically expressed in heart during murine and human development. Circ. Res. 1998, 82, 130–133. [Google Scholar] [CrossRef]
- Jia, W.; Shaffer, J.F.; Harris, S.P.; Leary, J.A. Identification of Novel Protein Kinase A Phosphorylation Sites in the M-domain of Human and Murine Cardiac Myosin Binding Protein-C Using Mass Spectrometry Analysis. J. Proteome Res. 2010, 9, 1843–1853. [Google Scholar] [CrossRef]
- Mohamed, A.S.; Dignam, J.D.; Schlender, K.K. Cardiac myosin-binding protein C (MyBP-C): Identification of protein kinase A and protein kinase C phosphorylation sites. Arch. Biochem. Biophys. 1998, 358, 313–319. [Google Scholar] [CrossRef]
- Sadayappan, S.; de Tombe, P.P. Cardiac myosin binding protein-C: Redefining its structure and function. Biophys. Rev. 2012, 4, 93–106. [Google Scholar] [CrossRef]
- Helms, A.S.; Tang, V.T.; O’Leary, T.S.; Friedline, S.; Wauchope, M.; Arora, A.; Wasserman, A.H.; Smith, E.D.; Lee, L.M.; Wen, X.W.; et al. Effects of MYBPC3 loss-of-function mutations preceding hypertrophic cardiomy-opathy. JCI Insight 2020, 5, 1–19. [Google Scholar] [CrossRef]
- Wijnker, P.J.; Friedrich, F.W.; Dutsch, A.; Reischmann, S.; Eder, A.; Mannhardt, I.; Mearini, G.; Eschenhagen, T.; van der Velden, J.; Carrier, L. Comparison of the effects of a truncating and a missense MYBPC3 mutation on contractile parameters of engineered heart tissue. J. Mol. Cell. Cardiol. 2016, 97, 82–92. [Google Scholar] [CrossRef]
- Sepp, R.; Hategan, L.; Csányi, B.; Borbás, J.; Tringer, A.; Pálinkás, E.D.; Nagy, V.; Takács, H.; Latinovics, D.; Nyolczas, N.; et al. The Genetic Architecture of Hypertrophic Cardiomyopathy in Hungary: Analysis of 242 Patients with a Panel of 98 Genes. Diagnostics 2022, 12, 1132. [Google Scholar] [CrossRef]
- Carrier, L.; Mearini, G.; Stathopoulou, K.; Cuello, F. Cardiac myosin-binding protein C (MYBPC3) in cardiac pathophysiology. Gene 2015, 573, 188–197. [Google Scholar] [CrossRef]
- Glazier, A.A.; Thompson, A.; Day, S.M. Allelic imbalance and haploinsufficiency in MYBPC3-linked hypertrophic cardiomyopathy. Pflug. Arch. Eur. J. Physiol. 2019, 471, 781–793. [Google Scholar] [CrossRef]
- Suay-Corredera, C.; Alegre-Cebollada, J. The mechanics of the heart: Zooming in on hypertrophic cardiomyopathy and cMyBP-C. FEBS Lett. 2022, 596, 703–746. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, T.; Piao, C.; Li, X.; Guo, J.; Zheng, S.; Zhang, X.; Cai, T.; Du, J. Screening Mutations of MYBPC3 in 114 Unrelated Patients with Hypertrophic Cardiomyopathy by Targeted Capture and Next-generation Sequencing. Sci. Rep. 2015, 5, 11411. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.P.; Lyons, R.G.; Bezold, K.L. In the thick of it: HCM-causing mutations in myosin binding proteins of the thick filament. Circ. Res. 2011, 108, 751–764. [Google Scholar] [CrossRef] [PubMed]
- Andersen, P.S.; Havndrup, O.; Bundgaard, H.; A Larsen, L.; Vuust, J.; Pedersen, A.K.; Kjeldsen, K.; Christiansen, M. Genetic and phenotypic characterization of mutations in myosin-binding protein C (MYBPC3) in 81 families with familial hypertrophic cardiomyopathy: Total or partial haploinsufficiency. Eur. J. Hum. Genet. 2004, 12, 673–677. [Google Scholar] [CrossRef]
- Suay-Corredera, C.; Pricolo, M.R.; Herrero-Galán, E.; Velázquez-Carreras, D.; Sánchez-Ortiz, D.; García-Giustiniani, D.; Delgado, J.; Galano-Frutos, J.J.; García-Cebollada, H.; Vilches, S.; et al. Protein haploinsufficiency drivers identify MYBPC3 variants that cause hypertrophic cardiomyopathy. J. Biol. Chem. 2021, 297, 100854. [Google Scholar] [CrossRef]
- Glazier, A.; Hafeez, N.; Mellacheruvu, D.; Basrur, V.; Nesvizhskii, A.I.; Lee, L.M.; Shao, H.; Tang, V.; Yob, J.M.; Gestwicki, J.E.; et al. HSC70 is a chaperone for wild-type and mutant cardiac myosin binding protein C. J. Clin. Investig. 2018, 3. [Google Scholar] [CrossRef]
- Marian, A.J. Molecular Genetic Basis of Hypertrophic Cardiomyopathy. Circ. Res. 2021, 128, 1533–1553. [Google Scholar] [CrossRef]
- Marston, S.; Copeland, O.; Jacques, A.; Livesey, K.; Tsang, V.; McKenna, W.J.; Jalilzadeh, S.; Carballo, S.; Redwood, C.; Watkins, H. Evidence from human myectomy samples that MYBPC3 mutations cause hy-pertrophic cardiomyopathy through haploinsufficiency. Circ. Res. 2009, 105, 219–222. [Google Scholar] [CrossRef]
- van Dijk, S.J.; Dooijes, D.; dos Remedios, C.; Michels, M.; Lamers, J.M.; Winegrad, S.; Schlossarek, S.; Carrier, L.; ten Cate, F.J.; Stienen, G.J.M.; et al. Cardiac myosin-binding protein C mutations and hypertrophic cardio-myopathy: Haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation 2009, 119, 1473–1483. [Google Scholar] [CrossRef]
- Deutschbauer, A.M.; Jaramillo, D.F.; Proctor, M.; Kumm, J.; E Hillenmeyer, M.; Davis, R.W.; Nislow, C.; Giaever, G. Mechanisms of Haploinsufficiency Revealed by Genome-Wide Profiling in Yeast. Genetics 2005, 169, 1915–1925. [Google Scholar] [CrossRef]
- Carrier, L.; Schlossarek, S.; Willis, M.; Eschenhagen, T. The ubiquitin-proteasome system and nonsense-mediated mRNA decay in hypertrophic cardiomyopathy. Cardiovasc. Res. 2009, 85, 330–338. [Google Scholar] [CrossRef]
- Kuster, D.W.D.; Lynch, T.L.; Barefield, D.Y.; Sivaguru, M.; Kuffel, G.; Zilliox, M.J.; Lee, K.H.; Craig, R.; Namakkal-Soorappan, R.; Sadayappan, S. Altered C10 domain in cardiac myosin binding protein-C results in hypertrophic cardiomyopathy. Cardiovasc. Res. 2019, 115, 1986–1997. [Google Scholar] [CrossRef]
- Kuster, D.W.; Govindan, S.; Springer, T.I.; Martin, J.L.; Finley, N.L.; Sadayappan, S. A Hypertrophic Cardiomyopathy-associated MYBPC3 Mutation Common in Populations of South Asian Descent Causes Contractile Dysfunction. J. Biol. Chem. 2015, 290, 5855–5867. [Google Scholar] [CrossRef]
- De Lange, W.J.; Farrell, E.T.; Hernandez, J.J.; Stempien, A.; Kreitzer, C.R.; Jacobs, D.R.; Petty, D.L.; Moss, R.L.; Crone, W.C.; Ralphe, J.C. cMyBP-C ablation in human engineered cardiac tissue causes progressive Ca2+-handling abnormalities. J. Gen. Physiol. 2023, 155, e202213204. [Google Scholar] [CrossRef]
- Barefield, D.; Kumar, M.; Gorham, J.; Seidman, J.G.; Seidman, C.E.; de Tombe, P.P.; Sadayappan, S.; Pedersen, E.; Lyons, R. Haploinsufficiency of MYBPC3 exacerbates the development of hypertrophic cardiomyopathy in heterozygous mice. J. Mol. Cell. Cardiol. 2014, 79, 234–243. [Google Scholar] [CrossRef]
- Helms, A.S.; Davis, F.M.; Coleman, D.; Bartolone, S.N.; Glazier, A.A.; Pagani, F.; Yob, J.M.; Sadayappan, S. Sarcomere mutation-specific expression patterns in human hypertrophic car-diomyopathy. Circ. Cardiovasc. Genet. 2014, 7, 434–443. [Google Scholar] [CrossRef]
- McNamara, J.W.; Li, A.; Lal, S.; Bos, J.M.; Harris, S.P.; van der Velden, J.; Ackerman, M.J.; Cooke, R.; dos Remedios, C.G. MYBPC3 mutations are associated with a reduced super-relaxed state in patients with hypertrophic cardiomyopathy. PLoS ONE 2017, 12, e0180064. [Google Scholar] [CrossRef]
- McNamara, J.W.; Li, A.; Smith, N.J.; Lal, S.; Graham, R.M.; Kooiker, K.B.; van Dijk, S.J.; dos Remedios, C.G.; Harris, S.P.; Cooke, R. Ablation of cardiac myosin binding protein-C disrupts the super-relaxed state of myosin in murine cardiomyocytes. J. Mol. Cell. Cardiol. 2016, 94, 65–71. [Google Scholar] [CrossRef]
- Toepfer, C.N.; Wakimoto, H.; Garfinkel, A.C.; McDonough, B.; Liao, D.; Jiang, J.; Tai, A.C.; Gorham, J.M.; Lunde, I.G.; Lun, M.; et al. Hypertrophic cardiomyopathy mutations in MYBPC3 dysregulate myosin. Sci. Transl. Med. 2019, 11, eaat1199. [Google Scholar] [CrossRef]
- Torrado, M.; Maneiro, E.; Junior, A.L.; Fernández-Burriel, M.; Giralt, S.S.; Martínez-Carapeto, A.; Cazón, L.; Santiago, E.; Ochoa, J.P.; McKenna, W.J.; et al. Identification of an elusive spliceogenic MYBPC3 variant in an otherwise genotype-negative hypertrophic cardiomyopathy pedigree. Sci. Rep. 2022, 12, 7284. [Google Scholar] [CrossRef]
- Lopes, L.R.; Barbosa, P.; Torrado, M.; Quinn, E.; Merino, A.; Ochoa, J.P.; Jager, J.; Futema, M.; Carmo-Fonseca, M.; Monserrat, L.; et al. Cryptic Splice-Altering Variants in MYBPC3 Are a Prevalent Cause of Hypertrophic Cardiomyopathy. Circ. Genom. Precis. Med. 2020, 13. [Google Scholar] [CrossRef]
- Singer, E.S.; Ingles, J.; Semsarian, C.; Bagnall, R.D. Key Value of RNA Analysis of MYBPC3 Splice-Site Variants in Hypertrophic Cardiomyopathy. Circ. Genom. Precis. Med. 2019, 12, e002368. [Google Scholar] [CrossRef]
- Petrich, J.; Marchese, D.; Jenkins, C.; Storey, M.; Blind, J. Gene Replacement Therapy: A Primer for the Health-system Pharmacist. J. Pharm. Pr. 2020, 33, 846–855. [Google Scholar] [CrossRef]
- Li, J.; Mamidi, R.; Doh, C.Y.; Holmes, J.B.; Bharambe, N.; Ramachandran, R.; Stelzer, J.E. AAV9 gene transfer of cMyBPC N-terminal domains ameliorates cardiomyopathy in cMyBPC-deficient mice. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Mearini, G.; Stimpel, D.; Geertz, B.; Weinberger, F.; Krämer, E.; Schlossarek, S.; Mourot-Filiatre, J.; Stoehr, A.; Dutsch, A.; Wijnker, P.J.M.; et al. Mybpc3 gene therapy for neonatal cardiomyopathy enables long-term disease prevention in mice. Nat. Commun. 2014, 5, 5515. [Google Scholar] [CrossRef]
- Monteiro da Rocha, A.; Guerrero-Serna, G.; Helms, A.; Luzod, C.; Mironov, S.; Russell, M.; Jalife, J.; Day, S.M.; Smith, G.D.; Herron, T.J. Deficient cMyBP-C protein expression during cardiomyocyte differentiation underlies human hypertrophic cardiomyopathy cellular phenotypes in disease specific human ES cell derived cardiomyocytes. J. Mol. Cell. Cardiol. 2016, 99, 197–206. [Google Scholar] [CrossRef]
- Tardiff, J.C.; Carrier, L.; Bers, D.; Poggesi, C.; Ferrantini, C.; Coppini, R.; Maier, L.S.; Ashrafian, H.; Huke, S.; Van Der Velden, J. Targets for therapy in sarcomeric cardiomyopathies. Cardiovasc. Res. 2015, 105, 457–470. [Google Scholar] [CrossRef]
- Prondzynski, M.; Krämer, E.; Laufer, S.D.; Shibamiya, A.; Pless, O.; Flenner, F.; Muller, O.J.; Munch, J.; Redwood, C.; Hansen, A.; et al. Evaluation of MYBPC3 trans-Splicing and Gene Replacement as Therapeutic Options in Human iPSC-Derived Cardiomyocytes. Mol. Ther. Nucleic Acids 2017, 7, 475–486. [Google Scholar] [CrossRef]
- Doroudgar, S.; Quijada, P.; Konstandin, M.; Ilves, K.; Broughton, K.; Khalafalla, F.G.; Casillas, A.; Nguyen, K.; Gude, N.; Toko, H.; et al. S100A4 protects the myocardium against ischemic stress. J. Mol. Cell. Cardiol. 2016, 100, 54–63. [Google Scholar] [CrossRef]
- Zhang, X.; Azhar, G.; Chai, J.; Sheridan, P.; Nagano, K.; Brown, T.; Yang, J.; Khrapko, K.; Borras, A.M.; Lawitts, J.; et al. Cardiomyopathy in transgenic mice with cardiac-specific overexpression of serum response factor. Am. J. Physiol. Circ. Physiol. 2001, 280, H1782–H1792. [Google Scholar] [CrossRef]
- Smelter, D.F.; de Lange, W.J.; Cai, W.; Ge, Y.; Ralphe, J.C. The HCM-linked W792R mutation in cardiac myosin-binding protein C reduces C6 FnIII domain stability. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H1179–H1191. [Google Scholar] [CrossRef]
- De Lange, W.J.; Grimes, A.C.; Hegge, L.F.; Spring, A.M.; Brost, T.M.; Ralphe, J.C. E258K HCM-causing mutation in cardiac MyBP-C reduces contractile force and accelerates twitch kinetics by disrupting the cMyBP-C and myosin S2 interaction. J. Gen. Physiol. 2013, 142, 241–255. [Google Scholar] [CrossRef]
- Watkins, H.; Ashrafian, H.; Redwood, C. Inherited cardiomyopathies. N. Engl. J. Med. 2011, 364, 1643–1656. [Google Scholar] [CrossRef]
- Gerasimavicius, L.; Livesey, B.J.; Marsh, J.A. Loss-of-function, gain-of-function and dominant-negative mutations have profoundly different effects on protein structure. Nat. Commun. 2022, 13, 3895. [Google Scholar] [CrossRef]
- Helms, A.S.; Thompson, A.D.; Glazier, A.A.; Hafeez, N.; Kabani, S.; Rodriguez, J.; Yob, J.M.; Woolcock, H.; Mazzarotto, F.; Lakdawala, N.K.; et al. Spatial and Functional Distribution of MYBPC3 Pathogenic Variants and Clinical Outcomes in Patients with Hypertrophic Cardiomyopathy. Circ. Genom. Precis. Med. 2020, 13, 396–405. [Google Scholar] [CrossRef]
- Page, S.P.; Kounas, S.; Syrris, P.; Christiansen, M.; Frank-Hansen, R.; Andersen, P.S.; Elliott, P.M.; McKenna, W.J. Cardiac myosin binding protein-C mutations in families with hypertrophic cardiomyopathy: Disease expression in relation to age, gender, and long term outcome. Circ. Cardiovasc. Genet. 2012, 5, 156–166. [Google Scholar] [CrossRef]
- Doh, C.Y.; Li, J.; Mamidi, R.; Stelzer, J.E. The HCM-causing Y235S cMyBPC mutation accelerates contractile function by altering C1 domain structure. Biochim. et Biophys. Acta (BBA) Mol. Basis Dis. 2019, 1865, 661–677. [Google Scholar] [CrossRef]
- Schuldt, M.; Pei, J.; Harakalova, M.; Dorsch, L.M.; Schlossarek, S.; Mokry, M.; Knol, J.C.; Pham, T.V.; Schelfhorst, T.; Piersma, S.R.; et al. Proteomic and Functional Studies Reveal Detyrosinated Tubulin as Treatment Target in Sarcomere Mutation-Induced Hypertrophic Cardiomyopathy. Circ. Hear. Fail. 2021, 14, e007022. [Google Scholar] [CrossRef]
- Chang, Y.-F.; Imam, J.S.; Wilkinson, M.F. The Nonsense-Mediated Decay RNA Surveillance Pathway. Annu. Rev. Biochem. 2007, 76, 51–74. [Google Scholar] [CrossRef]
- Siwaszek, A.; Ukleja, M.; Dziembowski, A. Proteins involved in the degradation of cytoplasmic mRNA in the major eukaryotic model systems. RNA Biol. 2014, 11, 1122–1136. [Google Scholar] [CrossRef]
- Mort, M.; Ivanov, D.; Cooper, D.N.; Chuzhanova, N.A. A meta-analysis of nonsense mutations causing human genetic disease. Hum. Mutat. 2008, 29, 1037–1047. [Google Scholar] [CrossRef]
- Moolman, J.A.; Reith, S.; Uhl, K.; Bailey, S.; Gautel, M.; Jeschke, B.; Fischer, C.; Ochs, J.; McKenna, W.J.; Klues, H.; et al. A newly created splice donor site in exon 25 of the MyBP-C gene is responsible for inherited hypertrophic cardiomyopathy with incomplete disease penetrance. Circulation 2000, 101, 1396–1402. [Google Scholar] [CrossRef] [PubMed]
- Zolk, O.; Schenke, C.; Sarikas, A. The ubiquitin–proteasome system: Focus on the heart. Cardiovasc. Res. 2006, 70, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Liu, Y.; Zhou, X.; Song, L. Progress in the Molecular Genetics of Hypertrophic Cardiomyopathy: A Mini-Review. Gerontology 2013, 59, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Schlossarek, S.; Englmann, D.R.; Sultan, K.R.; Sauer, M.; Eschenhagen, T.; Carrier, L. Defective proteolytic systems in Mybpc3-targeted mice with cardiac hypertrophy. Basic Res. Cardiol. 2012, 107, 235. [Google Scholar] [CrossRef]
- Vignier, N.; Schlossarek, S.; Fraysse, B.; Mearini, G.; Krämer, E.; Pointu, H.; Mougenot, N.; Guiard, J.; Reimer, R.; Hohenberg, H.; et al. Nonsense-Mediated mRNA Decay and Ubiquitin–Proteasome System Regulate Cardiac Myosin-Binding Protein C Mutant Levels in Cardiomyopathic Mice. Circ. Res. 2009, 105, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Carrier, L.; Knoll, R.; Vignier, N.; Keller, D.I.; Bausero, P.; Prudhon, B.; Isnard, R.; Ambroisine, M.-L.; Fiszman, M.; Ross, J., Jr.; et al. Asymmetric septal hypertrophy in heterozygous cMyBP-C null mice. Cardiovasc. Res. 2004, 63, 293–304. [Google Scholar] [CrossRef]
- Rivas, M.A.; Pirinen, M.; Conrad, D.F.; Lek, M.; Tsang, E.K.; Karczewski, K.J.; Maller, J.B.; Kukurba, K.R.; DeLuca, D.S.; Fromer, M.; et al. Human genomics. Effect of predicted protein-truncating genetic variants on the human transcriptome. Science 2015, 348, 666–669. [Google Scholar] [CrossRef]
- Vidal, D.O.; de Souza, J.E.S.; Pires, L.C.; Masotti, C.; Salim, A.C.M.; Costa, M.C.F.; Galante, P.A.F.; de Souza, S.J.; Camargo, A.A. Analysis of allelic differential expression in the human genome using allele-specific serial analysis of gene expression tags. Genome 2011, 54, 120–127. [Google Scholar] [CrossRef]
- Pricolo, M.R.; Herrero-Galan, E.; Mazzaccara, C.; Losi, M.A.; Alegre-Cebollada, J.; Frisso, G. Protein Thermodynamic Destabilization in the Assessment of Patho-genicity of a Variant of Uncertain Significance in Cardiac Myosin Binding Protein C. J. Cardiovasc. Transl. Res. 2020, 13, 867–877. [Google Scholar] [CrossRef]
- Suay-Corredera, C.; Pricolo, M.R.; Velazquez-Carreras, D.; Pathak, D.; Nandwani, N.; Pimenta-Lopes, C.; Sánchez-Ortiz, D.; Urrutia-Irazabal, I.; Vilches, S.; Dominguez, F.; et al. Nanomechanical Phenotypes in Cardiac Myo-sin-Binding Protein C Mutants That Cause Hypertrophic Cardiomyopathy. ACS Nano 2021, 15, 10203–10216. [Google Scholar] [CrossRef]
- Pioner, J.M.; Vitale, G.; Steczina, S.; Langione, M.; Margara, F.; Santini, L.; Giardini, F.; Lazzeri, E.; Piroddi, N.; Scellini, B.; et al. Slower Calcium Handling Balances Faster Cross-Bridge Cycling in Human MYBPC3 HCM. Circ. Res. 2023, 132, 628–644. [Google Scholar] [CrossRef]
- Méndez, I.; Fernández, A.I.; Espinosa, M.; Cuenca, S.; Lorca, R.; Rodríguez, J.F.; Tamargo, M.; García-Montero, M.; Gómez, C.; Vilches, S.; et al. Founder mutation in myosin-binding protein C with an early onset and a high penetrance in males. Open Hear. 2021, 8, e001789. [Google Scholar] [CrossRef]
- Robyns, T.; Breckpot, J.; Nuyens, D.; Vandenberk, B.; Corveleyn, A.; Kuiperi, C.; Aelst, L.V.; Van Cleemput, J.; Willems, R. Clinical and ECG variables to predict the outcome of genetic testing in hy-pertrophic cardiomyopathy. Eur. J. Med. Genet. 2020, 63, 103754. [Google Scholar] [CrossRef]
- Lopes, L.R.; Brito, D.; Belo, A.; Cardim, N. Genetic characterization and genotype-phenotype associations in a large cohort of patients with hypertrophic cardi-omyopathy—An ancillary study of the Portuguese registry of hypertrophic cardiomyopathy. Int. J. Cardiol. 2019, 278, 173–179. [Google Scholar] [CrossRef]
- Terauchi, Y.; Kubo, T.; Baba, Y.; Hirota, T.; Tanioka, K.; Yamasaki, N.; Furuno, T.; Kitaoka, H. Gender differences in the clinical features of hypertrophic cardiomyopathy caused by cardiac myosin-binding protein C gene mutations. J. Cardiol. 2015, 65, 423–428. [Google Scholar] [CrossRef]
- Adalsteinsdottir, B.; Burke, M.; Maron, B.J.; Danielsen, R.; Lopez, B.; Diez, J.; Jarolim, P.; Seidman, J.; Seidman, C.E.; Ho, C.Y.; et al. Hypertrophic cardiomyopathy in myosin-binding protein C (MYBPC3) Icelandic founder mutation carriers. Open Hear. 2020, 7, e001220. [Google Scholar] [CrossRef]
- Field, E.; Norrish, G.; Acquaah, V.; Dady, K.; Cicerchia, M.N.; Ochoa, J.P.; Syrris, P.; McLeod, K.; McGowan, R.; Fell, H.; et al. Cardiac myosin binding protein-C variants in paediatric-onset hypertrophic cardio-myopathy: Natural history and clinical outcomes. J. Med. Genet. 2022, 59, 768–775. [Google Scholar] [CrossRef]
- Haas, J.; Frese, K.S.; Peil, B.; Kloos, W.; Keller, A.; Nietsch, R.; Feng, Z.; Müller, S.; Kayvanpour, E.; Vogel, B.; et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur. Heart J. 2014, 36, 1123–1135. [Google Scholar] [CrossRef]
- Kissopoulou, A.; Trinks, C.; Green, A.; Karlsson, J.-E.; Jonasson, J.; Gunnarsson, C. Homozygous missense MYBPC3 Pro873His mutation associated with increased risk for heart failure development in hypertrophic cardiomyopathy. ESC Heart Fail. 2018, 5, 716–723. [Google Scholar] [CrossRef]
- Maron, B.J.; Maron, M.S.; Semsarian, C. Double or compound sarcomere mutations in hypertrophic cardiomyopathy: A potential link to sudden death in the absence of conventional risk factors. Hear. Rhythm. 2012, 9, 57–63. [Google Scholar] [CrossRef]
- Miller, R.J.H.; Heidary, S.; Pavlovic, A.; Schlachter, A.; Dash, R.; Fleischmann, D.; Ashley, E.A.; Wheeler, M.T.; Yang, P.C. Defining genotype-phenotype relationships in patients with hypertrophic cardiomyopathy using cardiovascular magnetic resonance imaging. PLoS ONE 2019, 14, e0217612. [Google Scholar] [CrossRef] [PubMed]
- Junquera, M.R.; Salgado, M.; González-Urbistondo, F.; Alén, A.; Rodríguez-Reguero, J.J.; Silva, I.; Coto, E.; Avanzas, P.; Morís, C.; Gómez, J.; et al. Different Phenotypes in Monozygotic Twins, Carriers of the Same Pathogenic Variant for Hypertrophic Cardiomyopathy. Life 2022, 12, 1346. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, J.-M.; Cho, J.S. Phenotypic Diversity of Cardiomyopathy Caused by an MYBPC3 Frameshift Mutation in a Korean Family: A Case Report. Medicina 2021, 57, 281. [Google Scholar] [CrossRef] [PubMed]
- Dhandapany, P.S.; Sadayappan, S.; Xue, Y.; Powell, G.T.; Rani, D.S.; Nallari, P.; Rai, T.S.; Khullar, M.; Soares, P.; Bahl, A.; et al. A common MYBPC3 (cardiac myosin binding protein C) variant associated with cardiomyopathies in South Asia. Nat. Genet. 2009, 41, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Waldmüller, S. Novel deletions in MYH7 and MYBPC3 identified in Indian families with familial hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 2003, 35, 623–636. [Google Scholar] [CrossRef]
- Wessels, M.W.; Herkert, J.C.; Frohn-Mulder, I.M.; Dalinghaus, M.; Wijngaard, A.V.D.; De Krijger, R.; Michels, M.; de Coo, I.F.; Hoedemaekers, Y.M.; Dooijes, D. Compound heterozygous or homozygous truncating MYBPC3 mutations cause lethal cardiomyopathy with features of noncompaction and septal defects. Eur. J. Hum. Genet. 2015, 23, 922–928. [Google Scholar] [CrossRef]
- Jansen, M.; Schuldt, M.; van Driel, B.O.; Schmidt, A.F.; Christiaans, I.; van der Crabben, S.N.; Hoedemaekers, Y.M.; Dooijes, D.; Jongbloed, J.D.H.; Boven, L.G.; et al. Untargeted Metabolomics Identifies Potential Hypertrophic Car-diomyopathy Biomarkers in Carriers of MYBPC3 Founder Variants. Int. J. Mol. Sci. 2023, 24, 4031. [Google Scholar] [CrossRef]
- Thompson, A.D.; Helms, A.S.; Kannan, A.; Yob, J.; Lakdawala, N.K.; Wittekind, S.G.; Pereira, A.C.; Jacoby, D.L.; Colan, S.D.; Ashley, E.A.; et al. Computational prediction of protein subdomain stability in MYBPC3 enables clinical risk stratification in hypertrophic cardiomyopathy and enhances variant interpretation. Anesthesia Analg. 2021, 23, 1281–1287. [Google Scholar] [CrossRef]
- Parbhudayal, R.Y.; Seegers, C.; Croisille, P.; Clarysse, P.; van Rossum, A.C.; Germans, T.; van der Velden, J. Regional myocardial function at preclinical disease stage of hypertrophic cardiomyopathy in female gene variant carriers. Int. J. Cardiovasc. Imaging 2021, 37, 2001–2010. [Google Scholar] [CrossRef]
- Tarkiainen, M.; Sipola, P.; Jalanko, M.; Heliö, T.; Jääskeläinen, P.; Kivelä, K.; Laine, M.; Lauerma, K.; Kuusisto, J. CMR derived left ventricular septal convexity in carriers of the hypertrophic cardiomyopathy-causing MYBPC3-Q1061X mutation. Sci. Rep. 2019, 9, 5960. [Google Scholar] [CrossRef]
- Grover, S.; Lloyd, R.; Perry, R.; Lou, P.W.; Haan, E.; Yeates, L.; Woodman, R.; Atherton, J.J.; Semsarian, C.; Selvanayagam, J.B. Assessment of myocardial oxygenation, strain, and diastology in MYBPC3-related hypertrophic cardiomyopathy: A cardiovascular magnetic resonance and echocardiography study. Eur. Hear. J. Cardiovasc. Imaging 2019, 20, 932–938. [Google Scholar] [CrossRef]
- Bazrafshan, S.; Sibilia, R.; Girgla, S.; Viswanathan, S.K.; Puckelwartz, M.J.; Sangha, K.S.; Singh, R.R.; Kakroo, M.; Jandarov, R.; Harris, D.M.; et al. South Asian-Specific MYBPC3Δ25bp Deletion Carriers Display Hy-percontraction and Impaired Diastolic Function Under Exercise Stress. Front. Cardiovasc. Med. 2021, 8, 766339. [Google Scholar] [CrossRef]
- Nakashima, Y.; Kubo, T.; Sugiura, K.; Ochi, Y.; Takahashi, A.; Baba, Y.; Hirota, T.; Yamasaki, N.; Kimura, A.; Doi, Y.L.; et al. Lifelong Clinical Impact of the Presence of Sarcomere Gene Mutation in Japanese Patients with Hypertrophic Cardiomyopathy. Circ. J. 2020, 84, 1846–1853. [Google Scholar] [CrossRef]
- Velicki, L.; Jakovljevic, D.G.; Preveden, A.; Golubovic, M.; Bjelobrk, M.; Ilic, A.; Stojsic, S.; Barlocco, F.; Tafelmeier, M.; Okwose, N.; et al. Genetic determinants of clinical phenotype in hypertrophic cardiomyopathy. BMC Cardiovasc. Disord. 2020, 20, 516. [Google Scholar] [CrossRef]
- Ho, C.Y.; Day, S.M.; Ashley, E.A.; Michels, M.; Pereira, A.C.; Jacoby, D.; Lakdawala, N.K.; Ware, J.S.; Helms, A.S.; Colan, S.D.; et al. Response by Ho et al to Letter Regarding Article, “Genotype and Lifetime Burden of Disease in Hypertrophic Cardiomyopathy: Insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe)”. Circulation 2019, 139, 1559–1560. [Google Scholar] [CrossRef]
- Williams, N.; Marion, R.; McDonald, T.V.; Wang, D.; Zhou, B.; Eng, L.S.; Um, S.Y.; Lin, Y.; Ruiter, K.; Rojas, L.; et al. Phenotypic variations in carriers of predicted protein-truncating genetic variants in MYBPC3: An autopsy-based case series. Cardiovasc. Pathol. 2018, 37, 30–33. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Study | Country | Population | Results |
|---|---|---|---|
| Viswanathan et al. [6] | USA | 150 individuals (80 symptomatic HCM patients, 35 asymptomatic carriers and 35 non-carriers) | HCM phenotype was independent of the specific causative mutation, despite MYH7 and MYBPC3 being the predominant ones |
| Page et al. [55] | UK | 585 consecutive, unrelated individuals fulfilling diagnostic criteria for HCM | Families with MYBPC3 mutations exhibited diverse clinical presentations and prognosis, with no clear correlation between mutation type or specific mutation and the observed phenotype, indicating marked phenotypic variability within and between families sharing the same mutation. |
| Robyns et al. [73] | Belgium | 378 HCM patients | Individuals carrying MYBPC3 mutations exhibited a higher incidence of sudden cardiac death in comparison to carriers of troponin complex mutations, MYH7 mutations and those without any mutations. |
| Lopes et al. [74] | Portugal | 528 patients from the Portuguese Registry of Hypertrophic Cardiomyopathy (PRo-HCM) | Sarcomere-positive patients had distinct demographics, ECG, imaging features, family history and an increased risk of sudden cardiac death. The presence of a MYH7 mutation was associated with a progression towards left ventricular systolic dysfunction. |
| Terauchi et al. [75] | Japan | 61 subjects (28 families) carrying MYBPC3 mutations | Females exhibited delayed onset of the disease, but increased symptomatology at the time of diagnosis and a higher frequency of heart failure events after the development of hypertrophy. |
| Adalsteinsdottir et al. [76] | Iceland | 60 probands with HCM caused by MYBPC3 c.927-2A>G founder mutation and 225 first-degree relatives | Phenotypic expression was varied based on age, sex, and proband status. Men were more likely to develop left ventricular hypertrophy at a younger age and probands showed more prominent disease manifestations compared to relatives, with carriers without hypertrophy displaying subtle clinical differences from unaffected relatives. |
| Field et al. [77] | UK | 62 consecutive patients diagnosed with HCM under 18 years of age and carrying at least one P/LP MYBPC3 variant | Significant and progressive LVH was observed in the cohort, with non-obstructive, arrhythmic disease phenotypes. Gender differences were observed, with worse clinical outcomes in male patients and earlier onset of the disease in males compared to females. |
| Miller et al. [81] | USA | 273 HCM patients | Patients with MYBPC3 variants had a higher risk of impaired ventricular function and arrhythmic events, with a higher proportion receiving ICDs. Patients with identifiable gene variants had a higher LGE burden. |
| Helms et al. [54] | International | 1316 HCM patients from the Sarcomeric Human Cardiomyopathy Registry | Cardiac structure and clinical outcomes were similar in patients with truncating versus nontruncating variants. |
| Dhandapany et al. [84] | India | 800 cardiomyopathy cases and 699 controls | The 25-bp deletion was linked to a persistent risk of heart failure, delayed symptom onset, and mild hypertrophy. |
| Thompson et al. [88] | International | 120 HCM patients from the Sarcomeric Human Cardiomyopathy Registry | Individuals carrying MYBPC3 missense STRUM+ VUS had a high incidence of adverse clinical outcomes, similar to those observed in patients with pathogenic MYBPC3 variants. |
| Méndez et al. [72] | Spain | 79 HCM patients with a pathogenic MYBPC3 variant | Male carriers of the MYBPC3 c.2149-1G>A founder pathogenic variant had earlier onset and higher penetrance of HCM. Compared to MYBPC3 p.Arg502Trp/Gln carriers, they showed better LVEF and functional class and similar rates of major adverse outcomes. |
| Tarkiainen et al. [90] | Finland | 67 individuals (47 subjects with the MYBPC3-Q1061X Finnish founder mutation and 20 healthy relatives without the mutation) | Subjects carrying the MYBPC3-Q10961X mutation showed higher septal convexity regardless of the presence of LVH. |
| Nakashima et al. [93] | Japan | 211 HCM patients | Sarcomere gene mutation carriers experienced more morbid events, notably lethal arrhythmias. At enrollment, they showed a higher prevalence of non-sustained ventricular tachycardia, dilated HCM, increased interventricular wall thickness, and a lower incidence of apical hypertrophy. |
| Velicki et al. [94] | International | 63 HCM patients (48 with a MYBPC3 mutation and 15 with a MYH7 mutation) | Patients with MYH7 gene mutations had greater disease severity compared to those with MYBPC3 mutations. |
| Ho et al. [95] | International | 4591 HCM patients from the Sarcomeric Human Cardiomyopathy Registry | HCM patients with sarcomere mutations had more adverse outcomes compared to HCM patients without such mutations. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tudurachi, B.-S.; Zăvoi, A.; Leonte, A.; Țăpoi, L.; Ureche, C.; Bîrgoan, S.G.; Chiuariu, T.; Anghel, L.; Radu, R.; Sascău, R.A.; et al. An Update on MYBPC3 Gene Mutation in Hypertrophic Cardiomyopathy. Int. J. Mol. Sci. 2023, 24, 10510. https://doi.org/10.3390/ijms241310510
Tudurachi B-S, Zăvoi A, Leonte A, Țăpoi L, Ureche C, Bîrgoan SG, Chiuariu T, Anghel L, Radu R, Sascău RA, et al. An Update on MYBPC3 Gene Mutation in Hypertrophic Cardiomyopathy. International Journal of Molecular Sciences. 2023; 24(13):10510. https://doi.org/10.3390/ijms241310510
Chicago/Turabian StyleTudurachi, Bogdan-Sorin, Alexandra Zăvoi, Andreea Leonte, Laura Țăpoi, Carina Ureche, Silviu Gabriel Bîrgoan, Traian Chiuariu, Larisa Anghel, Rodica Radu, Radu Andy Sascău, and et al. 2023. "An Update on MYBPC3 Gene Mutation in Hypertrophic Cardiomyopathy" International Journal of Molecular Sciences 24, no. 13: 10510. https://doi.org/10.3390/ijms241310510
APA StyleTudurachi, B.-S., Zăvoi, A., Leonte, A., Țăpoi, L., Ureche, C., Bîrgoan, S. G., Chiuariu, T., Anghel, L., Radu, R., Sascău, R. A., & Stătescu, C. (2023). An Update on MYBPC3 Gene Mutation in Hypertrophic Cardiomyopathy. International Journal of Molecular Sciences, 24(13), 10510. https://doi.org/10.3390/ijms241310510