
Target Identification in Anti-Tuberculosis Drug Discovery


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Challenges in TB Drug Discovery
3. Emerging Mtb Drug Targets
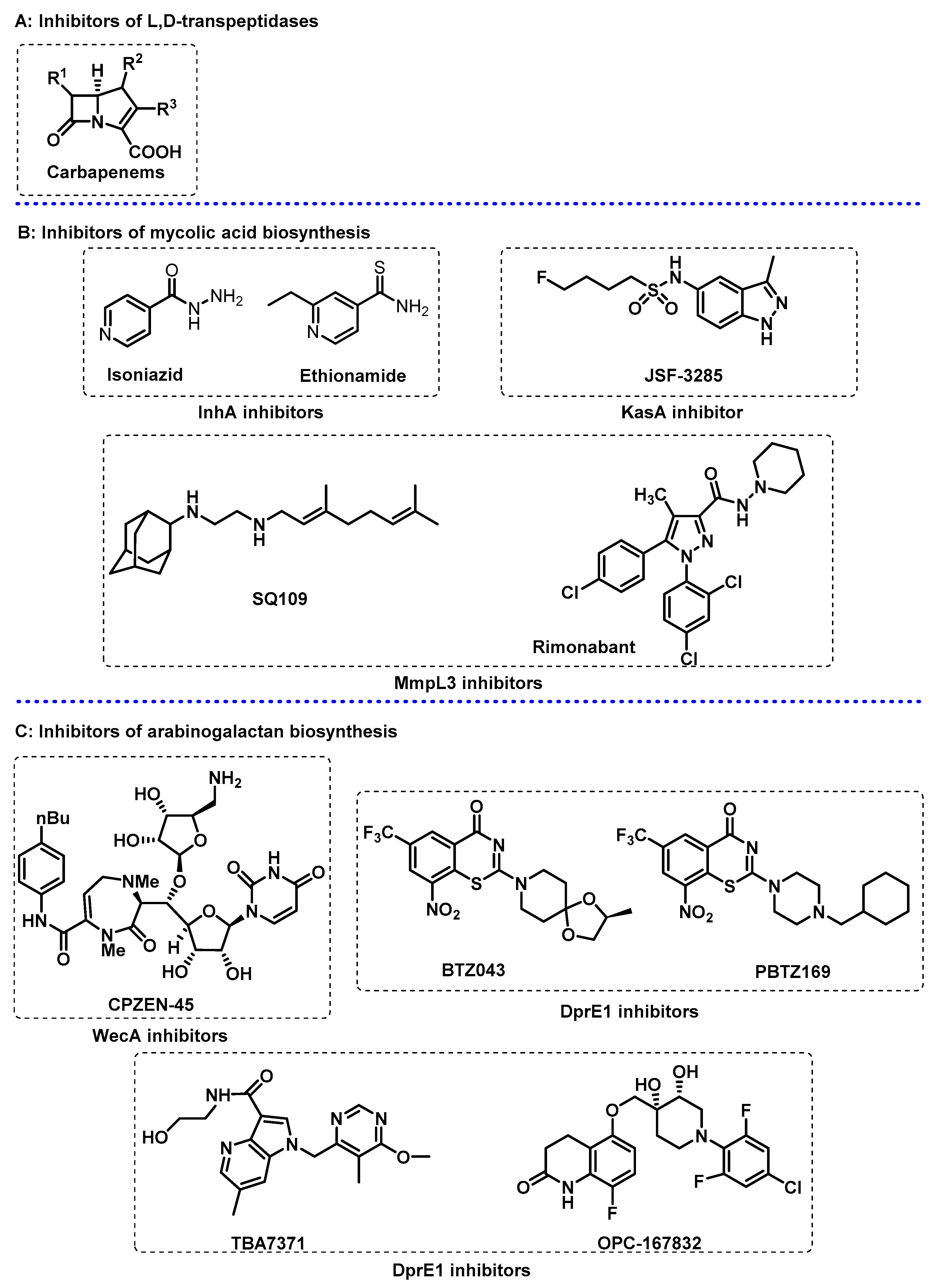
3.1. Cell Wall
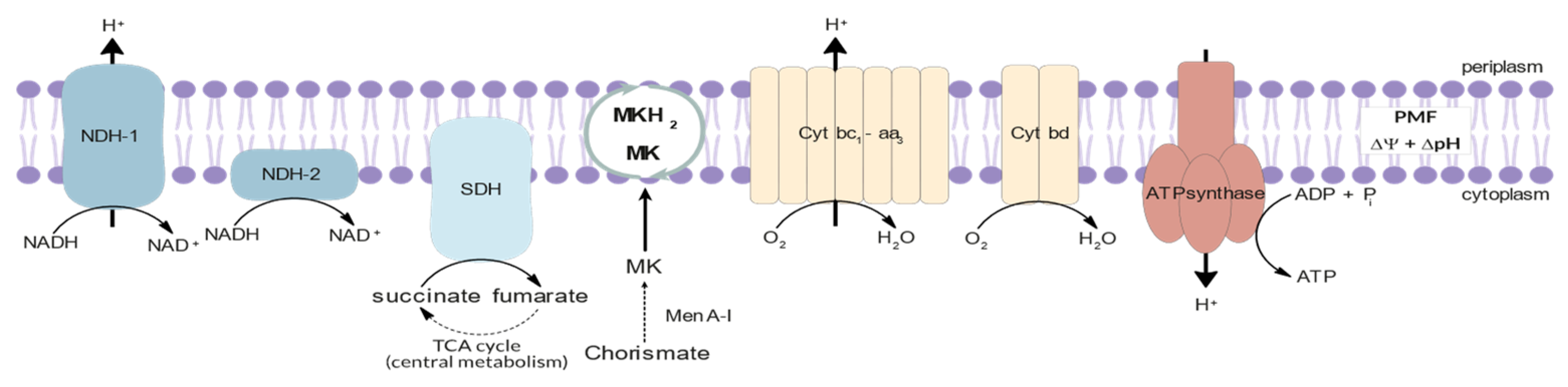
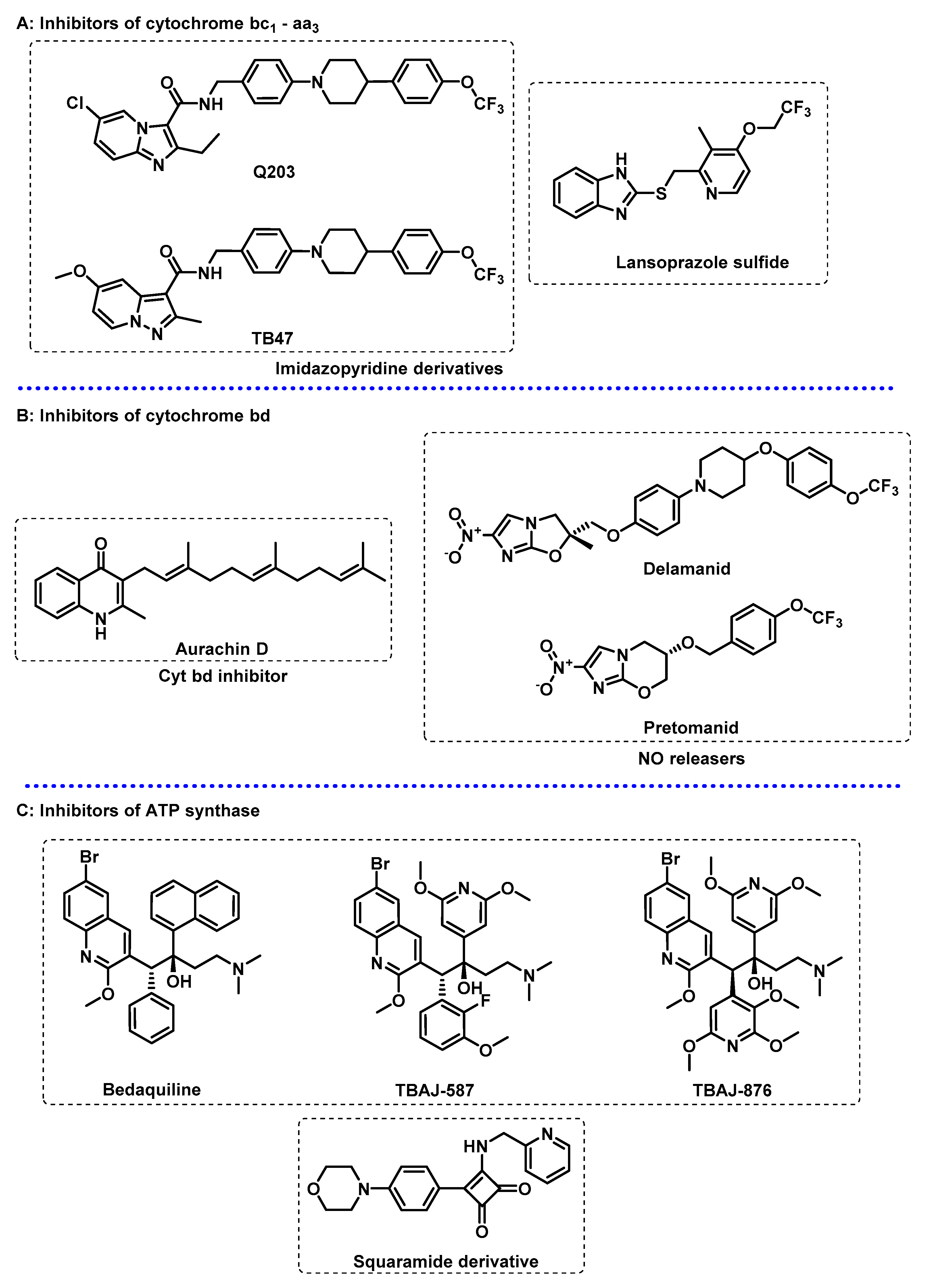
3.2. Energy Metabolism
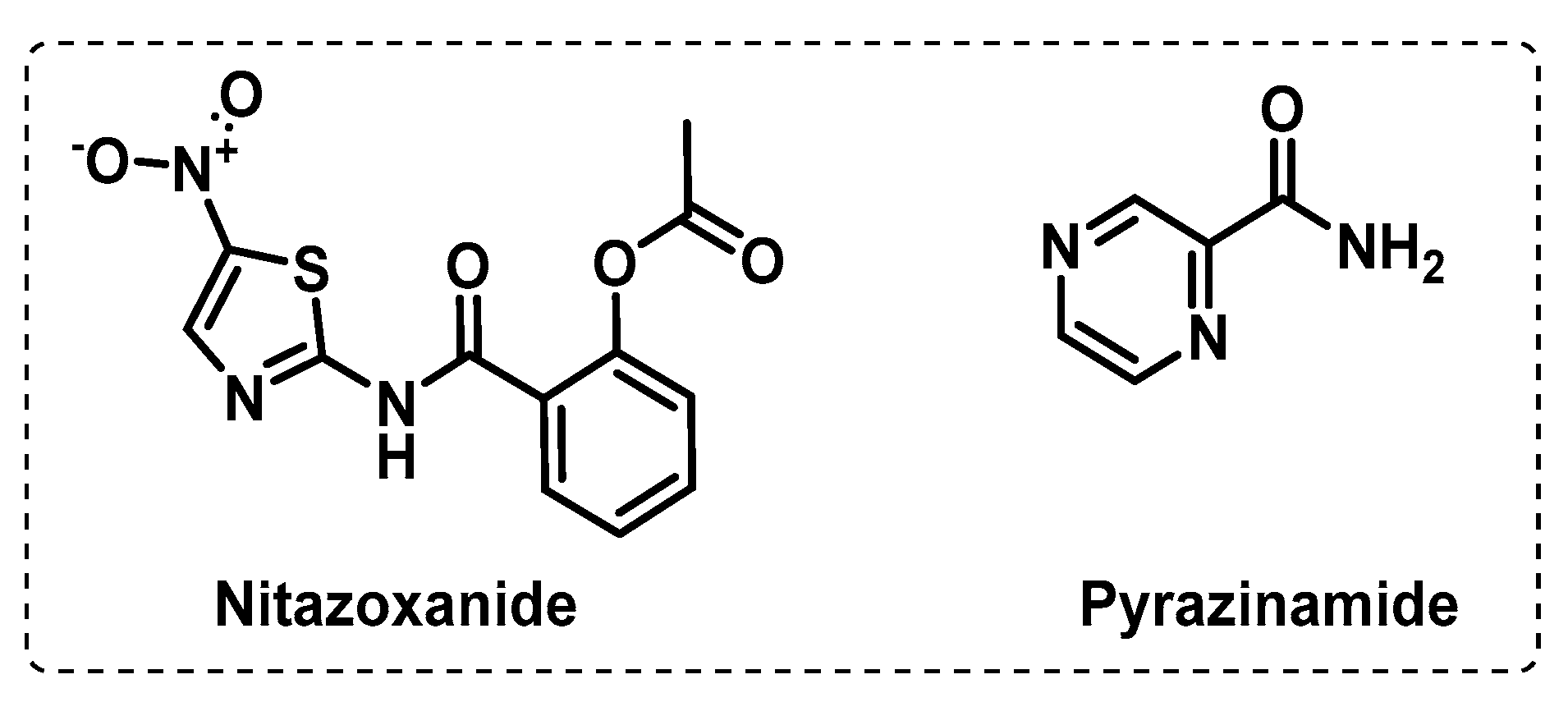
3.3. Other Targets
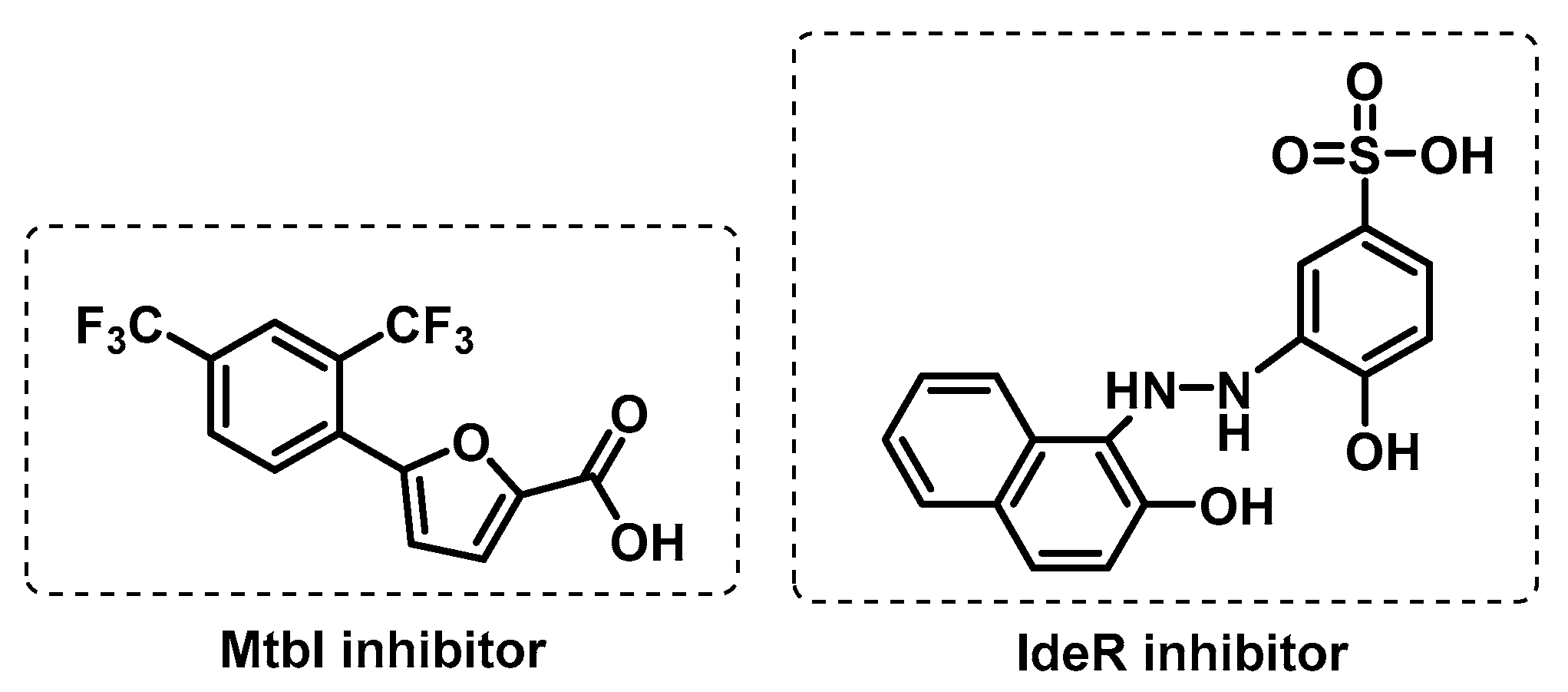
3.3.1. Iron Uptake
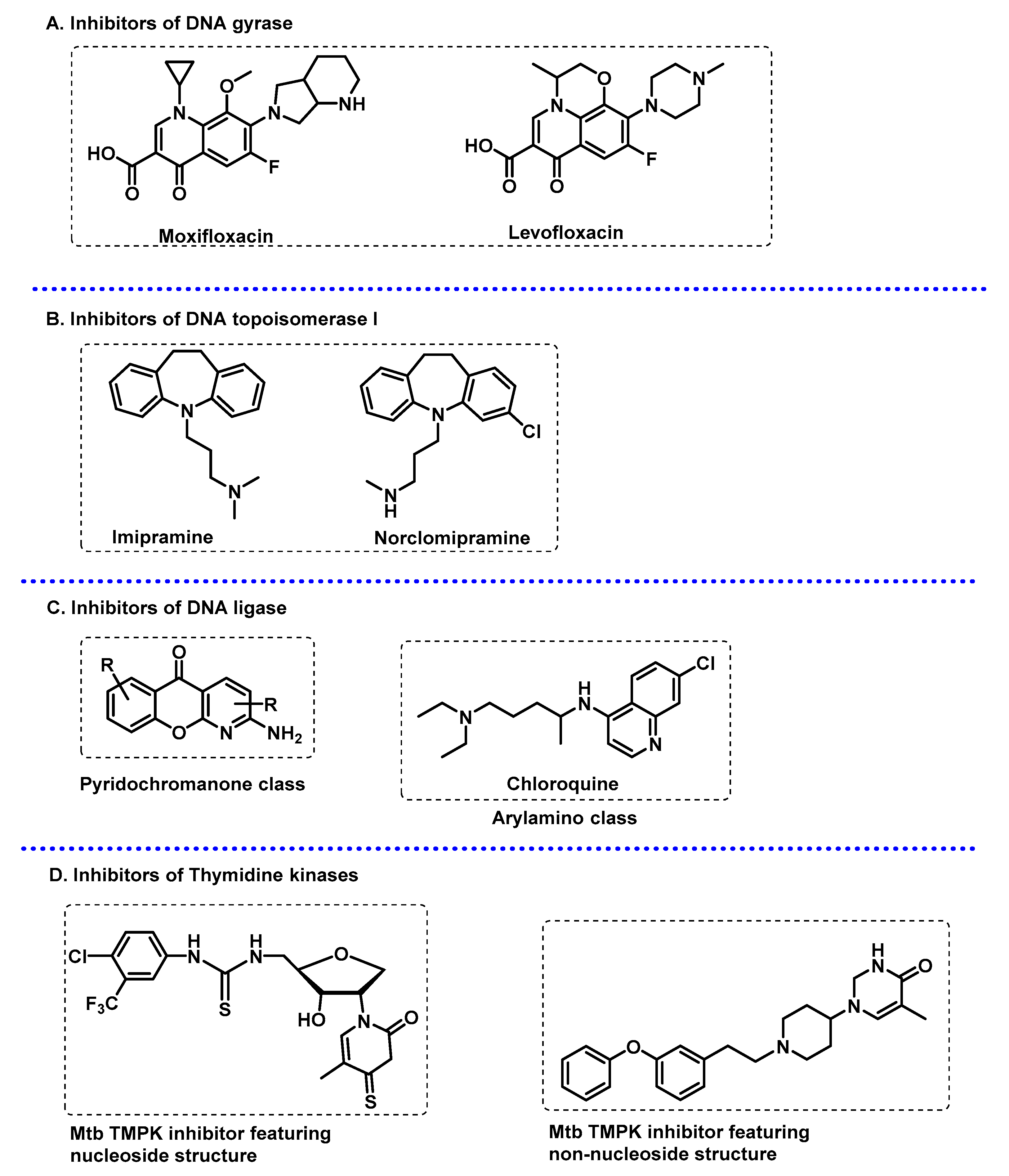
3.3.2. DNA-Related Enzymes
4. Chemical Probes for Target Identification in Mycobacteria
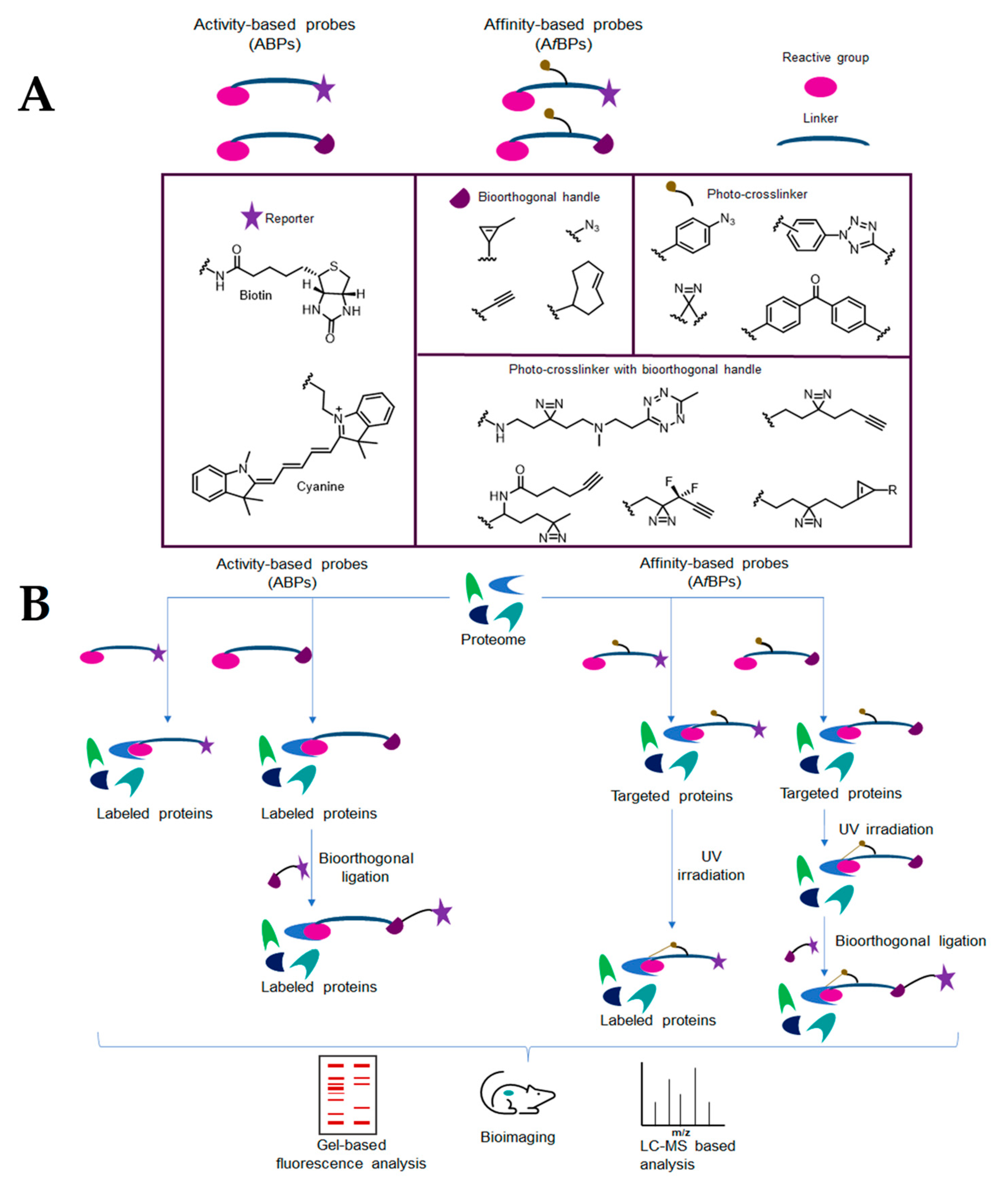
4.1. Activity-Based Protein Profiling (ABPP)
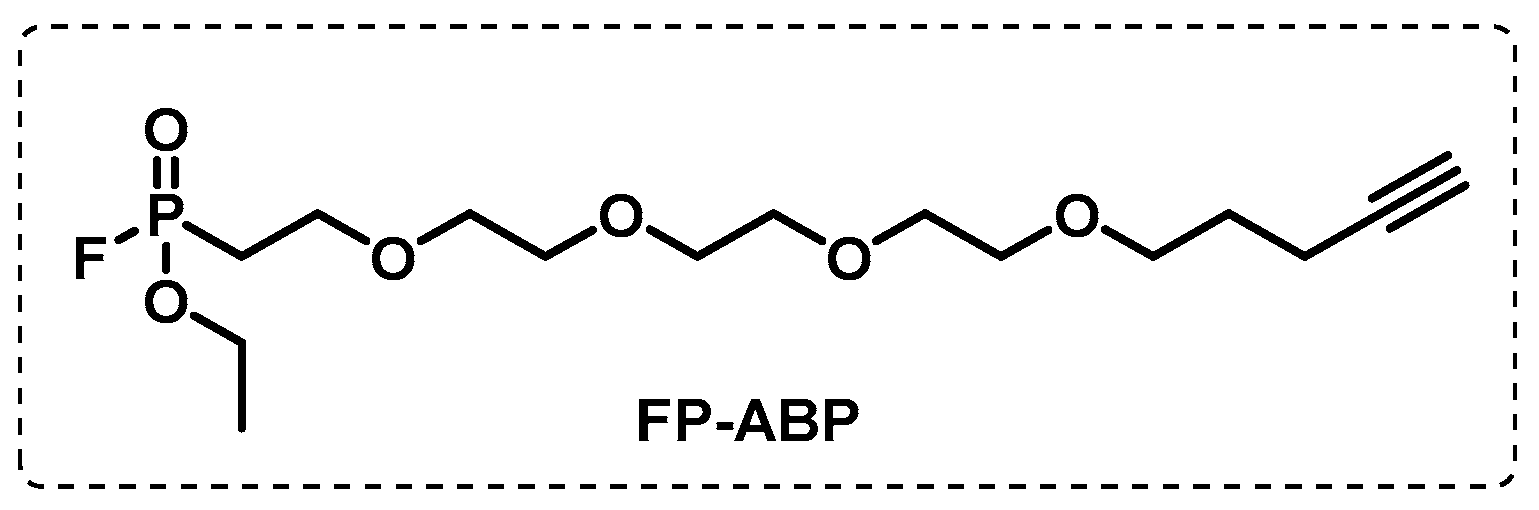
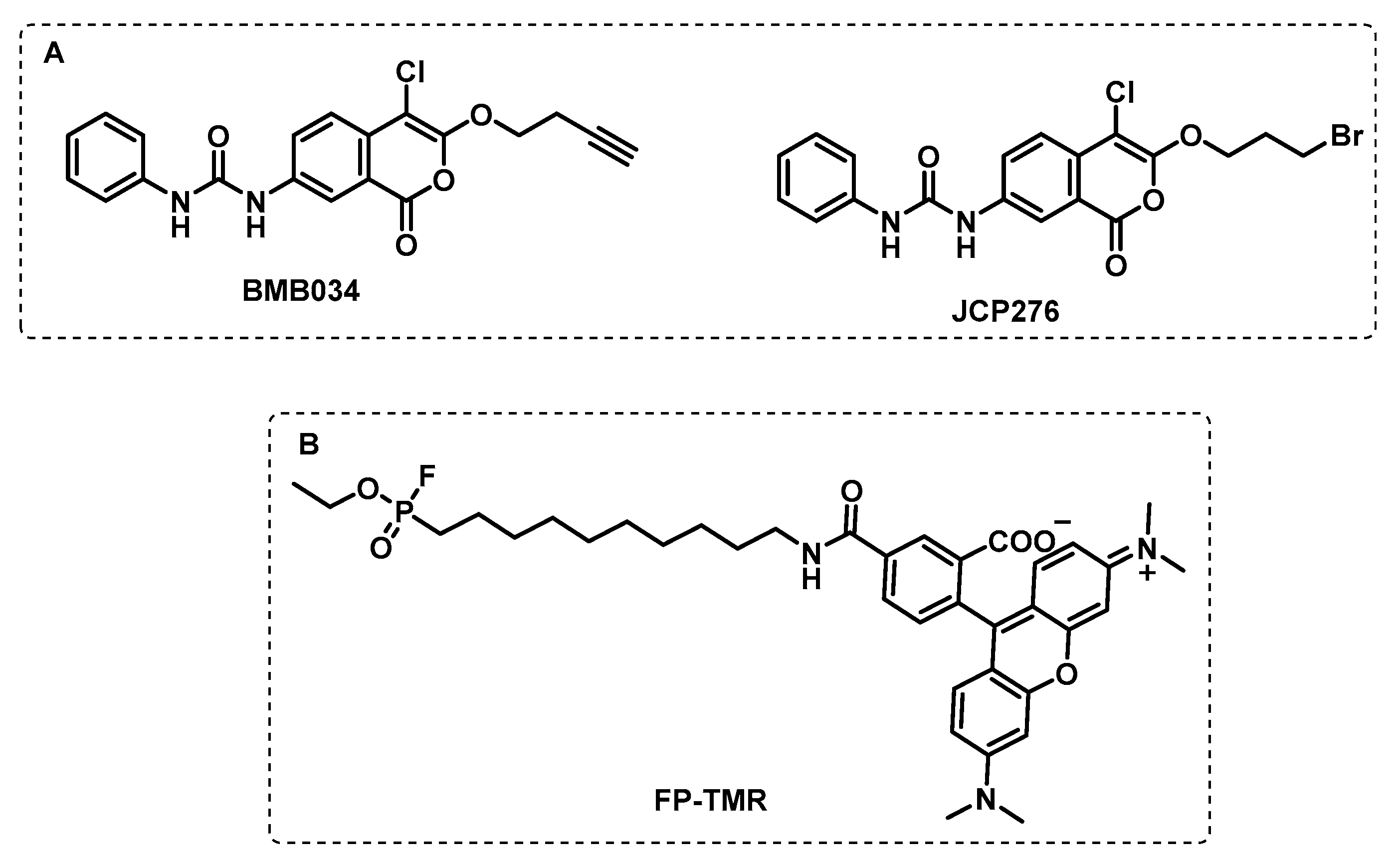
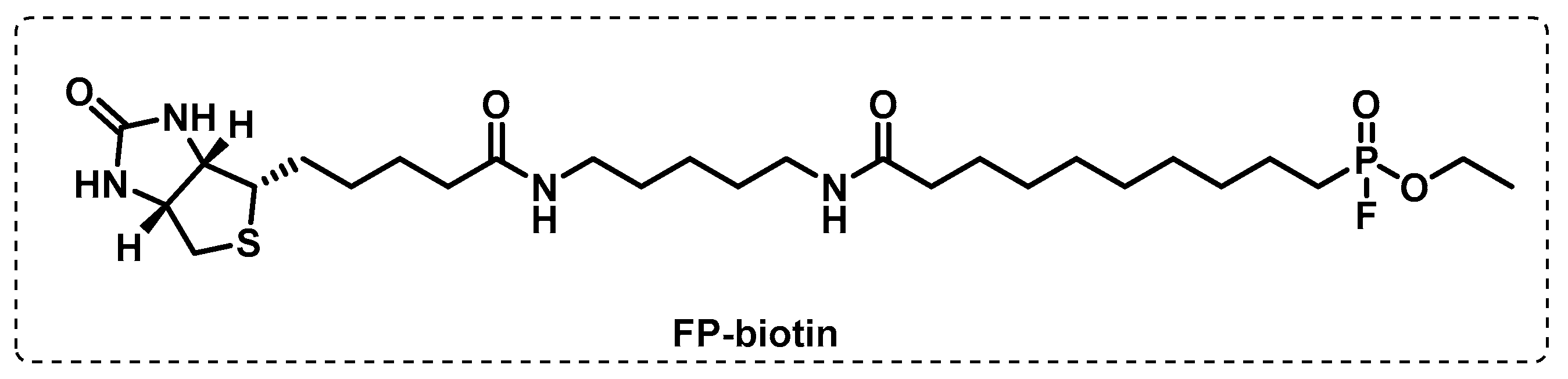
4.1.1. Cytosolic Serine Hydrolases
4.1.2. Membrane Serine and Cysteine Hydrolases
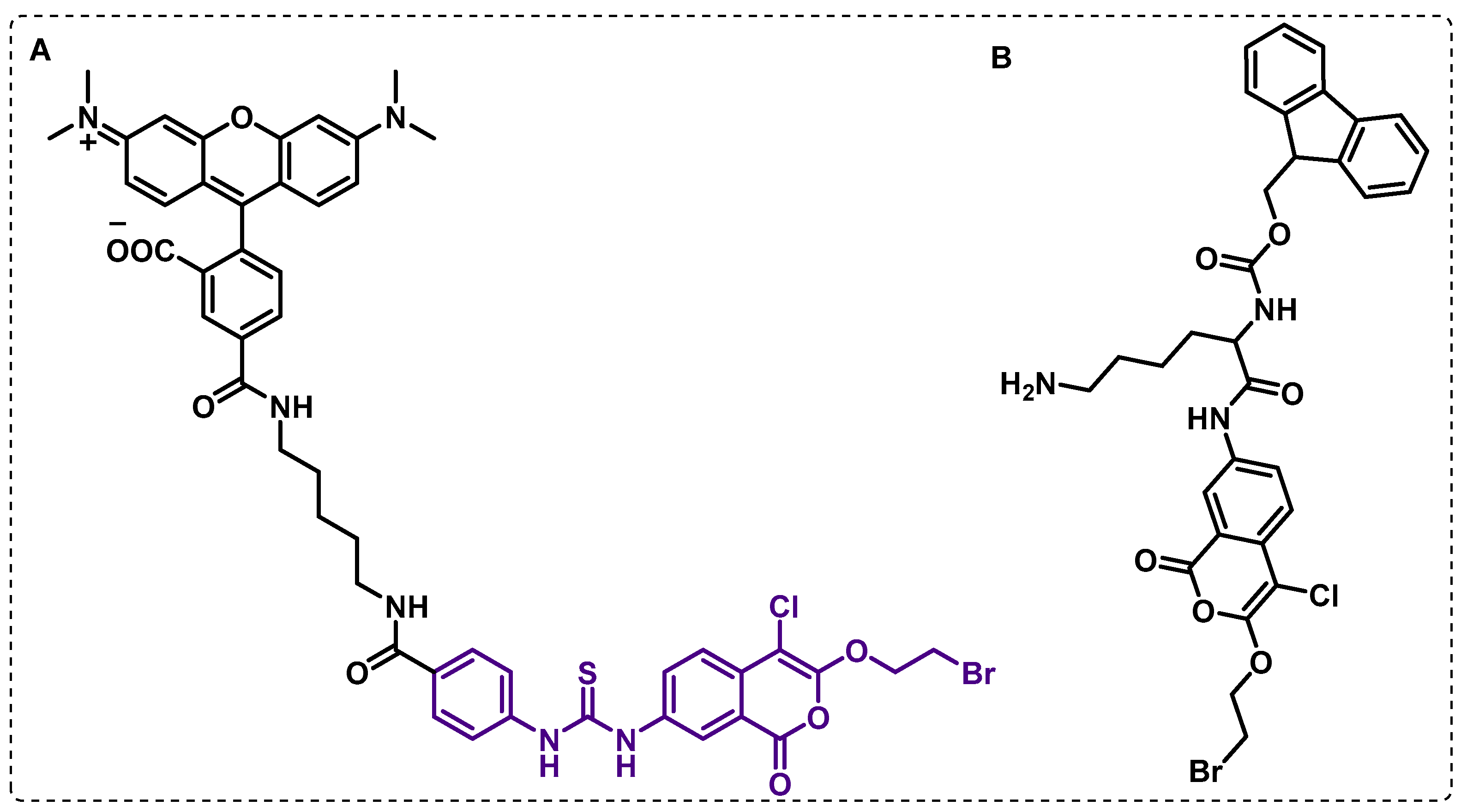
4.1.3. Other Membrane Targets
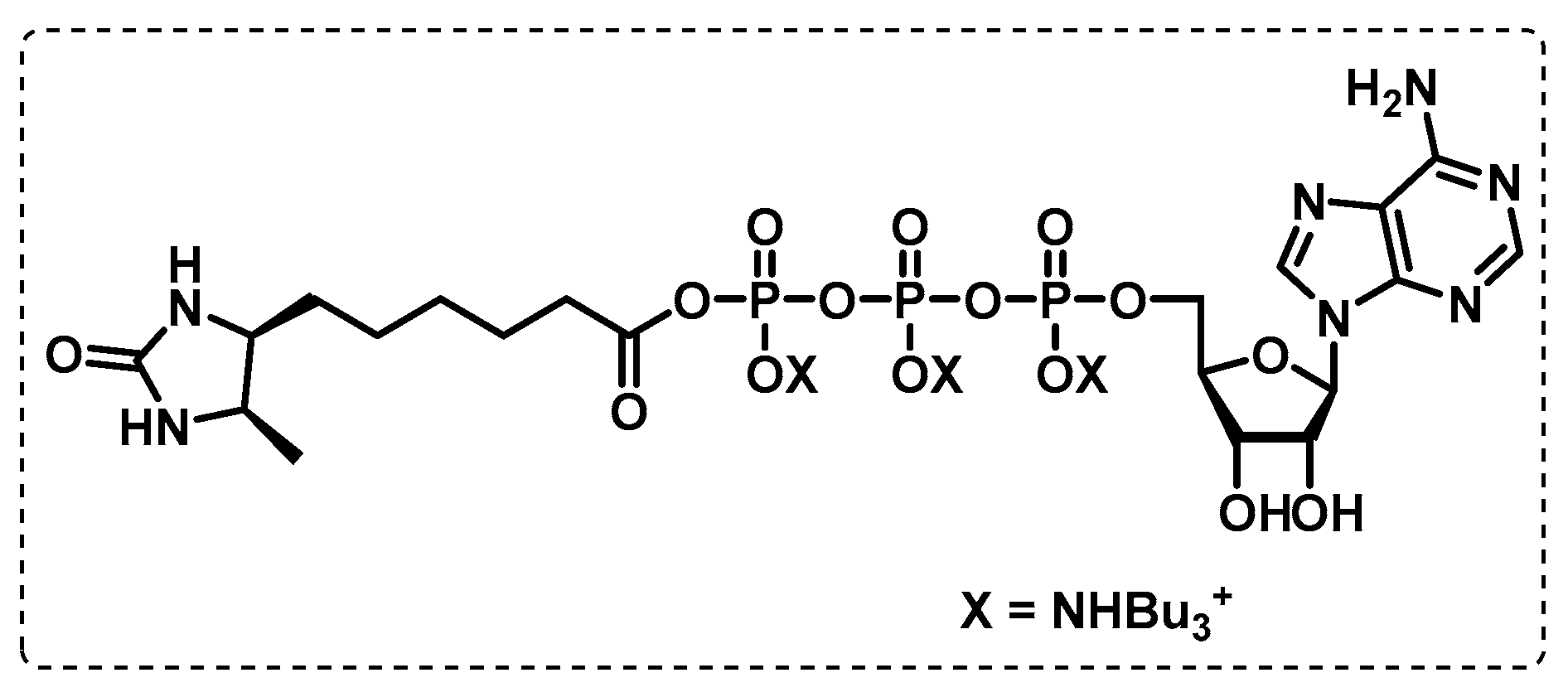
4.1.4. ATP-Binding Enzymes
4.2. Affinity-Based Probes (AfBPs)
5. Conclusions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Gagneux, S. Ecology and evolution of Mycobacterium tuberculosis. Nat. Rev. Microbiol. 2018, 16, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Mashabela Gabriel, T.; de Wet Timothy, J.; Warner Digby, F. Mycobacterium tuberculosis Metabolism. Microbiol. Spectr. 2019, 7, 7-4. [Google Scholar] [CrossRef] [PubMed]
- Woodman, M.; Haeusler, I.L.; Grandjean, L. Tuberculosis Genetic Epidemiology: A Latin American Perspective. Genes 2019, 10, 53. [Google Scholar] [CrossRef] [PubMed]
- Kiazyk, S.; Ball, T.B. Latent tuberculosis infection: An overview. Can. Commun. Dis. Rep. 2017, 43, 62–66. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Tuberculosis Report 2022; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- Campaniço, A.; Moreira, R.; Lopes, F. Drug discovery in tuberculosis. New drug targets and antimycobacterial agents. Eur. J. Med. Chem. 2018, 150, 525–545. [Google Scholar] [CrossRef]
- Tiberi, S.; du Plessis, N.; Walzl, G.; Vjecha, M.J.; Rao, M.; Ntoumi, F.; Mfinanga, S.; Kapata, N.; Mwaba, P.; McHugh, T.D.; et al. Tuberculosis: Progress and advances in development of new drugs, treatment regimens, and host-directed therapies. Lancet Infect. Dis. 2018, 18, 183–198. [Google Scholar] [CrossRef]
- Reid, M.J.A.; Arinaminpathy, N.; Bloom, A.; Bloom, B.R.; Boehme, C.; Chaisson, R.; Chin, D.P.; Churchyard, G.; Cox, H.; Ditiu, L.; et al. Building a tuberculosis-free world: The Lancet Commission on tuberculosis. Lancet 2019, 393, 1331–1384. [Google Scholar] [CrossRef]
- Nuermberger Eric, L. Preclinical Efficacy Testing of New Drug Candidates. Microbiol. Spectr. 2017, 5, 1–22. [Google Scholar] [CrossRef]
- Şenol, G. Recent and New Strategies for Extensively Drug-Resistant Tuberculosis. Mediterr. J. Infect. Microbes Antimicrob. 2018, 7, 22. [Google Scholar] [CrossRef]
- Gupta, V.K.; Kumar, M.M.; Singh, D.; Bisht, D.; Sharma, S. Drug targets in dormant Mycobacterium tuberculosis: Can the conquest against tuberculosis become a reality? Infect. Dis. 2018, 50, 81–94. [Google Scholar] [CrossRef]
- Reddy, D.S.; Sinha, A.; Kumar, A.; Saini, V.K. Drug re-engineering and repurposing: A significant and rapid approach to tuberculosis drug discovery. Arch. Der Pharm. 2022, 355, 1–26. [Google Scholar] [CrossRef]
- Macalino, S.J.Y.; Billones, J.B.; Organo, V.G.; Carrillo, M.C.O. In Silico Strategies in Tuberculosis Drug Discovery. Molecules 2020, 25, 665. [Google Scholar] [CrossRef] [PubMed]
- Huszár, S.; Chibale, K.; Singh, V. The quest for the holy grail: New antitubercular chemical entities, targets and strategies. Drug Discov. Today 2020, 25, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Velayati, A.A.; Farnia, P.; Hoffner, S. Drug-resistant Mycobacterium tuberculosis: Epidemiology and role of morphological alterations. J. Glob. Antimicrob. Resist. 2018, 12, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Sarathy, J.; Dartois, V.; Dick, T.; Gengenbacher, M. Reduced Drug Uptake in Phenotypically Resistant Nutrient-Starved Nonreplicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2013, 57, 1648–1653. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Chettiar, S.; Parish, T. Current challenges in drug discovery for tuberculosis. Expert Opin. Drug Discov. 2017, 12, 1–4. [Google Scholar] [CrossRef]
- da Silva, P.E.A.; Machado, D.; Ramos, D.; Couto, I.; Von Groll, A.; Viveiros, M. Efflux Pumps in Mycobacteria: Antimicrobial Resistance, Physiological Functions, and Role in Pathogenicity. In Efflux-Mediated Antimicrobial Resistance in Bacteria: Mechanisms, Regulation and Clinical Implications; Li, X.-Z., Elkins, C.A., Zgurskaya, H.I., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 527–559. [Google Scholar] [CrossRef]
- Eoh, H.; Wang, Z.; Layre, E.; Rath, P.; Morris, R.; Branch Moody, D.; Rhee, K.Y. Metabolic anticipation in Mycobacterium tuberculosis. Nat. Microbiol. 2017, 2, 17084. [Google Scholar] [CrossRef]
- Sharma, S.; Sharma, D.; Kalia, N.P. Editorial: Approaches to Address Resistance, Drug Discovery, and Vaccine Development in Mycobacterium tuberculosis: Challenges and Opportunities. Front. Microbiol. 2022, 13, 871464. [Google Scholar] [CrossRef]
- Tomasi, F.G.; Rubin, E.J. Failing upwards: Genetics-based strategies to improve antibiotic discovery and efficacy in Mycobacterium tuberculosis. Front. Cell. Infect. Microbiol. 2022, 12, 932556. [Google Scholar] [CrossRef] [PubMed]
- Mugumbate, G.; Mendes, V.; Blaszczyk, M.; Sabbah, M.; Papadatos, G.; Lelievre, J.; Ballell, L.; Barros, D.; Abell, C.; Blundell, T.L.; et al. Target Identification of Mycobacterium tuberculosis Phenotypic Hits Using a Concerted Chemogenomic, Biophysical, and Structural Approach. Front. Pharmacol. 2017, 8, 00681. [Google Scholar] [CrossRef]
- Lechartier, B.; Rybniker, J.; Zumla, A.; Cole, S.T. Tuberculosis drug discovery in the post-post-genomic era. EMBO Mol. Med. 2014, 6, 158–168. [Google Scholar] [CrossRef]
- Borsari, C.; Ferrari, S.; Venturelli, A.; Costi, M.P. Target-based approaches for the discovery of new antimycobacterial drugs. Drug Discov. Today 2017, 22, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Sang, H.C.; Warit, S.; Wan, B.; Chang, H.H.; Guido, F.P.; Scott, G.F. Low-Oxygen-Recovery Assay for High-Throughput Screening of Compounds against Nonreplicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2007, 51, 1380–1385. [Google Scholar] [CrossRef]
- Wayne, L.G.; Hayes, L.G. An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect. Immun. 1996, 64, 2062–2069. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Sala, C.; Hartkoorn Ruben, C.; Dhar, N.; Mendoza-Losana, A.; Cole Stewart, T. Streptomycin-Starved Mycobacterium tuberculosis 18b, a Drug Discovery Tool for Latent Tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 5782–5789. [Google Scholar] [CrossRef]
- Darby, C.M.; Ingólfsson, H.I.; Jiang, X.; Shen, C.; Sun, M.; Zhao, N.; Burns, K.; Liu, G.; Ehrt, S.; Warren, J.D.; et al. Whole Cell Screen for Inhibitors of pH Homeostasis in Mycobacterium tuberculosis. PLoS ONE 2013, 8, e68942. [Google Scholar] [CrossRef]
- Gold, B.; Warrier, T.; Nathan, C. A Multistress Model for High Throughput Screening (HTS) Against Nonreplicating Mycobacterium tuberculosis (M. tuberculosis). In Mycobacteria Protocols; Parish, T., Kumar, A., Eds.; Springer: New York, NY, USA, 2021; pp. 611–635. [Google Scholar] [CrossRef]
- Aguilar-Ayala, D.A.; Cnockaert, M.; Vandamme, P.; Palomino, J.C.; Martin, A.; Gonzalez-Y-Merchand, J. Antimicrobial activity against Mycobacterium tuberculosis under in vitro lipid-rich dormancy conditions. J. Med. Microbiol. 2018, 67, 282–285. [Google Scholar] [CrossRef]
- Wang, F.; Sambandan, D.; Halder, R.; Wang, J.; Batt, S.M.; Weinrick, B.; Ahmad, I.; Yang, P.; Zhang, Y.; Kim, J.; et al. Identification of a small molecule with activity against drug-resistant and persistent tuberculosis. Proc. Natl. Acad. Sci. USA 2013, 110, E2510–E2517. [Google Scholar] [CrossRef]
- Egorova, A.; Salina, E.G.; Makarov, V. Targeting Non-Replicating Mycobacterium tuberculosis and Latent Infection: Alternatives and Perspectives (Mini-Review). Int. J. Mol. Sci. 2021, 22, 13317. [Google Scholar] [CrossRef]
- Pai, M.; Behr, M.A.; Dowdy, D.; Dheda, K.; Divangahi, M.; Boehme, C.C.; Ginsberg, A.; Swaminathan, S.; Spigelman, M.; Getahun, H.; et al. Tuberculosis. Nat. Rev. Dis. Prim. 2016, 2, 16076. [Google Scholar] [CrossRef]
- Dartois, V.A.; Rubin, E.J. Anti-tuberculosis treatment strategies and drug development: Challenges and priorities. Nat. Rev. Microbiol. 2022, 20, 685–701. [Google Scholar] [CrossRef]
- Perveen, S.; Sharma, R. Screening approaches and therapeutic targets: The two driving wheels of tuberculosis drug discovery. Biochem. Pharmacol. 2022, 197, 114906. [Google Scholar] [CrossRef]
- Abrahams, K.A.; Besra, G.S. Mycobacterial drug discovery. RSC Med. Chem. 2020, 11, 1354–1365. [Google Scholar] [CrossRef]
- Xu, X.; Dong, B.; Peng, L.; Gao, C.; He, Z.; Wang, C.; Zeng, J. Anti-tuberculosis drug development via targeting the cell envelope of Mycobacterium tuberculosis. Front. Microbiol. 2022, 13, 1056608. [Google Scholar] [CrossRef]
- Shetye, G.S.; Franzblau, S.G.; Cho, S. New tuberculosis drug targets, their inhibitors, and potential therapeutic impact. Transl. Res. 2020, 220, 68–97. [Google Scholar] [CrossRef]
- Nataraj, V.; Varela, C.; Javid, A.; Singh, A.; Besra, G.S.; Bhatt, A. Mycolic acids: Deciphering and targeting the Achilles’ heel of the tubercle bacillus. Mol. Microbiol. 2015, 98, 7–16. [Google Scholar] [CrossRef]
- PaweŁczyk, J.; Kremer, L. The Molecular Genetics of Mycolic Acid Biosynthesis. Microbiol. Spectr. 2014, 2, 611–631. [Google Scholar] [CrossRef] [PubMed]
- DeJesus Michael, A.; Gerrick Elias, R.; Xu, W.; Park Sae, W.; Long Jarukit, E.; Boutte Cara, C.; Rubin Eric, J.; Schnappinger, D.; Ehrt, S.; Fortune Sarah, M.; et al. Comprehensive Essentiality Analysis of the Mycobacterium tuberculosis Genome via Saturating Transposon Mutagenesis. mBio 2017, 8, e02133-16. [Google Scholar] [CrossRef] [PubMed]
- Inoyama, D.; Awasthi, D.; Capodagli, G.C.; Tsotetsi, K.; Sukheja, P.; Zimmerman, M.; Li, S.-G.; Jadhav, R.; Russo, R.; Wang, X.; et al. A Preclinical Candidate Targeting Mycobacterium tuberculosis KasA. Cell Chem. Biol. 2020, 27, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, K.A.; Chung, C.-W.; Ghidelli-Disse, S.; Rullas, J.; Rebollo-López, M.J.; Gurcha, S.S.; Cox, J.A.G.; Mendoza, A.; Jiménez-Navarro, E.; Martínez-Martínez, M.S.; et al. Identification of KasA as the cellular target of an anti-tubercular scaffold. Nat. Commun. 2016, 7, 12581. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Capodagli Glenn, C.; Awasthi, D.; Shrestha, R.; Maharaja, K.; Sukheja, P.; Li, S.-G.; Inoyama, D.; Zimmerman, M.; Hsin, P.H.L.; et al. Synergistic Lethality of a Binary Inhibitor of Mycobacterium tuberculosis KasA. mBio 2018, 9, e02101–e02117. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, R.; Shingare, R.D.; Kumar, V.; Anand, A.B.S.; Veeraraghavan, S.; Viswanadha, S.; Ummanni, R.; Gokhale, R.; Srinivasa Reddy, D. Repurposing of a drug scaffold: Identification of novel sila analogues of rimonabant as potent antitubercular agents. Eur. J. Med. Chem. 2016, 122, 723–730. [Google Scholar] [CrossRef]
- Kwofie, S.K.; Hanson, G.; Sasu, H.; Enninful, K.S.; Mensah, F.A.; Nortey, R.T.; Yeboah, O.P.; Agoni, C.; Wilson, M.D. Molecular Modelling and Atomistic Insights into the Binding Mechanism of MmpL3 Mtb. Chem. Biodivers. 2022, 19, e202200160. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, K.A.; Besra, G.S. Mycobacterial cell wall biosynthesis: A multifaceted antibiotic target. Parasitology 2018, 145, 116–133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhao, Y.; Gao, Y.; Wu, L.; Gao, R.; Zhang, Q.; Wang, Y.; Wu, C.; Wu, F.; Gurcha, S.S.; et al. Structures of cell wall arabinosyltransferases with the anti-tuberculosis drug ethambutol. Science 2020, 368, 1211–1219. [Google Scholar] [CrossRef]
- Kastrinsky, D.B.; McBride, N.S.; Backus, K.M.; LeBlanc, J.J.; Barry, C.E. 1.04—Mycolic Acid/Cyclopropane Fatty Acid/Fatty Acid Biosynthesis and Health Relations. In Comprehensive Natural Products II; Liu, H.-W., Mander, L., Eds.; Elsevier: Oxford, UK, 2010; pp. 65–145. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, Y.; Gao, R.; Li, J.; Yang, X.; Gao, Y.; Zhao, W.; Gurcha, S.S.; Veerapen, N.; Batt, S.M.; et al. Cryo-EM snapshots of mycobacterial arabinosyltransferase complex EmbB2-AcpM2. Protein Cell 2020, 11, 505–517. [Google Scholar] [CrossRef]
- Goude, R.; Amin, A.G.; Chatterjee, D.; Parish, T. The Arabinosyltransferase EmbC Is Inhibited by Ethambutol in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2009, 53, 4138–4146. [Google Scholar] [CrossRef]
- Meniche, X.; de Sousa-d’Auria, C.; Van-der-Rest, B.; Bhamidi, S.; Huc, E.; Huang, H.; De Paepe, D.; Tropis, M.; McNeil, M.; Daffé, M.; et al. Partial redundancy in the synthesis of the d-arabinose incorporated in the cell wall arabinan of Corynebacterineae. Microbiology 2008, 154, 2315–2326. [Google Scholar] [CrossRef]
- Xu, L.; Qian, L.; Kang, J.; Sha, S.; Xin, Y.; Lu, S.; Ma, Y. Down-regulation of N-acetylglucosamine-1-phosphate transferase (WecA) enhanced the sensitivity of Mycobacterium smegmatis against rifampin. J. Appl. Microbiol. 2016, 121, 966–972. [Google Scholar] [CrossRef]
- Young, E.F.; Durham, P.G.; Perkowski, E.F.; Malik, S.; Hickey, A.J.; Braunstein, M. Efficacy of inhaled CPZEN-45 in treating tuberculosis in the guinea pig. Tuberculosis 2022, 135, 102207. [Google Scholar] [CrossRef]
- Sammartino, J.C.; Morici, M.; Stelitano, G.; Degiacomi, G.; Riccardi, G.; Chiarelli, L.R. Functional investigation of the antitubercular drug target Decaprenylphosphoryl-β-D-ribofuranose-2-epimerase DprE1/DprE2 complex. Biochem. Biophys. Res. Commun. 2022, 607, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, S.; Kumar, S.; Parkesh, R. Chemical Space Exploration of DprE1 Inhibitors Using Chemoinformatics and Artificial Intelligence. ACS Omega 2021, 6, 14430–14441. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, I.K.; Bajeli, S.; Akela, A.K.; Kumar, A. Bioenergetics of Mycobacterium: An Emerging Landscape for Drug Discovery. Pathogens 2018, 7, 24. [Google Scholar] [CrossRef]
- Wani, M.A.; Dhaked, D.K. Targeting the cytochrome bc1 complex for drug development in M. tuberculosis: Review. Mol. Divers. 2022, 26, 2949–2965. [Google Scholar] [CrossRef] [PubMed]
- Hasenoehrl, E.J.; Wiggins, T.J.; Berney, M. Bioenergetic Inhibitors: Antibiotic Efficacy and Mechanisms of Action in Mycobacterium tuberculosis. Front. Cell. Infect. Microbiol. 2021, 10, 611683. [Google Scholar] [CrossRef]
- Bajeli, S.; Baid, N.; Kaur, M.; Pawar, G.P.; Chaudhari, V.D.; Kumar, A. Terminal Respiratory Oxidases: A Targetables Vulnerability of Mycobacterial Bioenergetics? Front. Cell. Infect. Microbiol. 2020, 10, 589318. [Google Scholar] [CrossRef] [PubMed]
- Foo, C.S.; Pethe, K.; Lupien, A. Oxidative Phosphorylation—An Update on a New, Essential Target Space for Drug Discovery in Mycobacterium tuberculosis. Appl. Sci. 2020, 10, 2339. [Google Scholar] [CrossRef]
- Borisov, V.B.; Forte, E. Bioenergetics and Reactive Nitrogen Species in Bacteria. Int. J. Mol. Sci. 2022, 23, 7321. [Google Scholar] [CrossRef]
- Wiseman, B.; Nitharwal, R.G.; Fedotovskaya, O.; Schäfer, J.; Guo, H.; Kuang, Q.; Benlekbir, S.; Sjöstrand, D.; Ädelroth, P.; Rubinstein, J.L.; et al. Structure of a functional obligate complex III2IV2 respiratory supercomplex from Mycobacterium smegmatis. Nat. Struct. Mol. Biol. 2018, 25, 1128–1136. [Google Scholar] [CrossRef]
- Gregory, A.H.; Anne, E.M.B.; Singh, M.; Jayaraman, K.; Leslie, A.W.; Rachel, L.K.; Janessa, S.A.; Flentie, K.; Miranda, E.S.; Gaggioli, M.; et al. Identification of 4-Amino-Thieno [2,3-d]Pyrimidines as QcrB Inhibitors in Mycobacterium tuberculosis. mSphere 2019, 4, e00606–e00619. [Google Scholar] [CrossRef]
- Lupien, A.; Foo, C.S.-Y.; Savina, S.; Vocat, A.; Piton, J.; Monakhova, N.; Benjak, A.; Lamprecht, D.A.; Steyn, A.J.C.; Pethe, K.; et al. New 2-Ethylthio-4-methylaminoquinazoline derivatives inhibiting two subunits of cytochrome bc1 in Mycobacterium tuberculosis. PLoS Pathog. 2020, 16, e1008270. [Google Scholar] [CrossRef]
- Moraski, G.C.; Deboosère, N.; Marshall, K.L.; Weaver, H.A.; Vandeputte, A.; Hastings, C.; Woolhiser, L.; Lenaerts, A.J.; Brodin, P.; Miller, M.J. Intracellular and in vivo evaluation of imidazo[2,1-b]thiazole-5-carboxamide anti-tuberculosis compounds. PLoS ONE 2020, 15, e0227224. [Google Scholar] [CrossRef]
- Roy, K.K.; Wani, M.A. Emerging opportunities of exploiting mycobacterial electron transport chain pathway for drug-resistant tuberculosis drug discovery. Expert Opin. Drug Discov. 2020, 15, 231–241. [Google Scholar] [CrossRef]
- Thompson, A.M.; Denny, W.A. Chapter Four—Inhibitors of enzymes in the electron transport chain of Mycobacterium tuberculosis. In Annual Reports in Medicinal Chemistry; Chibale, K., Ed.; Academic Press: Cambridge, MA, USA, 2019; Volume 52, pp. 97–130. [Google Scholar]
- Lee, B.S.; Kalia, N.P.; Jin, X.E.F.; Hasenoehrl, E.J.; Berney, M.; Pethe, K. Inhibitors of energy metabolism interfere with antibiotic-induced death in mycobacteria. J. Biol. Chem. 2019, 294, 1936–1943. [Google Scholar] [CrossRef]
- Yu, W.; Chiwala, G.; Gao, Y.; Liu, Z.; Sapkota, S.; Lu, Z.; Guo, L.; Khan, S.A.; Zhong, N.; Zhang, T. TB47 and clofazimine form a highly synergistic sterilizing block in a second-line regimen for tuberculosis in mice. Biomed. Pharmacother. 2020, 131, 110782. [Google Scholar] [CrossRef]
- Yu, W.; Yusuf, B.; Wang, S.; Tian, X.; Hameed, H.M.A.; Lu, Z.; Chiwala, G.; Alam Md, S.; Cook Gregory, M.; Maslov Dmitry, A.; et al. Sterilizing Effects of Novel Regimens Containing TB47, Clofazimine, and Linezolid in a Murine Model of Tuberculosis. Antimicrob. Agents Chemother. 2021, 65, e00706–e00721. [Google Scholar] [CrossRef]
- Cai, Y.; Jaecklein, E.; Mackenzie, J.S.; Papavinasasundaram, K.; Olive, A.J.; Chen, X.; Steyn, A.J.C.; Sassetti, C.M. Host immunity increases Mycobacterium tuberculosis reliance on cytochrome bd oxidase. PLoS Pathog. 2021, 17, e1008911. [Google Scholar] [CrossRef]
- Harikishore, A.; Chong, S.S.M.; Ragunathan, P.; Bates, R.W.; Grüber, G. Targeting the menaquinol binding loop of mycobacterial cytochrome bd oxidase. Mol. Divers. 2021, 25, 517–524. [Google Scholar] [CrossRef]
- Friedrich, T.; Wohlwend, D.; Borisov, V.B. Recent Advances in Structural Studies of Cytochrome bd and Its Potential Application as a Drug Target. Int. J. Mol. Sci. 2022, 23, 3166. [Google Scholar] [CrossRef]
- Foo Caroline, S.; Lupien, A.; Kienle, M.; Vocat, A.; Benjak, A.; Sommer, R.; Lamprecht Dirk, A.; Steyn Adrie, J.C.; Pethe, K.; Piton, J.; et al. Arylvinylpiperazine Amides, a New Class of Potent Inhibitors Targeting QcrB of Mycobacterium tuberculosis. mBio 2018, 9, e01276-18. [Google Scholar] [CrossRef]
- Moosa, A.; Lamprecht Dirk, A.; Arora, K.; Barry Clifton, E.; Boshoff Helena, I.M.; Ioerger Thomas, R.; Steyn Adrie, J.C.; Mizrahi, V.; Warner Digby, F. Susceptibility of Mycobacterium tuberculosis Cytochrome bd Oxidase Mutants to Compounds Targeting the Terminal Respiratory Oxidase, Cytochrome c. Antimicrob. Agents Chemother. 2017, 61, e01338-17. [Google Scholar] [CrossRef] [PubMed]
- Sarathy, J.P.; Gruber, G.; Dick, T. Re-Understanding the Mechanisms of Action of the Anti-Mycobacterial Drug Bedaquiline. Antibiotics 2019, 8, 261. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Fatima, Z.; Kumawat, S. Study of the bioenergetics to identify the novel pathways as a drug target against Mycobacterium tuberculosis using Petri net. Biosystems 2021, 209, 104509. [Google Scholar] [CrossRef] [PubMed]
- Jeon, A.B.; Ackart, D.F.; Li, W.; Jackson, M.; Melander, R.J.; Melander, C.; Abramovitch, R.B.; Chicco, A.J.; Basaraba, R.J.; Obregón-Henao, A. 2-aminoimidazoles collapse mycobacterial proton motive force and block the electron transport chain. Sci. Rep. 2019, 9, 1513. [Google Scholar] [CrossRef]
- Odingo, J.; Bailey, M.A.; Files, M.; Early, J.V.; Alling, T.; Dennison, D.; Bowman, J.; Dalai, S.; Kumar, N.; Cramer, J.; et al. In Vitro Evaluation of Novel Nitazoxanide Derivatives against Mycobacterium tuberculosis. ACS Omega 2017, 2, 5873–5890. [Google Scholar] [CrossRef]
- Chao, A.; Sieminski, P.J.; Owens, C.P.; Goulding, C.W. Iron Acquisition in Mycobacterium tuberculosis. Chem. Rev. 2019, 119, 1193–1220. [Google Scholar] [CrossRef]
- Chiarelli, L.R.; Mori, M.; Barlocco, D.; Beretta, G.; Gelain, A.; Pini, E.; Porcino, M.; Mori, G.; Stelitano, G.; Costantino, L.; et al. Discovery and development of novel salicylate synthase (MbtI) furanic inhibitors as antitubercular agents. Eur. J. Med. Chem. 2018, 155, 754–763. [Google Scholar] [CrossRef]
- Chiarelli, L.R.; Mori, M.; Beretta, G.; Gelain, A.; Pini, E.; Sammartino, J.C.; Stelitano, G.; Barlocco, D.; Costantino, L.; Lapillo, M.; et al. New insight into structure-activity of furan-based salicylate synthase (MbtI) inhibitors as potential antitubercular agents. J. Enzym. Inhib. Med. Chem. 2019, 34, 823–828. [Google Scholar] [CrossRef]
- Rohilla, A.; Khare, G.; Tyagi, A.K. Virtual Screening, pharmacophore development and structure based similarity search to identify inhibitors against IdeR, a transcription factor of Mycobacterium tuberculosis. Sci. Rep. 2017, 7, 4653. [Google Scholar] [CrossRef]
- Yang, L.; Hu, X.; Chai, X.; Ye, Q.; Pang, J.; Li, D.; Hou, T. Opportunities for overcoming tuberculosis: Emerging targets and their inhibitors. Drug Discov. Today 2022, 27, 326–336. [Google Scholar] [CrossRef]
- Khisimuzi, M.; Zhenkun, M. Mycobacterium tuberculosis DNA Gyrase as a Target for Drug Discovery. Infect. Disord. Drug Targets 2007, 7, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.; Sankhe, K.; Suvarna, V.; Sherje, A.; Patel, K.; Dravyakar, B. DNA gyrase inhibitors: Progress and synthesis of potent compounds as antibacterial agents. Biomed. Pharmacother. 2018, 103, 923–938. [Google Scholar] [CrossRef] [PubMed]
- Godbole Adwait, A.; Ahmed, W.; Bhat Rajeshwari, S.; Bradley Erin, K.; Ekins, S.; Nagaraja, V. Targeting Mycobacterium tuberculosis Topoisomerase I by Small-Molecule Inhibitors. Antimicrob. Agents Chemother. 2015, 59, 1549–1557. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, N.; Dube, D.; Pandey, J.; Singh, B.; Kukshal, V.; Ramachandran, R.; Tripathi, R.P. NAD+-Dependent DNA Ligase: A novel target waiting for the right inhibitor. Med. Res. Rev. 2008, 28, 545–568. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.K.; Tripathi, R.P.; Ramachandran, R. NAD+-dependent DNA Ligase (Rv3014c) from Mycobacterium tuberculosis: Crystal structure of the adenylation domain and identification of novel inhibitors*. J. Biol. Chem. 2005, 280, 30273–30281. [Google Scholar] [CrossRef]
- Jian, Y.; Risseeuw, M.D.P.; Froeyen, M.; Song, L.; Cappoen, D.; Cos, P.; Munier-Lehmann, H.; van Calenbergh, S. 1-(Piperidin-3-yl)thymine amides as inhibitors of M. tuberculosis thymidylate kinase. J. Enzym. Inhib. Med. Chem. 2019, 34, 1730–1739. [Google Scholar] [CrossRef]
- Fang, H.; Peng, B.; Ong, S.Y.; Wu, Q.; Li, L.; Yao, S.Q. Recent advances in activity-based probes (ABPs) and affinity-based probes (AfBPs) for profiling of enzymes. Chem. Sci. 2021, 12, 8288–8310. [Google Scholar] [CrossRef]
- Conlon, B.P.; Nakayasu, E.S.; Fleck, L.E.; LaFleur, M.D.; Isabella, V.M.; Coleman, K.; Leonard, S.N.; Smith, R.D.; Adkins, J.N.; Lewis, K. Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature 2013, 503, 365–370. [Google Scholar] [CrossRef]
- Vandal, O.H.; Pierini, L.M.; Schnappinger, D.; Nathan, C.F.; Ehrt, S. A membrane protein preserves intrabacterial pH in intraphagosomal Mycobacterium tuberculosis. Nat. Med. 2008, 14, 849–854. [Google Scholar] [CrossRef]
- Simon, G.M.; Cravatt, B.F. Activity-based Proteomics of Enzyme Superfamilies: Serine Hydrolases as a Case Study. J. Biol. Chem. 2010, 285, 11051–11055. [Google Scholar] [CrossRef]
- Ortega, C.; Lindsey, N.A.; Frando, A.; Natalie, C.S.; Robert, W.B.; Richard, D.S.; Aaron, T.W.; Grundner, C. Systematic Survey of Serine Hydrolase Activity in Mycobacterium tuberculosis Defines Changes Associated with Persistence. Cell Chem. Biol. 2016, 23, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Bachovchin, D.A.; Ji, T.; Li, W.; Simon, G.M.; Blankman, J.L.; Adibekian, A.; Hoover, H.; Niessen, S.; Cravatt, B.F. Superfamily-wide portrait of serine hydrolase inhibition achieved by library-versus-library screening. Proc. Natl. Acad. Sci. USA 2010, 107, 20941–20946. [Google Scholar] [CrossRef] [PubMed]
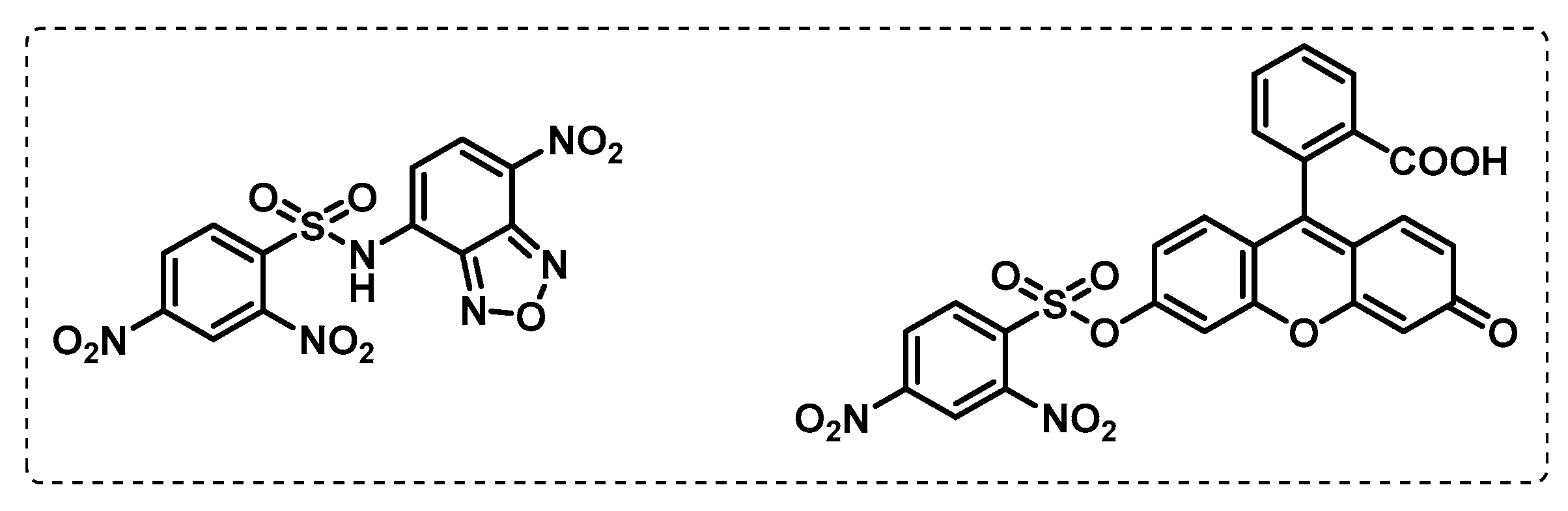
- Lentz, C.S.; Ordonez, A.A.; Kasperkiewicz, P.; La Greca, F.; O’Donoghue, A.J.; Schulze, C.J.; Powers, J.C.; Craik, C.S.; Drag, M.; Jain, S.K.; et al. Design of Selective Substrates and Activity-Based Probes for Hydrolase Important for Pathogenesis 1 (HIP1) from Mycobacterium tuberculosis. ACS Infect. Dis. 2016, 2, 807–815. [Google Scholar] [CrossRef] [PubMed]
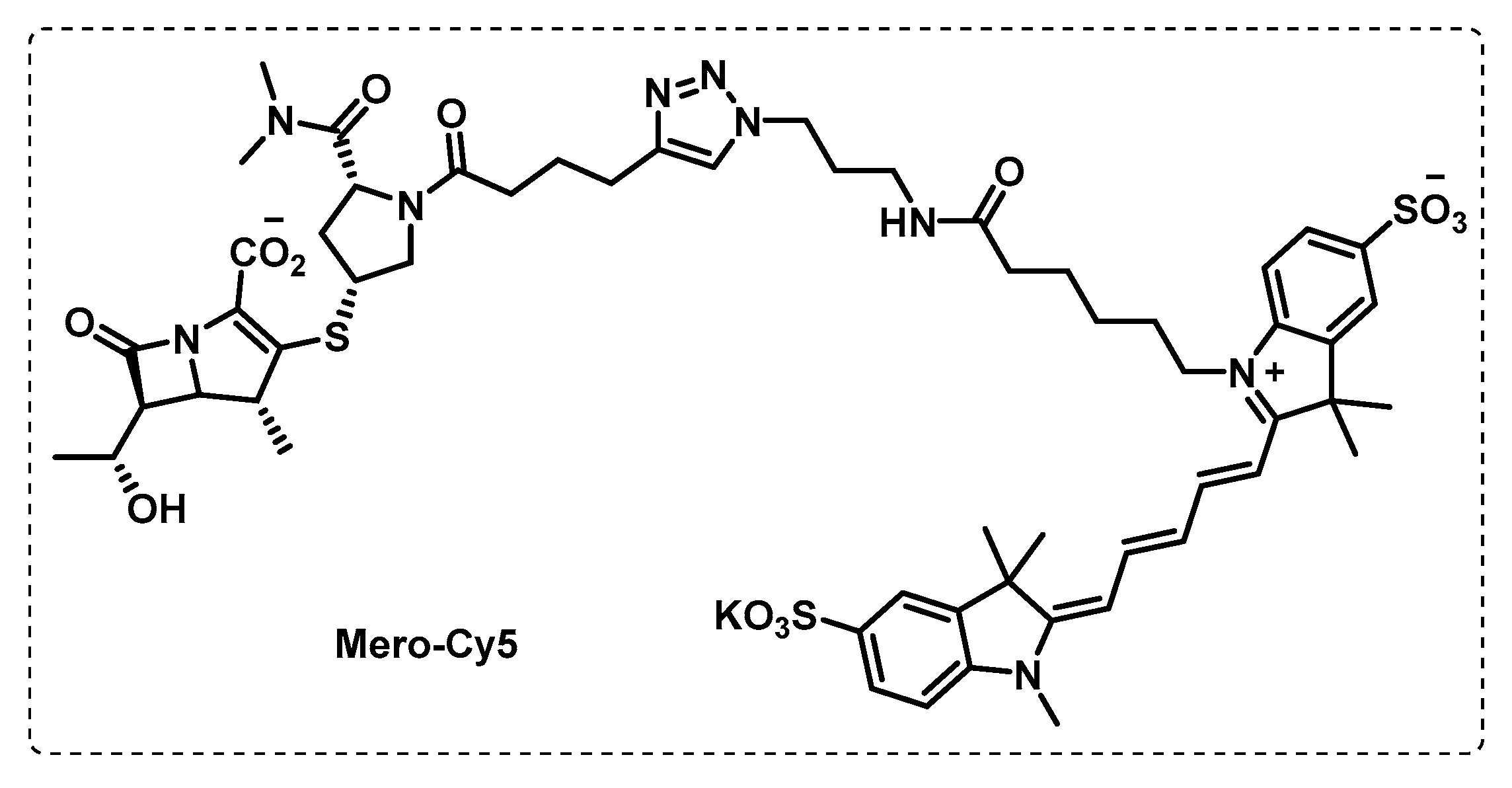
- Babin, B.M.; Keller, L.J.; Pinto, Y.; Li, V.L.; Eneim, A.S.; Vance, S.E.; Terrell, S.M.; Bhatt, A.S.; Long, J.Z.; Bogyo, M. Identification of covalent inhibitors that disrupt M. tuberculosis growth by targeting multiple serine hydrolases involved in lipid metabolism. Cell Chem. Biol. 2022, 29, 897–909.e897. [Google Scholar] [CrossRef]
- Li, M.; Patel, H.V.; Cognetta, A.B., III; Smith, T.C., II; Mallick, I.; Cavalier, J.-F.; Previti, M.L.; Canaan, S.; Aldridge, B.B.; Cravatt, B.F.; et al. Identification of cell wall synthesis inhibitors active against Mycobacterium tuberculosis by competitive activity-based protein profiling. Cell Chem. Biol. 2022, 29, 883–896.e885. [Google Scholar] [CrossRef] [PubMed]
- Gun, M.A.; Bozdogan, B.; Coban, A.Y. Tuberculosis and beta-lactam antibiotics. Future Microbiol. 2020, 15, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.; Muraoka, A.; Bedenbaugh, M.; Childress, B.; Pernot, L.; Wiencek, M.; Peterson, Y.K. The Chemical Relationship Among Beta-Lactam Antibiotics and Potential Impacts on Reactivity and Decomposition. Front. Microbiol. 2022, 13, 807955. [Google Scholar] [CrossRef]
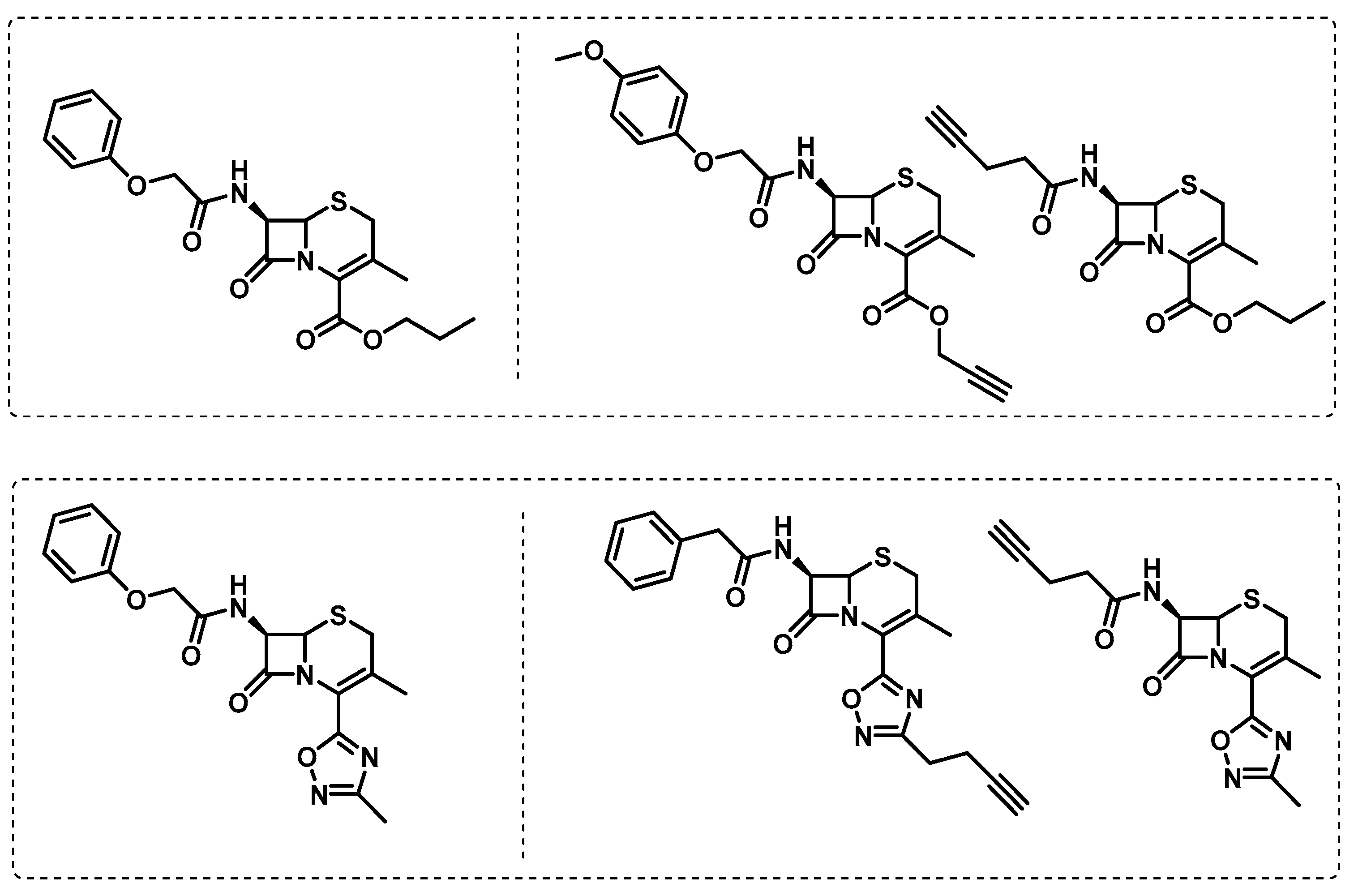
- Lopez Quezada, L.; Smith, R.; Lupoli, T.J.; Edoo, Z.; Li, X.; Gold, B.; Roberts, J.; Ling, Y.; Park, S.W.; Nguyen, Q.; et al. Activity-Based Protein Profiling Reveals That Cephalosporins Selectively Active on Non-replicating Mycobacterium tuberculosis Bind Multiple Protein Families and Spare Peptidoglycan Transpeptidases. Front. Microbiol. 2020, 11, 1248. [Google Scholar] [CrossRef]
- de Munnik, M.; Lohans, C.T.; Langley, G.W.; Bon, C.; Brem, J.; Schofield, C.J. A Fluorescence-Based Assay for Screening β-Lactams Targeting the Mycobacterium tuberculosis Transpeptidase LdtMt2. ChemBioChem 2020, 21, 368–372. [Google Scholar] [CrossRef]
- Levine, S.R.; Beatty, K.E. Investigating β-Lactam Drug Targets in Mycobacterium tuberculosis Using Chemical Probes. ACS Infect. Dis. 2021, 7, 461–470. [Google Scholar] [CrossRef]
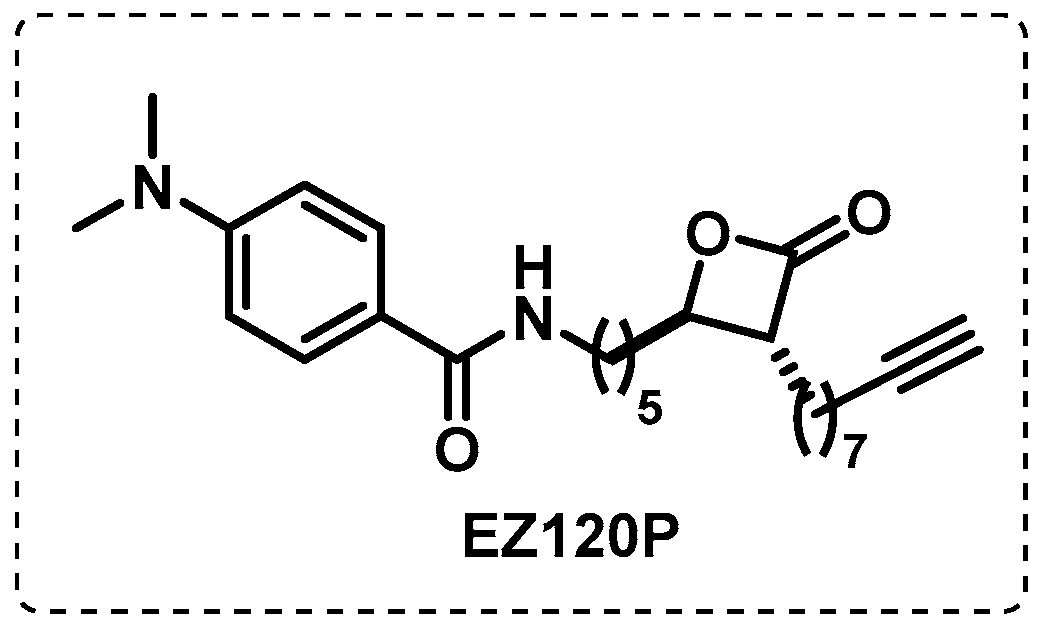
- Lehmann, J.; Cheng, T.-Y.; Aggarwal, A.; Park, A.S.; Zeiler, E.; Raju, R.M.; Akopian, T.; Kandror, O.; Sacchettini, J.C.; Moody, D.B.; et al. An Antibacterial β-Lactone Kills Mycobacterium tuberculosis by Disrupting Mycolic Acid Biosynthesis. Angew. Chem. Int. Ed. 2018, 57, 348–353. [Google Scholar] [CrossRef] [PubMed]
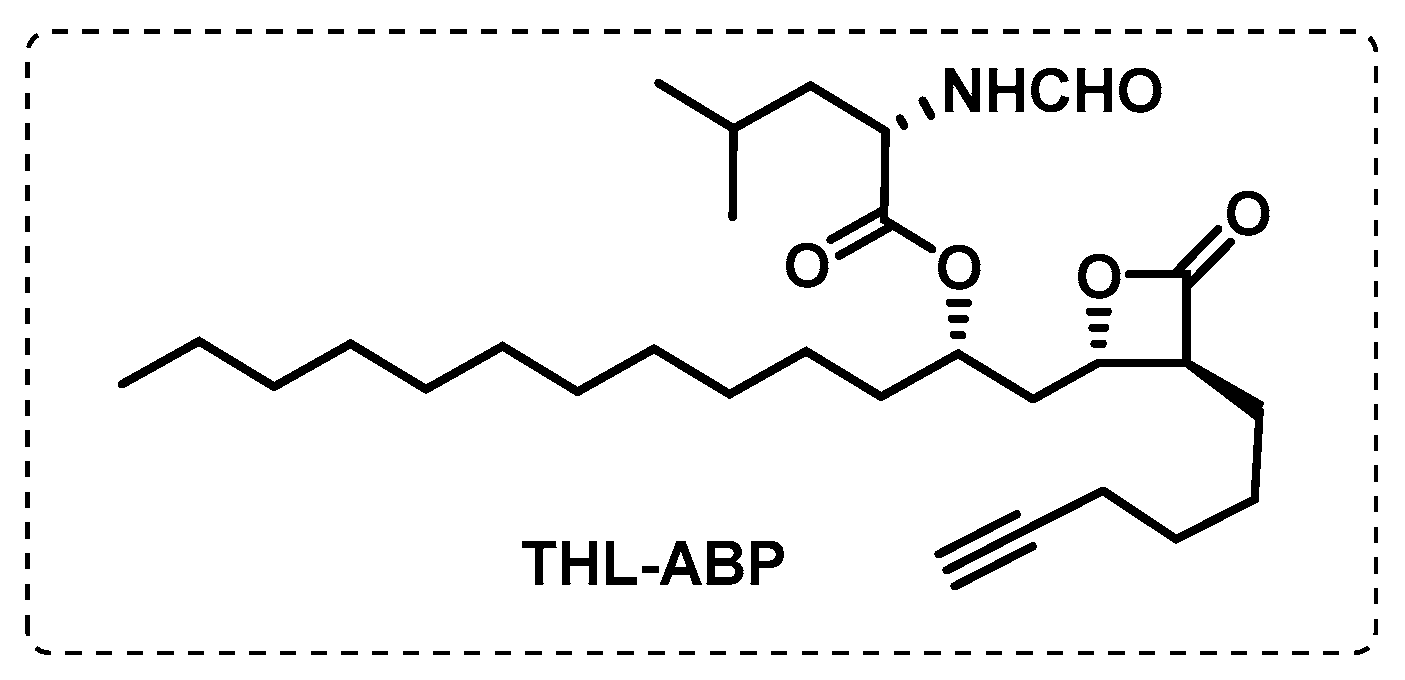
- Ravindran, M.S.; Rao, S.P.S.; Cheng, X.; Shukla, A.; Cazenave-Gassiot, A.; Yao, S.Q.; Wenk, M.R. Targeting Lipid Esterases in Mycobacteria Grown Under Different Physiological Conditions Using Activity-based Profiling with Tetrahydrolipstatin (THL). Mol. Cell. Proteom. 2014, 13, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Tallman, K.R.; Beatty, K.E. Far-red fluorogenic probes for esterase and lipase detection. Chembiochem A Eur. J. Chem. Biol. 2015, 16, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Tallman, K.R.; Levine, S.R.; Beatty, K.E. Small-Molecule Probes Reveal Esterases with Persistent Activity in Dormant and Reactivating Mycobacterium tuberculosis. ACS Infect. Dis. 2016, 2, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Tallman, K.R.; Levine, S.R.; Beatty, K.E. Profiling Esterases in Mycobacterium tuberculosis Using Far-Red Fluorogenic Substrates. ACS Chem. Biol. 2016, 11, 1810–1815. [Google Scholar] [CrossRef] [PubMed]
- Touchette Megan, H.; Holsclaw Cynthia, M.; Previti Mary, L.; Solomon Viven, C.; Leary Julie, A.; Bertozzi Carolyn, R.; Seeliger Jessica, C. The rv1184c Locus Encodes Chp2, an Acyltransferase in Mycobacterium tuberculosis Polyacyltrehalose Lipid Biosynthesis. J. Bacteriol. 2015, 197, 201–210. [Google Scholar] [CrossRef]
- Belardinelli, J.M.; Larrouy-Maumus, G.; Jones, V.; de Carvalho, L.P.S.; McNeil, M.R.; Jackson, M. Biosynthesis and Translocation of Unsulfated Acyltrehaloses in Mycobacterium tuberculosis. J. Biol. Chem. 2014, 289, 27952–27965. [Google Scholar] [CrossRef]
- Seeliger, J.C.; Holsclaw, C.M.; Schelle, M.W.; Botyanszki, Z.; Gilmore, S.A.; Tully, S.E.; Niederweis, M.; Cravatt, B.F.; Leary, J.A.; Bertozzi, C.R. Elucidation and Chemical Modulation of Sulfolipid-1 Biosynthesis in Mycobacterium tuberculosis*. J. Biol. Chem. 2012, 287, 7990–8000. [Google Scholar] [CrossRef]
- Ishikawa, F.; Tanabe, G.; Kakeya, H. Activity-Based Protein Profiling of Non-ribosomal Peptide Synthetases. In Activity-Based Protein Profiling; Cravatt, B.F., Hsu, K.-L., Weerapana, E., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 321–349. [Google Scholar] [CrossRef]
- Li, W.; Deng, G.; Li, M.; Liu, X.; Wang, Y. Roles of Mucosal Immunity against Mycobacterium tuberculosis Infection. Tuberc. Res. Treat. 2012, 2012, 791728. [Google Scholar] [CrossRef]
- Derrien, M.; van Passel, M.W.J.; van de Bovenkamp, J.H.B.; Schipper, R.; de Vos, W.; Dekker, J. Mucin-bacterial interactions in the human oral cavity and digestive tract. Gut Microbes 2010, 1, 254–268. [Google Scholar] [CrossRef]
- Mougous, J.D.; Leavell, M.D.; Senaratne, R.H.; Leigh, C.D.; Williams, S.J.; Riley, L.W.; Leary, J.A.; Bertozzi, C.R. Discovery of sulfated metabolites in mycobacteria with a genetic and mass spectrometric approach. Proc. Natl. Acad. Sci. USA 2002, 99, 17037–17042. [Google Scholar] [CrossRef] [PubMed]
- Mougous, J.D.; Lee, D.H.; Hubbard, S.C.; Schelle, M.W.; Vocadlo, D.J.; Berger, J.M.; Bertozzi, C.R. Molecular Basis for G Protein Control of the Prokaryotic ATP Sulfurylase. Mol. Cell 2006, 21, 109–122. [Google Scholar] [CrossRef] [PubMed]
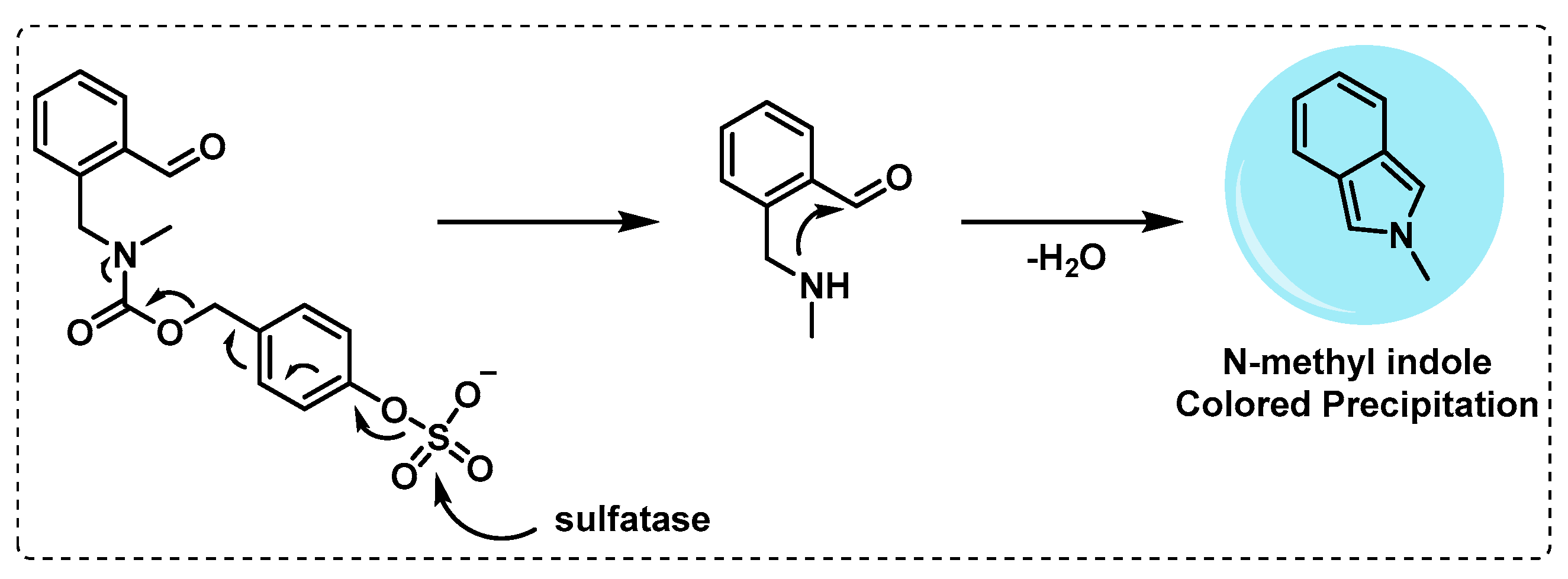
- Yoon, H.Y.; Kim, H.J.; Jang, S.; Hong, J.-I. Detection of bacterial sulfatase activity through liquid- and solid-phase colony-based assays. AMB Express 2017, 7, 150. [Google Scholar] [CrossRef] [PubMed]
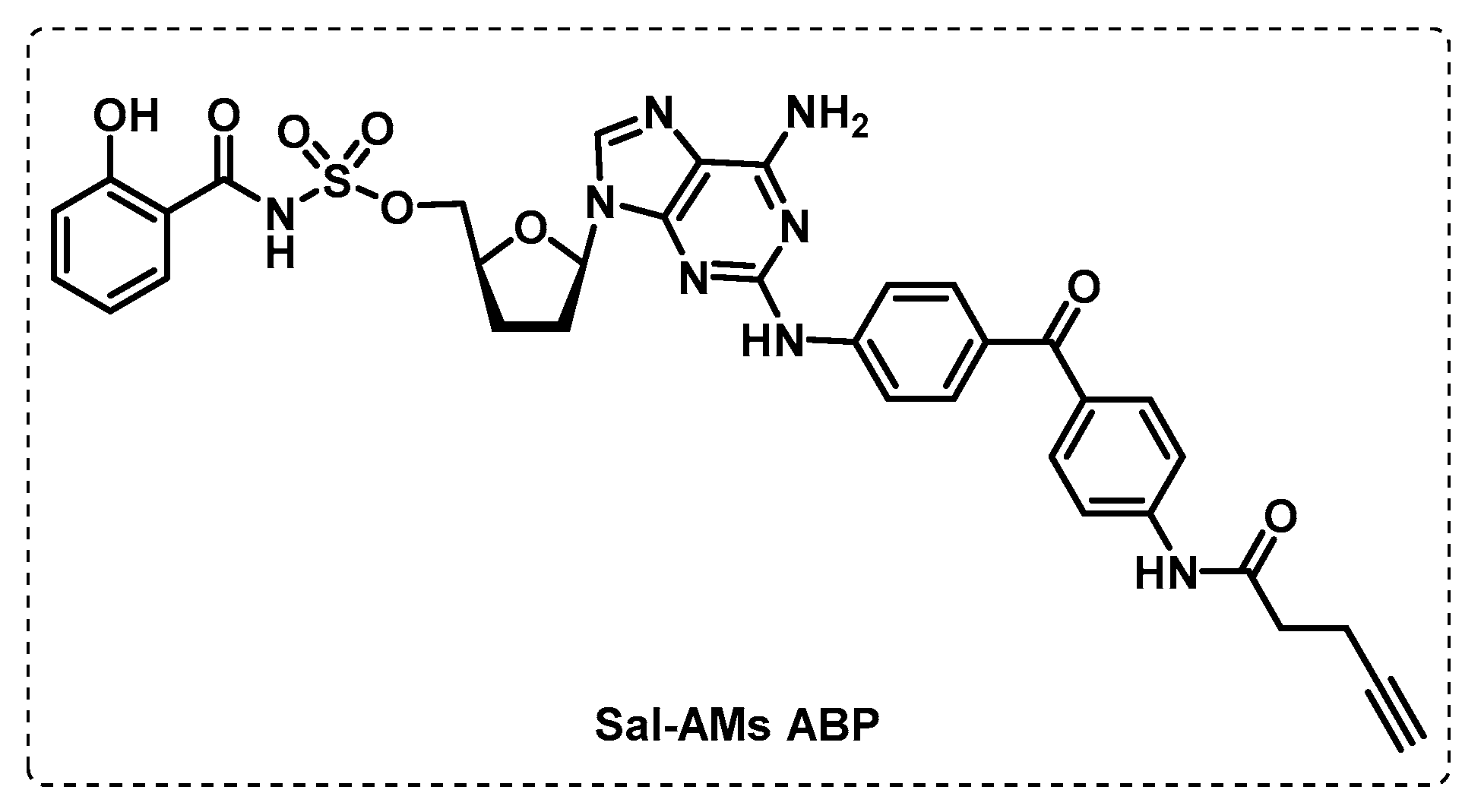
- Duckworth, B.P.; Wilson, D.J.; Nelson, K.M.; Boshoff, H.I.; Barry, C.E., III; Aldrich, C.C. Development of a Selective Activity-Based Probe for Adenylating Enzymes: Profiling MbtA Involved in Siderophore Biosynthesis from Mycobacterium tuberculosis. ACS Chem. Biol. 2012, 7, 1653–1658. [Google Scholar] [CrossRef]
- Wolfe, L.M.; Veeraraghavan, U.; Idicula-Thomas, S.; Schürer, S.; Wennerberg, K.; Reynolds, R.; Besra, G.S.; Dobos, K.M. A chemical proteomics approach to profiling the ATP-binding proteome of Mycobacterium tuberculosis. Mol. Cell. Proteom. 2013, 12, 1644–1660. [Google Scholar] [CrossRef]
- Batt, S.M.; Minnikin, D.E.; Besra, G.S. The thick waxy coat of mycobacteria, a protective layer against antibiotics and the host’s immune system. Biochem. J. 2020, 477, 1983–2006. [Google Scholar] [CrossRef]
- Vilchèze, C. Mycobacterial Cell Wall: A Source of Successful Targets for Old and New Drugs. Appl. Sci. 2020, 10, 2278. [Google Scholar] [CrossRef]
- Grzegorzewicz, A.E.; Eynard, N.; Quémard, A.; North, E.J.; Margolis, A.; Lindenberger, J.J.; Jones, V.; Korduláková, J.; Brennan, P.J.; Lee, R.E.; et al. Covalent Modification of the Mycobacterium tuberculosis FAS-II Dehydratase by Isoxyl and Thiacetazone. ACS Infect. Dis. 2015, 1, 91–97. [Google Scholar] [CrossRef]
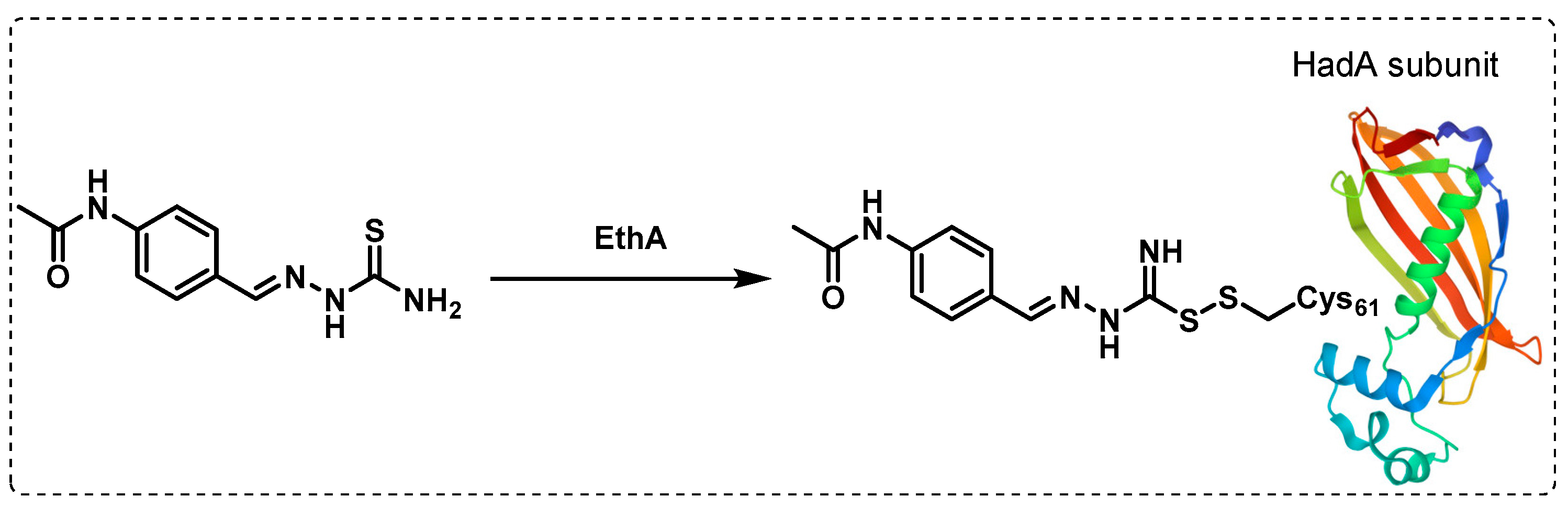
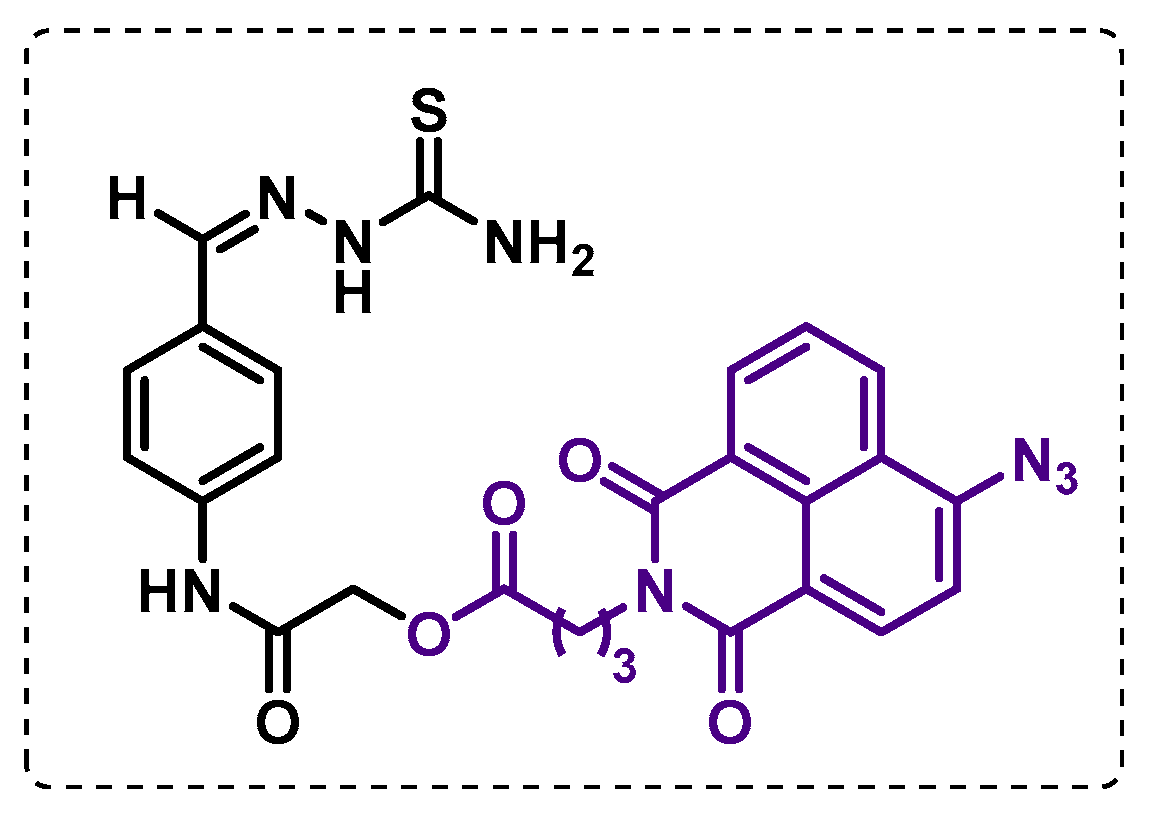
- Singh, B.K.; Singha, M.; Basak, S.; Biswas, R.; Das, A.K.; Basak, A. Fluorescently labelled thioacetazone for detecting the interaction with Mycobacterium dehydratases HadAB and HadBC. Org. Biomol. Chem. 2022, 20, 1444–1452. [Google Scholar] [CrossRef]
- Rahlwes, K.C.; Dias, B.R.S.; Campos, P.C.; Alvarez-Arguedas, S.; Shiloh, M.U. Pathogenicity and virulence of Mycobacterium tuberculosis. Virulence 2023, 14, 2150449. [Google Scholar] [CrossRef]
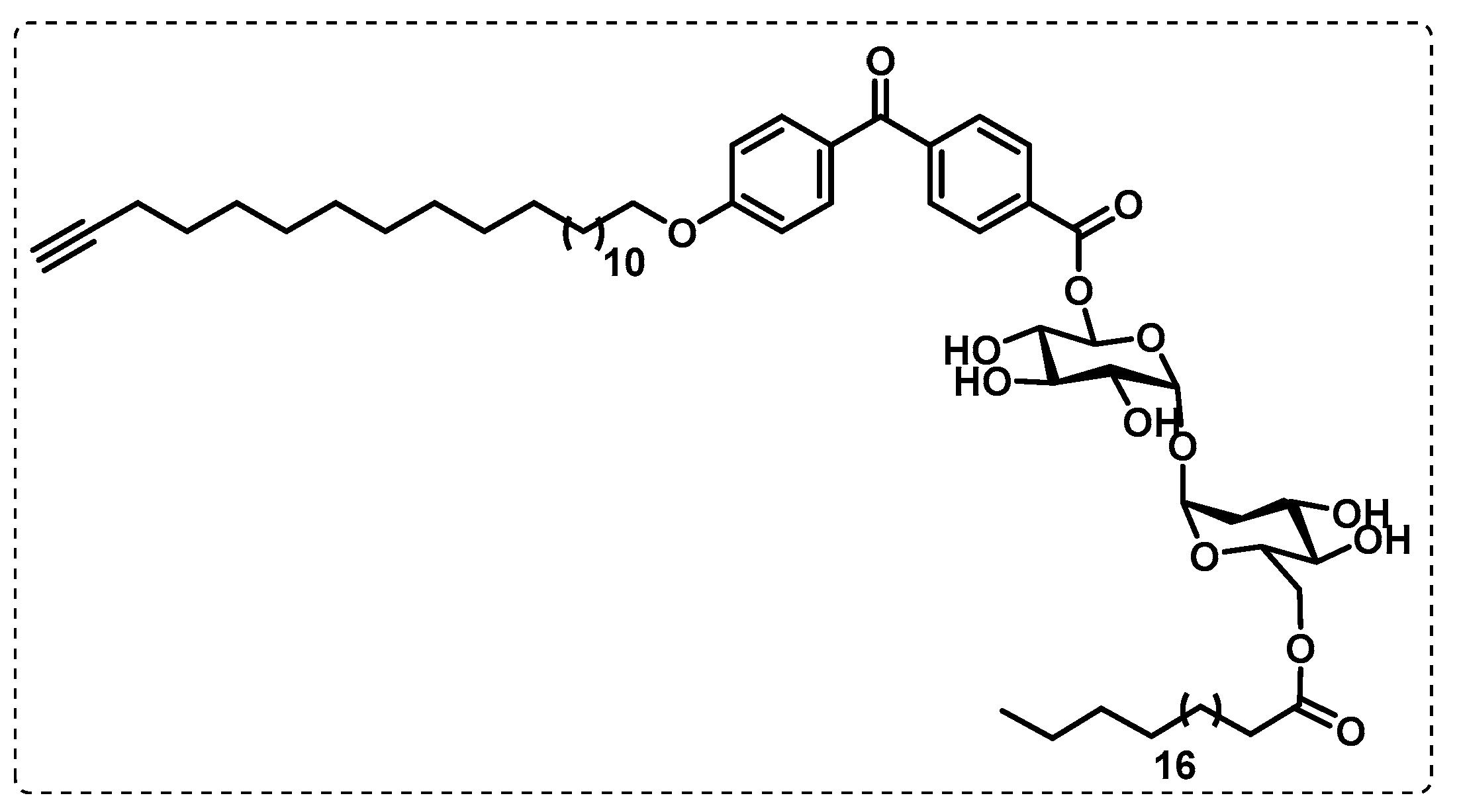
- Khan, A.A.; Kamena, F.; Timmer, M.S.M.; Stocker, B.L. Development of a benzophenone and alkyne functionalised trehalose probe to study trehalose dimycolate binding proteins. Org. Biomol. Chem. 2013, 11, 881–885. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions, and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions, or products referred to in the content. |

























Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capela, R.; Félix, R.; Clariano, M.; Nunes, D.; Perry, M.d.J.; Lopes, F. Target Identification in Anti-Tuberculosis Drug Discovery. Int. J. Mol. Sci. 2023, 24, 10482. https://doi.org/10.3390/ijms241310482
Capela R, Félix R, Clariano M, Nunes D, Perry MdJ, Lopes F. Target Identification in Anti-Tuberculosis Drug Discovery. International Journal of Molecular Sciences. 2023; 24(13):10482. https://doi.org/10.3390/ijms241310482
Chicago/Turabian StyleCapela, Rita, Rita Félix, Marta Clariano, Diogo Nunes, Maria de Jesus Perry, and Francisca Lopes. 2023. "Target Identification in Anti-Tuberculosis Drug Discovery" International Journal of Molecular Sciences 24, no. 13: 10482. https://doi.org/10.3390/ijms241310482
APA StyleCapela, R., Félix, R., Clariano, M., Nunes, D., Perry, M. d. J., & Lopes, F. (2023). Target Identification in Anti-Tuberculosis Drug Discovery. International Journal of Molecular Sciences, 24(13), 10482. https://doi.org/10.3390/ijms241310482