
Multi-Organ Morphological Findings in a Humanized Murine Model of Sickle Cell Trait

 , , , ,
, , , ,  ,
,  and
and 
Abstract
1. Introduction
2. Results
2.1. Hematological Values
2.2. Erythropoiesis, Spleen Size, and Organ Iron Content
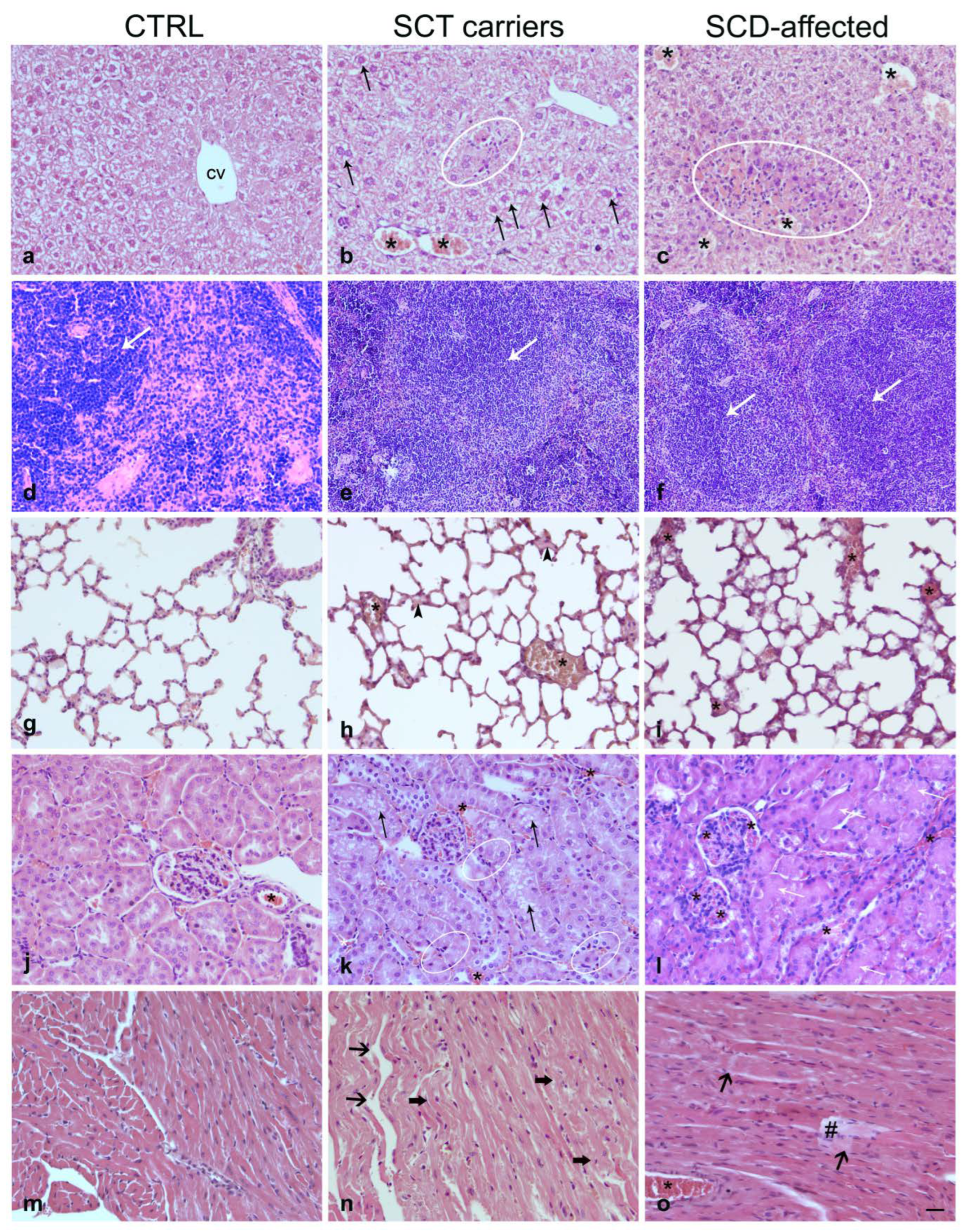
2.3. Histopathology
2.3.1. Liver
2.3.2. Spleen
2.3.3. Lung
2.3.4. Kidney
2.3.5. Heart
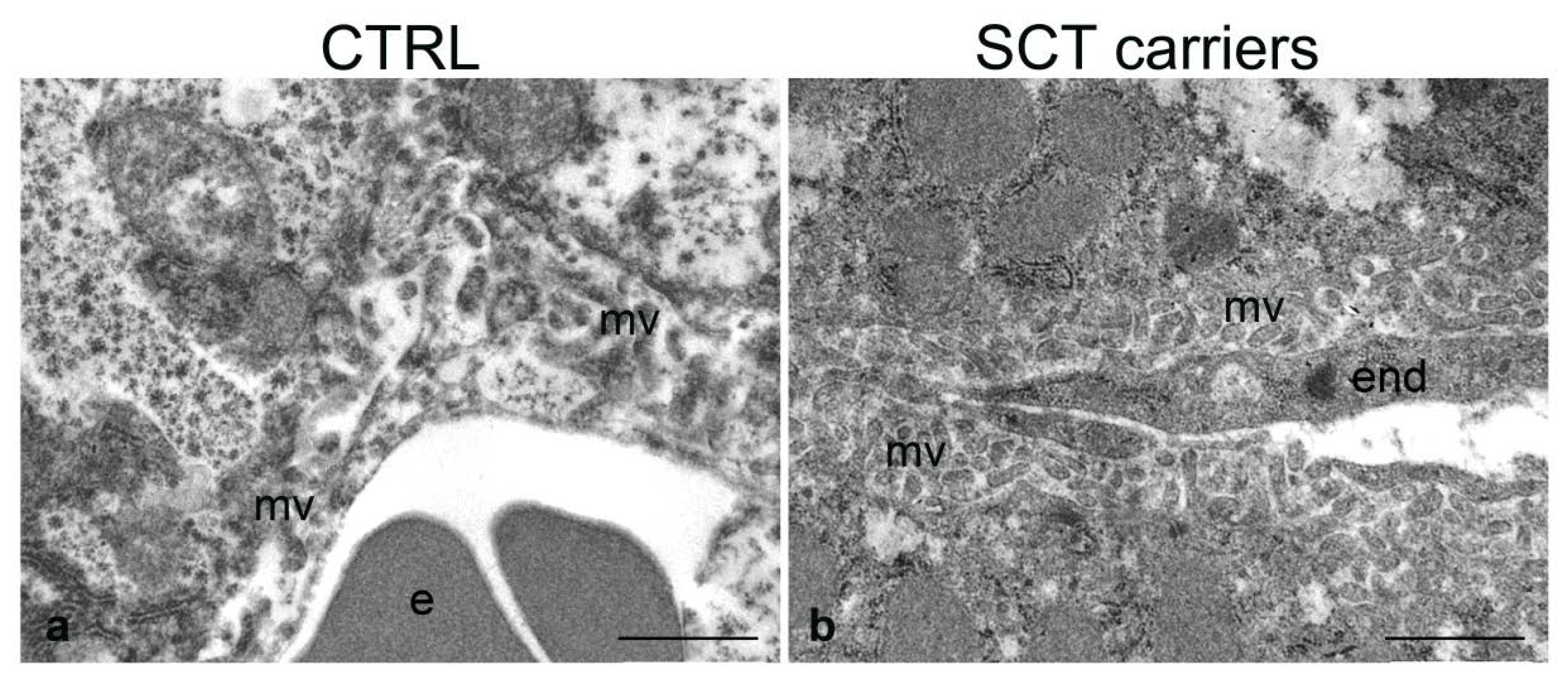
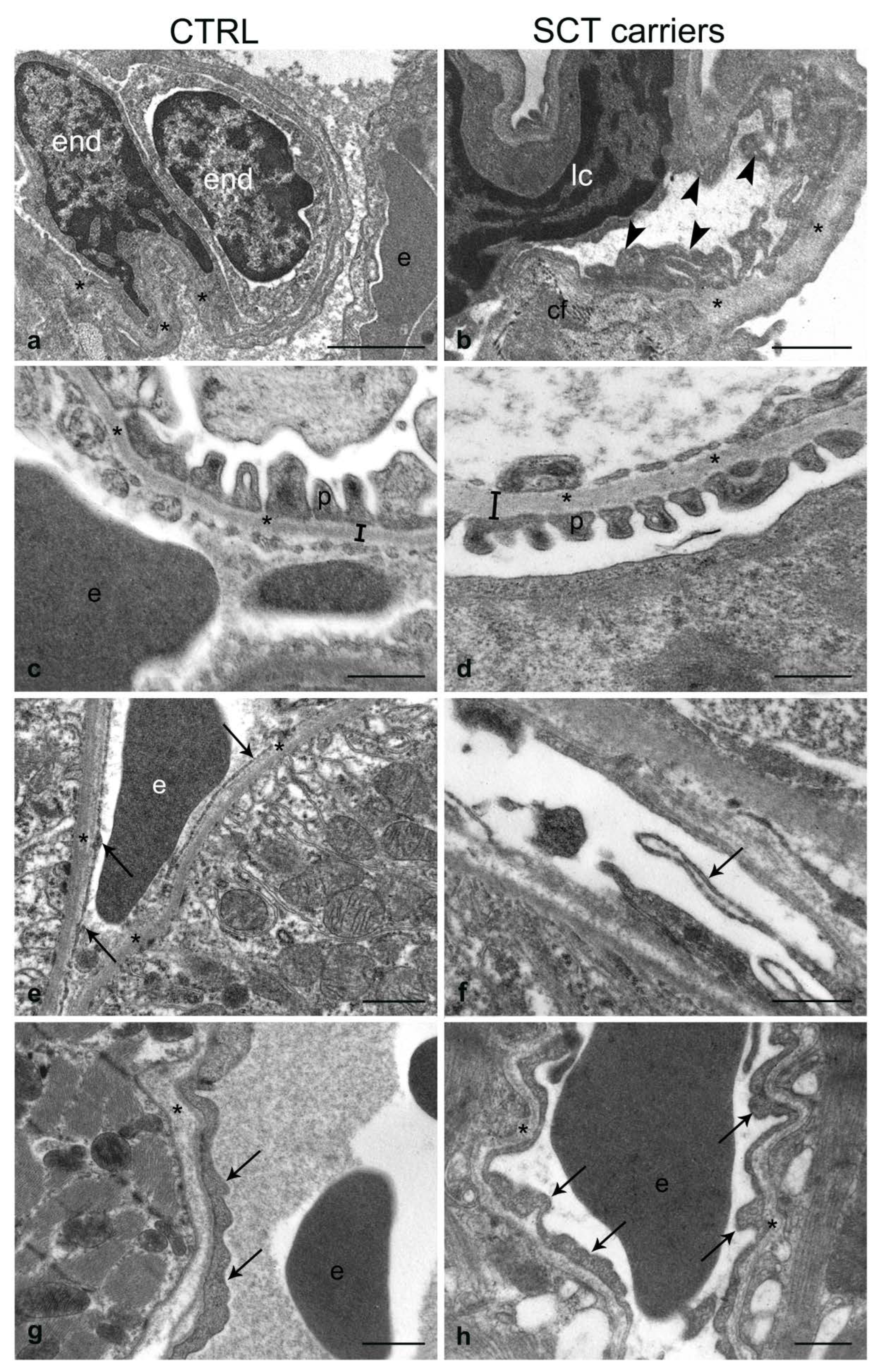
2.4. Transmission Electron Microscopy
2.4.1. Liver
2.4.2. Spleen
2.4.3. Lung
2.4.4. Kidney
2.4.5. Heart
3. Discussion
4. Materials and Methods
4.1. Animal Model
4.1.1. Hematological Values
4.1.2. Flow Cytometry
4.1.3. Spleen Size and Organ Iron Content
4.2. Histopathology
- -
- normal/minimal damage when the fields involved were from 0 to 30%
- -
- mild/moderate damage when the fields involved were from 30 to 70%
- -
- severe damage when the fields involved were from 70 to 100%
4.3. Transmission Electron Microscopy
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rees, D.C.; Williams, T.N.; Gladwin, M.T. Sickle-cell disease. Lancet 2010, 376, 2018–2031. [Google Scholar] [CrossRef]
- Porcu, S.; Simbula, M.; Marongiu, M.F.; Perra, A.; Poddie, D.; Perseu, L.; Kowalik, M.A.; Littera, R.; Barella, S.; Caria, C.A.; et al. Delta-globin gene expression improves sickle cell disease in a humanised mouse model. Br. J. Haematol. 2021, 193, 1228–1237. [Google Scholar] [CrossRef]
- Bender, M.A.; Carlberg, K. Sickle Cell Disease. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993–2023. [Google Scholar]
- Pawloski, J.R.; Hess, D.T.; Stamler, J.S. Impaired vasodilation by red blood cells in sickle cell disease. Proc. Natl. Acad. Sci. USA 2005, 102, 2531–2536. [Google Scholar] [CrossRef]
- Palomo, M.; Diaz-Ricart, M.; Carreras, E. Is sickle cell disease-related neurotoxicity a systemic endotheliopathy? Hematol. Oncol. Stem Cell Ther. 2020, 13, 111–115. [Google Scholar] [CrossRef]
- Aguilar, C.; Vichinsky, E.; Neumayr, L. Bone and joint disease in sickle cell disease. Hematol. Oncol. Clin. N. Am. 2005, 19, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Onimoe, G.; Rotz, S. Sickle cell disease: A primary care update. Clevel. Clin. J. Med. 2020, 87, 19–27. [Google Scholar] [CrossRef]
- Naik, R.P.; Haywood, C., Jr. Sickle cell trait diagnosis: Clinical and social implications. Hematol. Am. Soc. Hematol. Educ. Program 2015, 2015, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Gibson, J.S.; Rees, D.C. How benign is sickle cell trait? EBioMedicine 2016, 11, 21–22. [Google Scholar] [CrossRef] [PubMed]
- Shephard, R.J. Sickle cell trait: What are the costs and benefits of screening? J. Sports Med. Phys. Fit. 2016, 56, 1562–1573. [Google Scholar]
- Pecker, L.H.; Naik, R.P. The current state of sickle cell trait: Implications for reproductive and genetic counseling. Blood 2018, 132, 2331–2338. [Google Scholar] [CrossRef] [PubMed]
- Tsaras, G.; Owusu-Ansah, A.; Boateng, F.O.; Amoateng-Adjepong, Y. Complications Associated with Sickle Cell Trait: A Brief Narrative Review. Am. J. Med. 2009, 122, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Pecker, L.H.; Naik, R.P. The current state of sickle cell trait: Implications for reproductive and genetic counseling. Hematol. Am. Soc. Hematol. Educ. Program 2018, 2018, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.Z.; Thein, S.L. The carrier state for sickle cell disease is not completely harmless. Haematologica 2019, 104, 1106–1111. [Google Scholar] [CrossRef]
- Ramos, R.P. How can anemia negatively influence gas exchange? J. Bras. Pneumol. 2017, 43, 1–2. [Google Scholar] [CrossRef]
- Runkel, B.; Klüppelholz, B.; Rummer, A.; Sieben, W.; Lampert, U.; Bollig, C.; Markes, M.; Paschen, U.; Angelescu, K. Screening for sickle cell disease in newborns: A systematic review. Syst. Rev. 2020, 9, 250. [Google Scholar] [CrossRef]
- Webber, B.J.; Witkop, C.T. Screening for Sickle-Cell Trait at Accession to the United States Military. Mil. Med. 2014, 179, 1184–1189. [Google Scholar] [CrossRef]
- Decastel, M.; Leborgne-Samuel, Y.; Alexandre, L.; Merault, G.; Berchel, C. Morphological features of the human umbilical vein in normal, sickle cell trait, and sickle cell disease pregnancies. Hum. Pathol. 1999, 30, 13–20. [Google Scholar] [CrossRef]
- Saraf, S.L.; Sysol, J.R.; Susma, A.; Setty, S.; Zhang, X.; Gudehithlu, K.P.; Arruda, J.A.; Singh, A.K.; Machado, R.F.; Gordeuk, V.R. Progressive glomerular and tubular damage in sickle cell trait and sickle cell anemia mouse models. Transl. Res. 2018, 197, 1–11. [Google Scholar] [CrossRef]
- Pászty, C.; Brion, C.M.; Manci, E.; Witkowska, H.E.; Stevens, M.E.; Mohandas, N.; Rubin, E.M. Transgenic knockout mice with exclusively human sickle hemoglobin and sickle cell disease. Science 1997, 278, 876–878. [Google Scholar] [CrossRef] [PubMed]
- Manci, E.A.; Hillery, C.A.; Bodian, C.A.; Zhang, Z.G.; Lutty, G.A.; Coller, B.S. Pathology of Berkeley sickle cell mice: Similarities and differences with human sickle cell disease. Blood 2006, 107, 1651–1658. [Google Scholar] [CrossRef] [PubMed]
- Beuzard, Y. Mouse models of sickle cell disease. Transfus. Clin. Biol. 2008, 15, 7–11. [Google Scholar] [CrossRef] [PubMed]
- De Paepe, M.E.; Trudel, M. The transgenic SAD mouse: A model of human sickle cell glomerulopathy. Kidney Int. 1994, 46, 1337–1345. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.C.; Lu, R.; Lin, C.; Xu, S.M.; Kan, Y.W.; Porcu, S.; Carlson, E.; Kitamura, M.; Yang, S.; Flebbe-Rehwaldt, L.; et al. Transgenic knockout mice exclusively expressing human hemoglobin S after transfer of a 240-kb betas-globin yeast artificial chromosome: A mouse model of sickle cell anemia. Proc. Natl. Acad. Sci. USA 1998, 8, 14886–14890. [Google Scholar] [CrossRef] [PubMed]
- Ryan, T.M.; Ciavatta, D.J.; Townes, T.M. Knockout-transgenic mouse model of sickle cell disease. Science 1997, 278, 873–876. [Google Scholar] [CrossRef] [PubMed]
- Pradhan-Sundd, T.; Kato, G.J.; Novelli, E.M. Molecular mechanisms of hepatic dysfunction in sickle cell disease: Lessons from Townes mouse model. Am. J. Physiol. Cell Physiol. 2022, 323, C494–C504. [Google Scholar] [CrossRef]
- Woodard, K.J.; Doerfler, P.A.; Mayberry, K.D.; Sharma, A.; Levine, R.; Yen, J.; Valentine, V.; Palmer, L.E.; Valentine, M.; Weiss, M.J. Limitations of mouse models for sickle cell disease conferred by their human globin transgene configurations. Dis. Model. Mech. 2022, 15, dmm049463. [Google Scholar] [CrossRef]
- Skinner, S.; Liu, K.L.; Lo, M.; Josset-Lamaugarny, A.; Charrin, E.; Martin, C.; Pialoux, V.; Fromy, B.; Connes, P.; Sigaudo-Roussel, D. Alterations in vascular reactivity in a transgenic mouse model of sickle cell trait. Br. J. Haematol. 2020, 189, e154–e157. [Google Scholar] [CrossRef]
- Alvarez-Argote, J.; Dlugi, T.A.; Sundararajan, T.; Kleynerman, A.; Faber, M.L.; McKillop, W.M.; Medin, J.A. Pathophysiological characterization of the Townes mouse model for sickle cell disease. Transl. Res. 2022, 254, 77–91. [Google Scholar] [CrossRef]
- Nguyen, J.; Abdulla, F.; Chen, C.; Nguyen, P.; Nguyen, M.; Tittle, B.; O’Sullivan, G.; Belcher, J.D.; Vercellotti, G.M. Phenotypic Characterization the Townes Sickle Mice. Blood 2014, 124, 4916. [Google Scholar] [CrossRef]
- Ataga, K.I.; Saraf, S.L.; Derebail, V.K. The nephropathy of sickle cell trait and sickle cell disease. Nat. Rev. Nephrol. 2022, 18, 361–377. [Google Scholar] [CrossRef]
- Belhassen, L.; Pelle, G.; Sediame, S.; Bachir, D.; Carville, C.; Bucherer, C.; Lacombe, C.; Galacteros, F.; Adnot, S. Endothelial dysfunction in patients with sickle cell disease is related to selective impairment of shear stress–mediated vasodilation. Blood 2001, 97, 1584–1589. [Google Scholar] [CrossRef]
- Hebbel, R.P.; Vercellotti, G.M. Multiple inducers of endothelial NOS (eNOS) dysfunction in sickle cell disease. Am. J. Hematol. 2021, 96, 1505–1517. [Google Scholar] [CrossRef]
- Charrin, E.; Ofori-Acquah, S.F.; Nader, E.; Skinner, S.; Connes, P.; Pialoux, V.; Joly, P.; Martin, C. Inflammatory and oxidative stress phenotypes in transgenic sickle cell mice. Blood Cells Mol. Dis. 2016, 62, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Aufradet, E.; Monchanin, G.; Oyonno-Engelle, S.; Feasson, L.; Messonnier, L.; Francina, A.; Bezin, L.; Serpero, L.D.; Gozal, D.; Dodogba, M.; et al. Habitual Physical Activity and Endothelial Activation in Sickle Cell Trait Carriers. Med. Sci. Sports Exerc. 2010, 42, 1987–1994. [Google Scholar] [CrossRef] [PubMed]
- Tripette, J.; Connes, P.; Beltan, E.; Chalabi, T.; Marlin, L.; Chout, R.; Baskurt, O.K.; Hue, O.; Hardy-Dessources, M.-D. Red blood cell deformability and aggregation, cell adhesion molecules, oxidative stress and nitric oxide markers after a short term, submaximal, exercise in sickle cell trait carriers. Clin. Hemorheol. Microcirc. 2010, 45, 39–52. [Google Scholar] [CrossRef]
- Zappia, K.J.; Guo, Y.; Retherford, D.; Wandersee, N.J.; Stucky, C.L.; Hillery, C.A. Characterization of a mouse model of sickle cell trait: Parallels to human trait and a novel finding of cutaneous sensitization. Br. J. Haematol. 2017, 179, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Dei-Adomakoh, Y.A.; Afriyie-Mensah, J.S.; Forson, A.; Adadey, M.; Ndanu, T.A.; Acquaye, J.K. Lung Function Abnormalities in Sickle Cell Anaemia. Adv. Hematol. 2019, 2019, 1783240. [Google Scholar] [CrossRef]
- Kassim, A.A.; Payne, A.B.; Rodeghier, M.; Macklin, E.A.; Strunk, R.C.; DeBaun, M.R. Low forced expiratory volume is associated with earlier death in sickle cell anemia. Blood 2015, 126, 1544–1550. [Google Scholar] [CrossRef]
- Dillard, T.A.; Kark, J.A.; Rajagopal, K.R.; Key, J.A.; Canik, J.J.; Ruehle, C.J. Pulmonary Function in Sickle Cell Trait. Ann. Intern. Med. 1987, 106, 191–196. [Google Scholar] [CrossRef]
- Saad, E.J.; Tarditi Barra, A.; Monzoni, G.; Villegas, C.; Tabares, A.H. Sickle cell trait: A cause of abdominal pain and pulmonary embolism. Rev. La Fac. Cienc. Médicas Córdoba 2020, 77, 360–362. [Google Scholar] [CrossRef]
- Ataga, K.I.; Derebail, V.K.; Archer, D.R. The glomerulopathy of sickle cell disease. Am. J. Hematol. 2014, 89, 907–914. [Google Scholar] [CrossRef]
- Bábíčková, J.; Klinkhammer, B.M.; Buhl, E.M.; Djudjaj, S.; Hoss, M.; Heymann, F.; Tacke, F.; Floege, J.; Becker, J.U.; Boor, P. Regardless of etiology, progressive renal disease causes ultrastructural and functional alterations of peritubular capillaries. Kidney Int. 2017, 91, 70–85. [Google Scholar] [CrossRef]
- Eardley, K.S.; Kubal, C.; Zehnder, D.; Quinkler, M.; Lepenies, J.; Savage, C.O.; Howie, A.J.; Kaur, K.; Cooper, M.S.; Adu, D.; et al. The role of capillary density, macrophage infiltration and interstitial scarring in the pathogenesis of human chronic kidney disease. Kidney Int. 2008, 74, 495–504. [Google Scholar] [CrossRef]
- Kida, Y.; Tchao, B.N.; Yamaguchi, I. Peritubular capillary rarefaction: A new therapeutic target in chronic kidney disease. Pediatr. Nephrol. 2014, 29, 333–342. [Google Scholar] [CrossRef]
- Lee, S.P.; Cunningham, M.L.; Hines, P.C.; Joneckis, C.C.; Orringer, E.P.; Parise, L.V. Sickle cell adhesion to laminin: Potential role for the alpha5 chain. Blood 1998, 92, 2951–2958. [Google Scholar] [CrossRef]
- Johnson, L.D. The biochemical properties of basement membrane components in health and disease. Clin. Biochem. 1980, 13, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Isaji, T.; Xu, Q.; Kariya, Y.; Gu, W.; Fukuda, T.; Du, Y. Potential roles of N-glycosylation in cell adhesion. Glycoconj. J. 2012, 29, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Kariya, Y.; Kato, R.; Itoh, S.; Fukuda, T.; Shibukawa, Y.; Sanzen, N.; Sekiguchi, K.; Wada, Y.; Kawasaki, N.; Gu, J. N-Glycosylation of laminin-332 regulates its biological functions. A novel function of the bisecting GlcNAc. J. Biol. Chem. 2008, 283, 33036–33045. [Google Scholar] [CrossRef] [PubMed]
- Bennmann, D.; Horstkorte, R.; Hofmann, B.; Jacobs, K.; Navarrete-Santos, A.; Simm, A.; Bork, K.; Gnanapragassam, V.S. Advanced Glycation Endproducts Interfere with Adhesion and Neurite Outgrowth. PLoS ONE 2014, 9, 112–115. [Google Scholar] [CrossRef]
- Maciaszek, J.L.; Lykotrafitis, G. Sickle cell trait human erythrocytes are significantly stiffer than normal. J. Biomech. 2011, 44, 657–661. [Google Scholar] [CrossRef]
- You, Q.; Kong, L.-J.; Li, F.-D.; Wang, H.-Y.; Liu, D.-G.; Pei, F.-H.; Song, J.-T.; Xu, J.; Chen, J. Human recombinant endostatin Endostar attenuates hepatic sinusoidal endothelial cell capillarization in CCl4-induced fibrosis in mice. Mol. Med. Rep. 2015, 12, 5594–5600. [Google Scholar] [CrossRef]
- Sanz-García, C.; Fernández-Iglesias, A.; Gracia-Sancho, J.; Arráez-Aybar, L.A.; Nevzorova, Y.A.; Cubero, F.J. The Space of Disse: The Liver Hub in Health and Disease. Livers 2021, 1, 3–26. [Google Scholar] [CrossRef]
- Wang, Q.; Zennadi, R. The Role of RBC Oxidative Stress in Sickle Cell Disease: From the Molecular Basis to Pathologic Implications. Antioxidants 2021, 10, 1608. [Google Scholar] [CrossRef] [PubMed]
- Lamarre, Y.; Nader, E.; Connes, P.; Romana, M.; Garnier, Y. Extracellular Vesicles in Sickle Cell Disease: A Promising Tool. Bioengineering 2022, 9, 439. [Google Scholar] [CrossRef]
- Romana, M.; Connes, P.; Key, N.S. Microparticles in sickle cell disease. Clin. Hemorheol. Microcirc. 2018, 68, 319–329. [Google Scholar] [CrossRef]
- Stephens, O.R.; Grant, D.; Frimel, M.; Wanner, N.; Yin, M.; Willard, B.; Erzurum, S.C.; Asosingh, K. Characterization and origins of cell-free mitochondria in healthy murine and human blood. Mitochondrion 2020, 54, 102–112. [Google Scholar] [CrossRef] [PubMed]
- She, Z.; Xie, M.; Hun, M.; Abdirahman, A.S.; Li, C.; Wu, F.; Luo, S.; Wan, W.; Wen, C.; Tian, J. Immunoregulatory Effects of Mitochondria Transferred by Extracellular Vesicles. Front. Immunol. 2021, 11, 628576. [Google Scholar] [CrossRef] [PubMed]
- Bello, N.A.; Hyacinth, H.I.; Roetker, N.S.; Seals, S.R.; Naik, R.P.; Derebail, V.K.; Kshirsagar, A.V.; Key, N.S.; Wilson, J.G.; Correa, A.; et al. Sickle cell trait is not associated with an increased risk of heart failure or abnormalities of cardiac structure and function. Blood 2017, 129, 799–801. [Google Scholar] [CrossRef] [PubMed]
- Hyacinth, H.I.; Franceschini, N.; Seals, S.R.; Irvin, M.R.; Chaudhary, N.; Naik, R.P.; Alonso, A.; Carty, C.L.; Burke, G.L.; Zakai, N.A.; et al. Association of Sickle Cell Trait with Incidence of Coronary Heart Disease among African American Individuals. JAMA Netw. Open 2021, 4, e2030435. [Google Scholar] [CrossRef]
- Wu, L.C.; Sun, C.W.; Ryan, T.M.; Pawlik, K.M.; Ren, J.; Townes, T.M. Correction of sickle cell disease by homologous recombination in embryonic stem cells. Blood 2006, 15, 1183–1188. [Google Scholar] [CrossRef]
- Burattini, S.; Salucci, S.; Baldassarri, V.; Accorsi, A.; Piatti, E.; Madrona, A.; Espartero, J.L.; Candiracci, M.; Zappia, G.; Falcieri, E. Anti-apoptotic activity of hydroxytyrosol and hydroxytyrosyl laurate. Food Chem. Toxicol. 2013, 55, 248–256. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mice | RBC (M/mm3) | Hb (g/dL) | Hct (%) | MCV (fl) | MCH (pg) | MCHC (g/dL) | RDW |
|---|---|---|---|---|---|---|---|
| HBBA/A | 9.7 ± 0.3 | 12.8 ± 1.3 | 32.2 ± 1.7 | 33.2 ± 1.7 | 13.0 ± 0.3 | 39.9 ± 2.2 | 17.5 ± 2.8 |
| HBBA/S | 9.6 ± 0.6 ### | 11.7 ± 0.5 ### | 31.1 ± 1.0 | 32.5 ± 2.0 ## | 12.2 ± 0.5 * | 37.7 ± 1.0 # | 17.6 ± 2.3 # |
| HBBS/S | 6.2 ± 1.5 | 8.5 ± 0.8 | 26.3 ± 7.3 | 42.4 ± 4.6 | 15.2 ± 2.3 | 33.3 ± 5.0 | 20.6 ± 2.2 |
| Mean (DS): | |||||
|---|---|---|---|---|---|
| Ter119+ CD71+/Ter119+ CD71− % | |||||
| Mice | BM Cells | Spleen Index | Iron Liver, μg | Iron Spleen, μg | Iron Heart, μg |
| HBBA/A | 1.97 ± 0.53 | 895 ± 272 | 122.57± 70.4 | 95.8 ± 55.6 | 9.3 ± 4.6 |
| HBBA/S | 2.68 ± 0.92 ## | 1245.7 ± 203.6 ### | 106.9 ± 30.8 ### | 94.2 ± 28.4 ## | 8.6 ± 4.4 |
| HBBS/S | 6.67 ± 2.03 | 6414 ± 684 | 896.4 ± 112.65 | 1409.76 ± 378.1 | 19.45 ± 8.4 |
| Liver | Spleen | Lung | Kidney | Heart | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Grade | n | n | n | n | n | |||||
| Normal/ Minimal | minimal cell stress, preserved tissue architecture | 2 | minimal increase in the white pulp | 0 | absent to low inflammation, minimal capillary congestion | 5 | mononuclear infiltrates, swelling of tubular cells, vascular congestion | 0 | normally stained fibers, clearly visible nuclei, normal vessels, no connective fiber deposits, focal weaving | 5 |
| Mild/ moderate | mild ischemic lesion, hepatocyte vacuolization, light mononuclear infiltrates, pericentral vein tissue pallor | 3 | increase in white pulp, vascular ectasia | 5 | mononuclear infiltrates, capillary congestion, edema, dilated capillaries, thickness of intima and media of pulmonary vessels | 0 | mononuclear infiltrates, steatosis and swelling of tubular cells, vascular congestion | 5 | pale staining of myocardiocytes, ischemic swelling, disorganization of histoarchitecture | 0 |
| Severe | large areas of necrosis, infarcts, mononuclear infiltrates, endovascular sickle cells | 0 | gross infarcts, increased spleen diameter, pigment deposits | 0 | infarcts, emphysema, hemorrhage, edema, thickness of intima and media of pulmonary vessels | 0 | gross infarcts, glomerular necrosis, papillary necrosis | 0 | Fragmentation and pale staining of myocardiocytes, nuclei not clearly visible, large areas of coagulative necrosis | 0 |
| Genotype | Sex | |
|---|---|---|
| Control 1 (C1) | HBBA/A | Male |
| Control 2 (C2) | HBBA/A | Male |
| Mouse 1 (M1) | HBBA/S | Male |
| Mouse 2 (M2) | HBBA/S | Male |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trucas, M.; Burattini, S.; Porcu, S.; Simbula, M.; Ristaldi, M.S.; Kowalik, M.A.; Serra, M.P.; Gobbi, P.; Battistelli, M.; Perra, A.; et al. Multi-Organ Morphological Findings in a Humanized Murine Model of Sickle Cell Trait. Int. J. Mol. Sci. 2023, 24, 10452. https://doi.org/10.3390/ijms241310452
Trucas M, Burattini S, Porcu S, Simbula M, Ristaldi MS, Kowalik MA, Serra MP, Gobbi P, Battistelli M, Perra A, et al. Multi-Organ Morphological Findings in a Humanized Murine Model of Sickle Cell Trait. International Journal of Molecular Sciences. 2023; 24(13):10452. https://doi.org/10.3390/ijms241310452
Chicago/Turabian StyleTrucas, Marcello, Sabrina Burattini, Susanna Porcu, Michela Simbula, Maria Serafina Ristaldi, Marta Anna Kowalik, Maria Pina Serra, Pietro Gobbi, Michela Battistelli, Andrea Perra, and et al. 2023. "Multi-Organ Morphological Findings in a Humanized Murine Model of Sickle Cell Trait" International Journal of Molecular Sciences 24, no. 13: 10452. https://doi.org/10.3390/ijms241310452
APA StyleTrucas, M., Burattini, S., Porcu, S., Simbula, M., Ristaldi, M. S., Kowalik, M. A., Serra, M. P., Gobbi, P., Battistelli, M., Perra, A., & Quartu, M. (2023). Multi-Organ Morphological Findings in a Humanized Murine Model of Sickle Cell Trait. International Journal of Molecular Sciences, 24(13), 10452. https://doi.org/10.3390/ijms241310452