
α-Aminobutyric Acid Constrains Macrophage-Associated Inflammatory Diseases through Metabolic Reprogramming and Epigenetic Modification

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
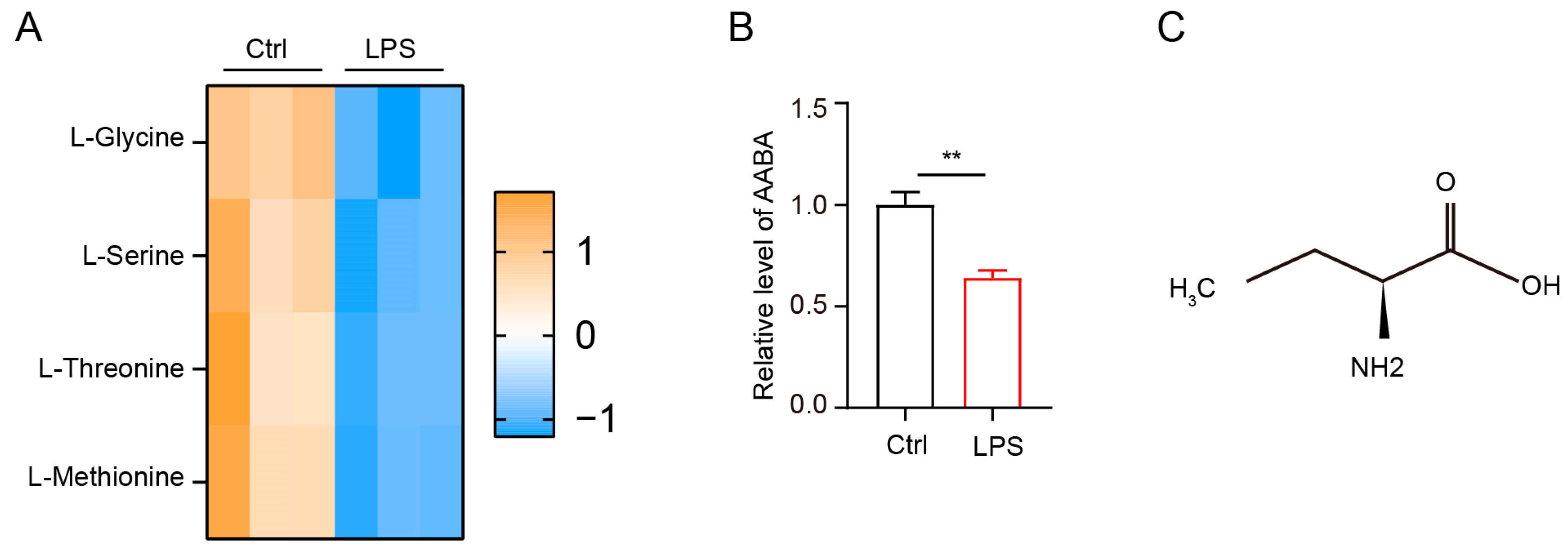
2.1. LPS Stimulation Impacts AABA Metabolism in Macrophages
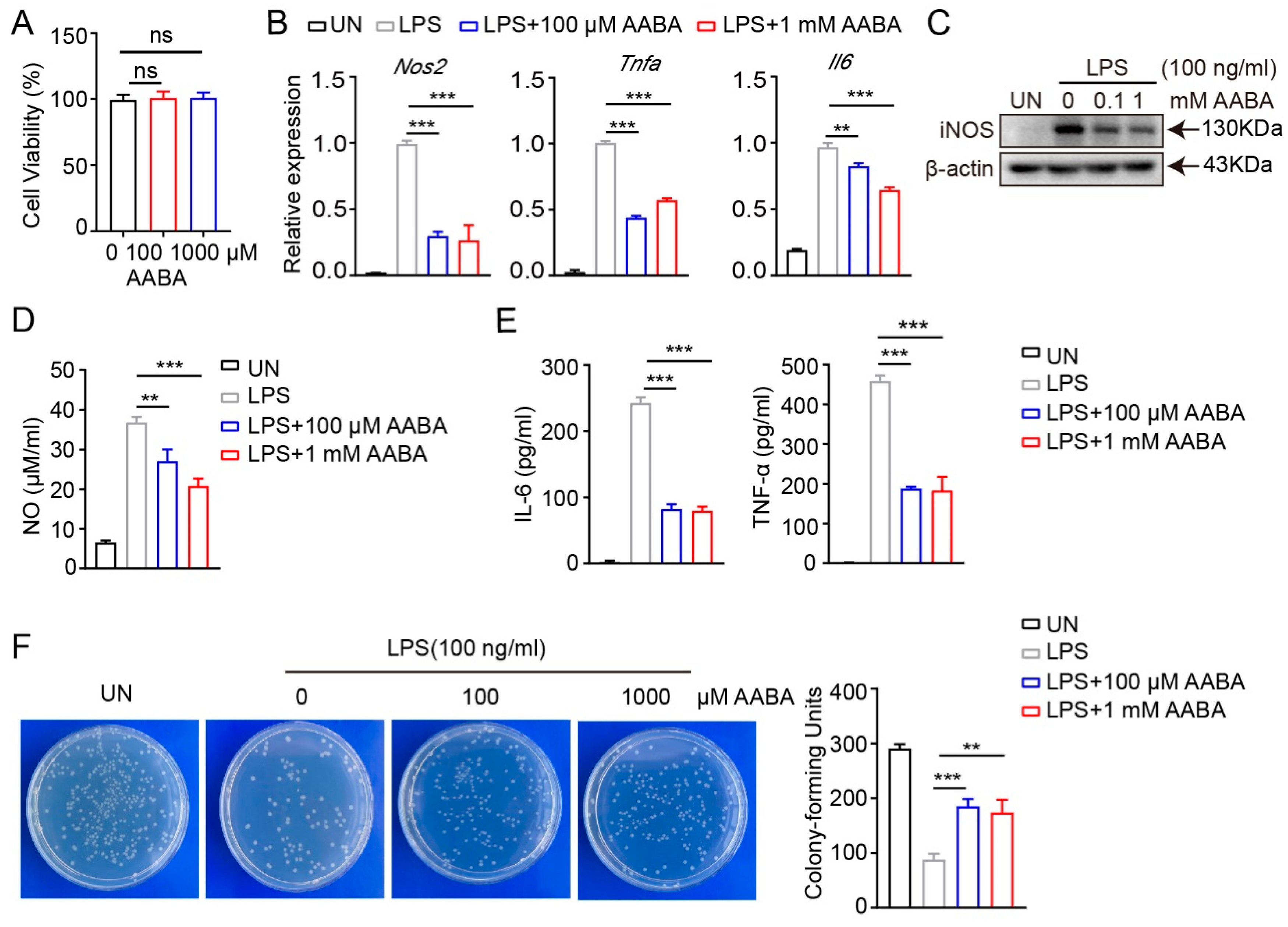
2.2. AABA Impairs Activation and Function of M1 Macrophages
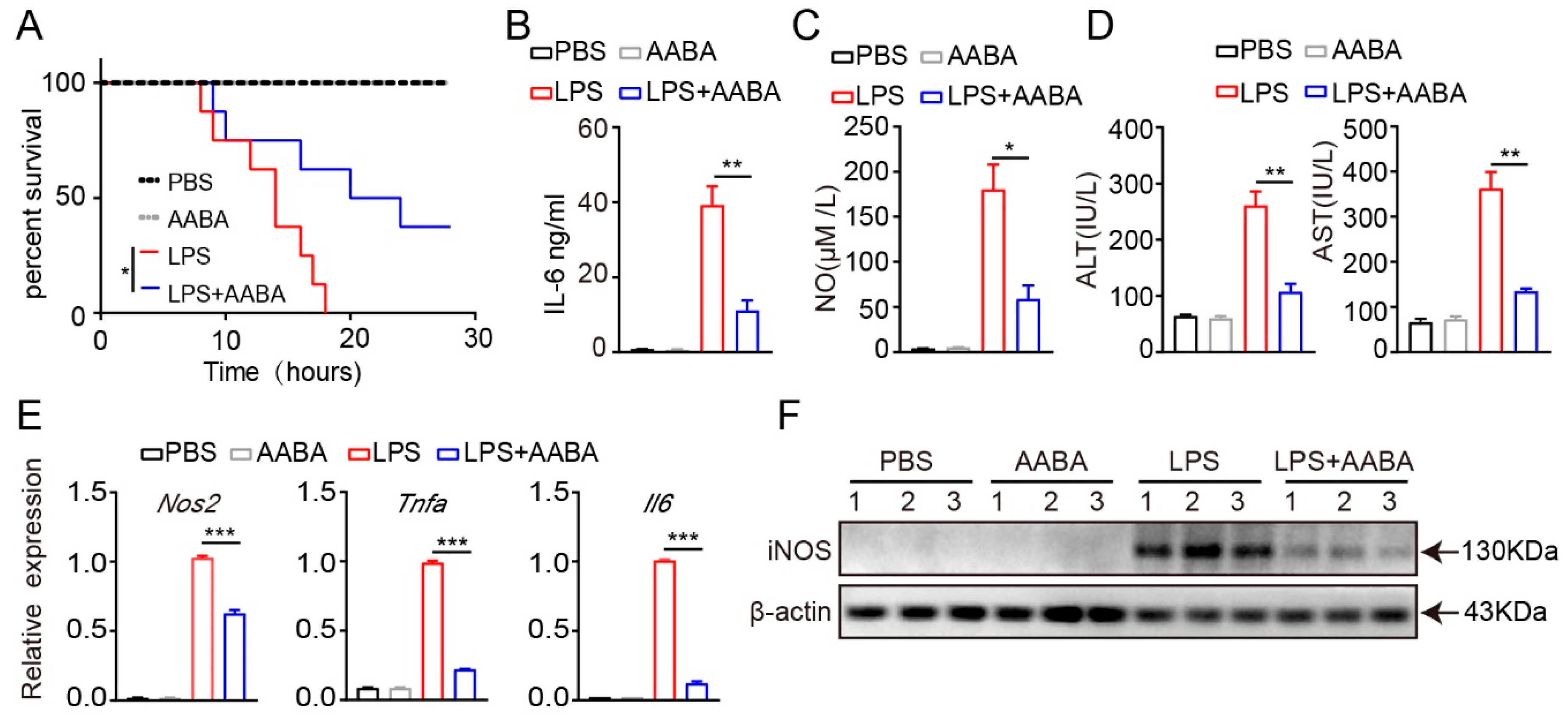
2.3. AABA Protects Mice from LPS-Induced Sepsis
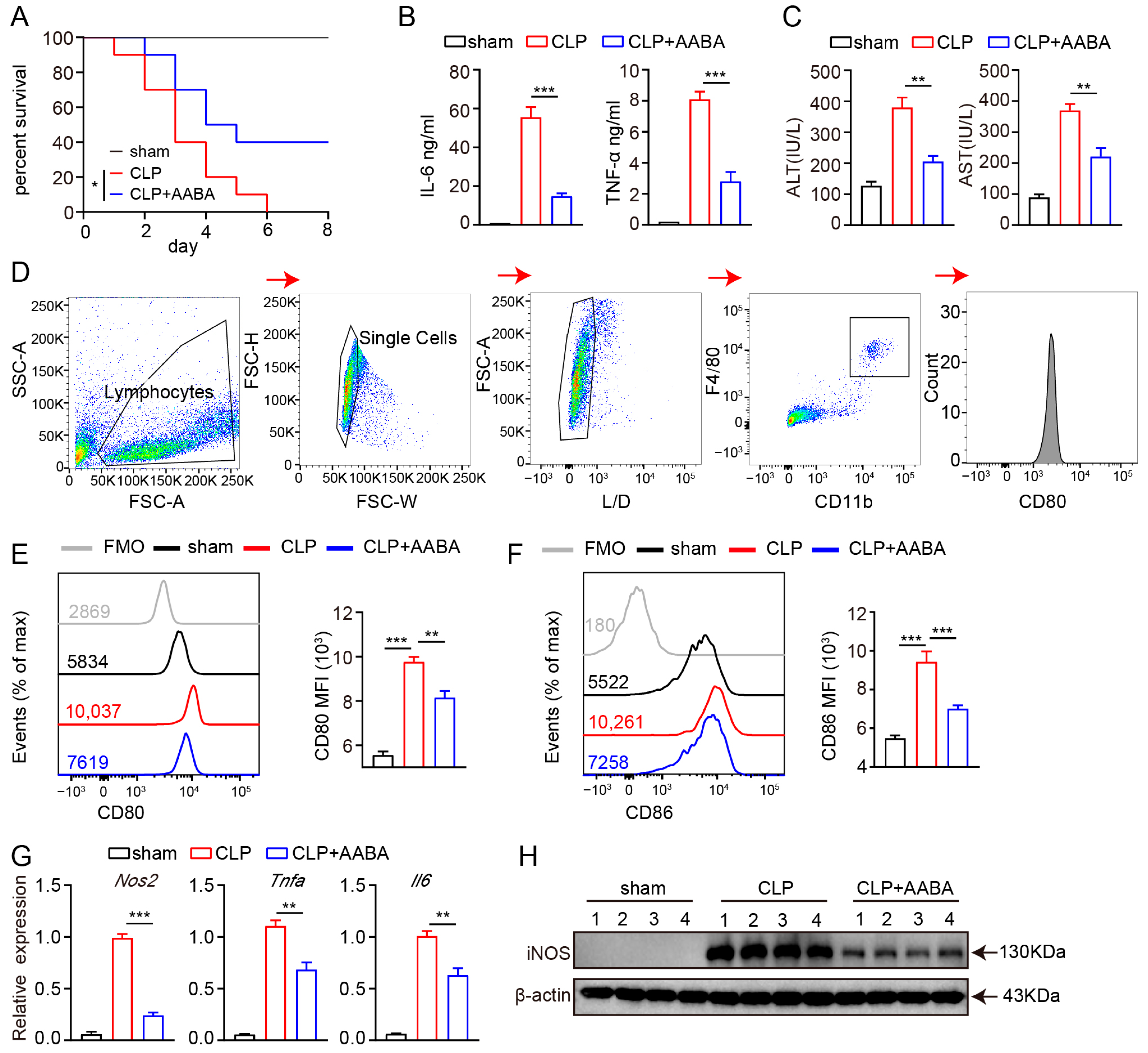
2.4. AABA Relieves the Development of CLP-Induced Sepsis
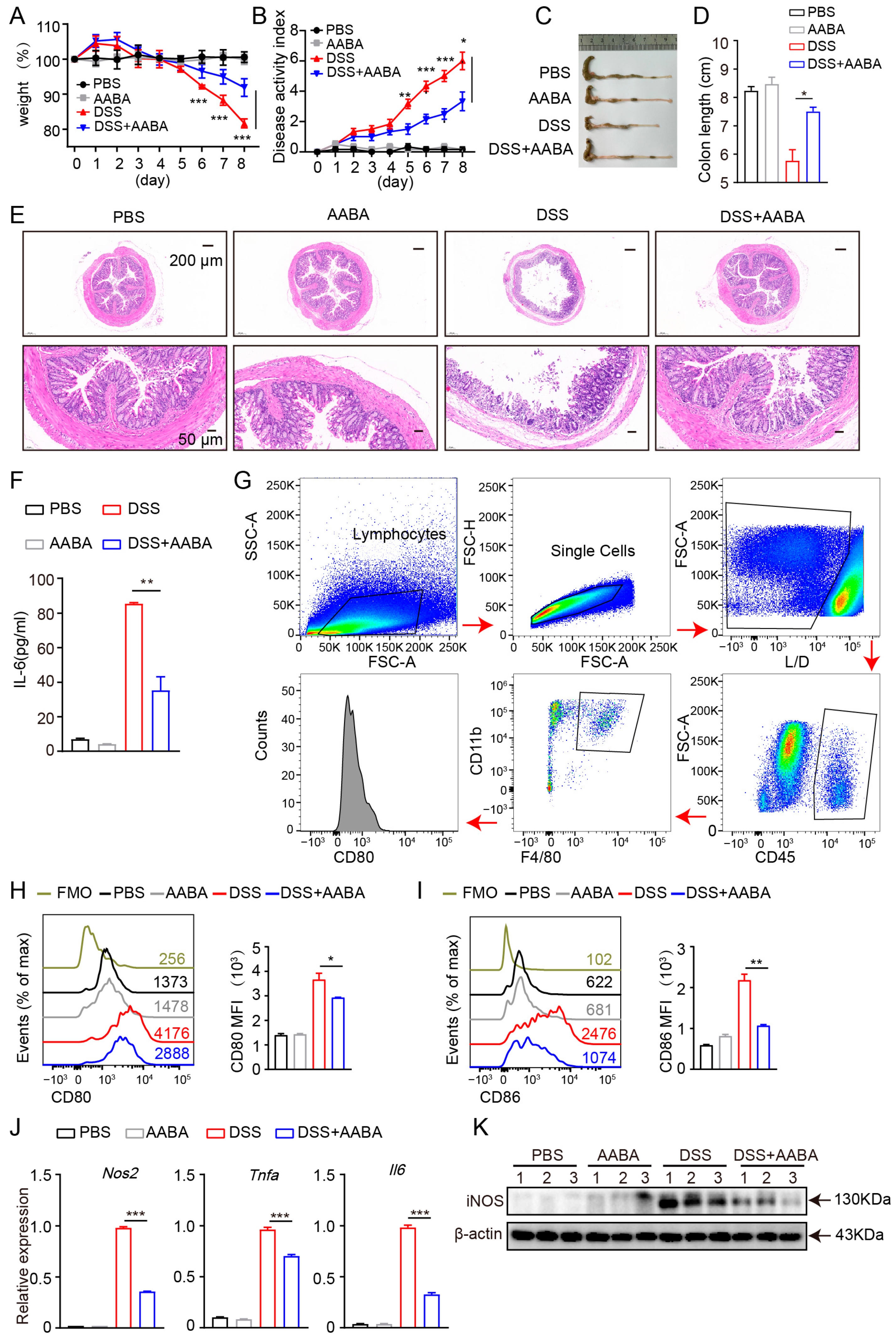
2.5. AABA Protects Mice against DSS-Induced Inflammatory Bowel Disease
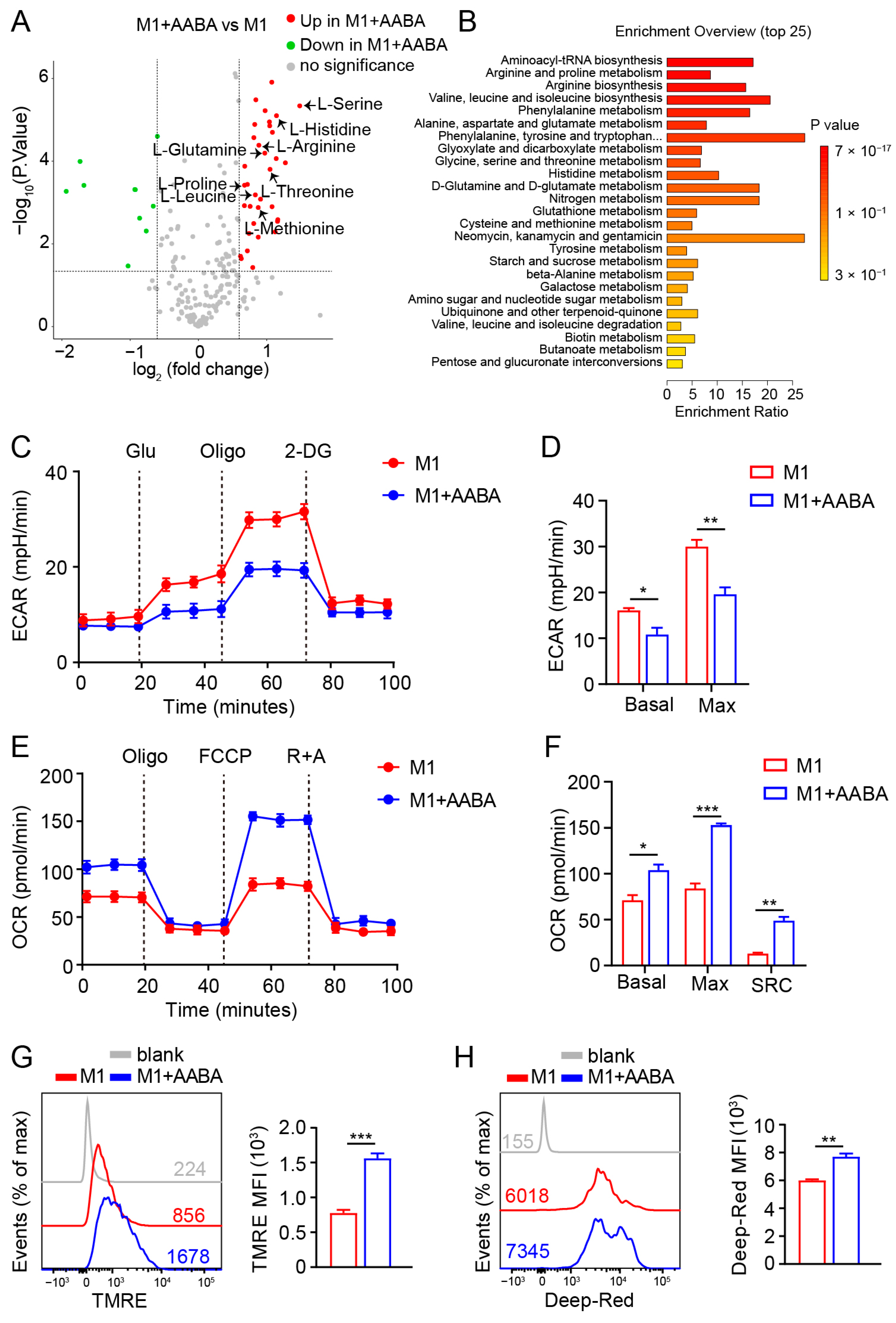
2.6. AABA Reprograms Metabolism of M1 Macrophages
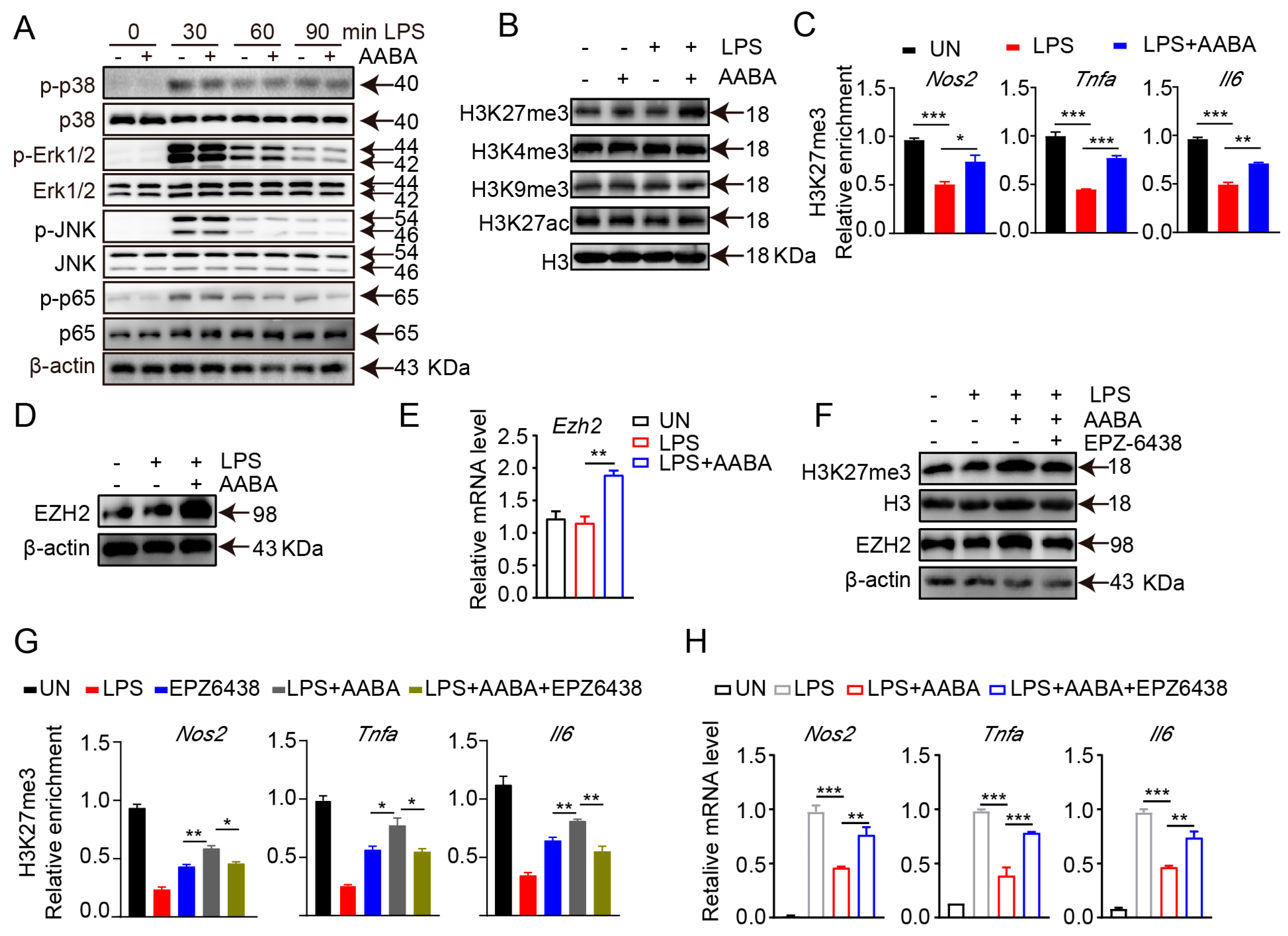
2.7. AABA Enhances EZH2 Expression to Improve H3K27me3 Chromatin Occupancy in M1 Macrophage-Associated Inflammatory Genes
3. Discussion
4. Materials and Methods
4.1. Mice and Generation of BMDMs
4.2. LPS-Induced Sepsis Model
4.3. Cecal Ligation and Puncture (CLP)
4.4. The Development and Evaluation of DSS-Induced Colitis in Mice
4.5. Peritoneal Macrophage Isolation
4.6. Intestinal Macrophage Sorting
4.7. Measurement of IL-6 and TNF-α
4.8. Measurement of NO
4.9. Measurement of ALT and AST
4.10. ECAR and OCR Analysis
4.11. LC-MS/MS
4.12. Reagents and Complete Methods
4.13. Data Analysis
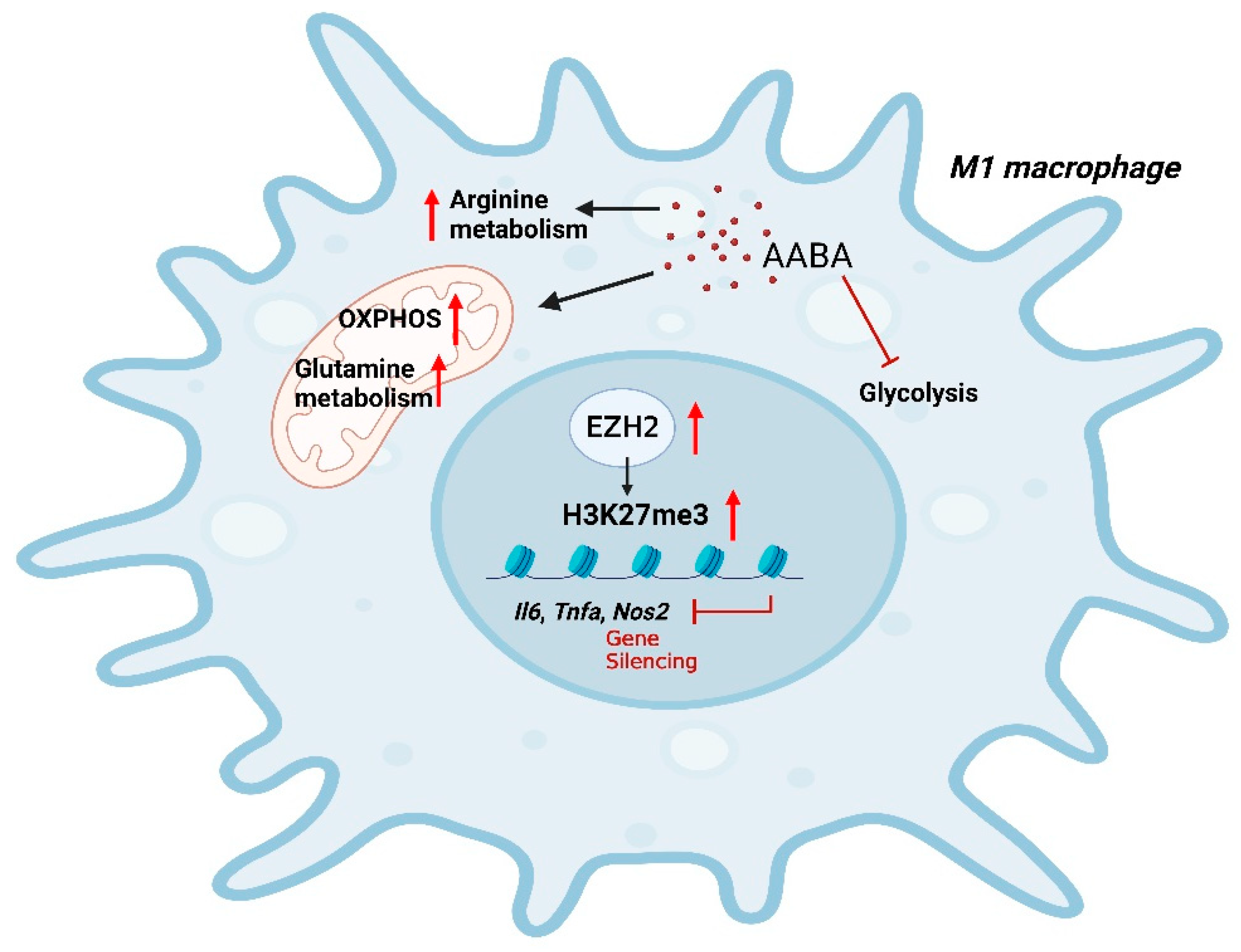
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Locati, M.; Curtale, G.; Mantovani, A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu. Rev. Pathol. 2020, 15, 123–147. [Google Scholar] [CrossRef] [PubMed]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Chen, S.; Saeed, A.F.U.H.; Liu, Q.; Jiang, Q.; Xu, H.; Xiao, G.G.; Rao, L.; Duo, Y. Macrophages in immunoregulation and therapeutics. Signal Transduct. Target. Ther. 2023, 8, 207. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef]
- Juhas, U.; Ryba-Stanisławowska, M.; Szargiej, P.; Myśliwska, J. Different pathways of macrophage activation and polarization. Adv. Hyg. Exp. Med. 2015, 69, 496–502. [Google Scholar] [CrossRef]
- Chen, X.; Liu, Y.; Gao, Y.; Shou, S.; Chai, Y. The roles of macrophage polarization in the host immune response to sepsis. Int. Immunopharmacol. 2021, 96, 107791. [Google Scholar] [CrossRef] [PubMed]
- Na, Y.R.; Stakenborg, M.; Seok, S.H.; Matteoli, G. Macrophages in intestinal inflammation and resolution: A potential therapeutic target in IBD. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 531–543. [Google Scholar] [CrossRef]
- Liu, X.; Ren, X.; Zhou, L.; Liu, K.; Deng, L.; Qing, Q.; Li, J.; Zhi, F.; Li, M. Tollip Orchestrates Macrophage Polarization to Alleviate Intestinal Mucosal Inflammation. J. Crohn’s Colitis 2022, 16, 1151–1167. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, T.; Prange, K.H.M.; Glass, C.K.; de Winther, M.P.J. Transcriptional and epigenetic regulation of macrophages in atherosclerosis. Nat. Rev. Cardiol. 2020, 17, 216–228. [Google Scholar] [CrossRef]
- El Kasmi, K.C.; Stenmark, K.R. Contribution of metabolic reprogramming to macrophage plasticity and function. Semin. Immunol. 2015, 27, 267–275. [Google Scholar] [CrossRef]
- Wculek, S.K.; Heras-Murillo, I.; Mastrangelo, A.; Mañanes, D.; Galán, M.; Miguel, V.; Curtabbi, A.; Barbas, C.; Chandel, N.S.; Enríquez, J.A.; et al. Oxidative phosphorylation selectively orchestrates tissue macrophage homeostasis. Immunity 2023, 56, 516–530.e9. [Google Scholar] [CrossRef] [PubMed]
- Van den Bossche, J.; O’Neill, L.A.; Menon, D. Macrophage Immunometabolism: Where Are We (Going)? Trends Immunol. 2017, 38, 395–406. [Google Scholar] [CrossRef]
- Van den Bossche, J.; Baardman, J.; Otto, N.A.; van der Velden, S.; Neele, A.E.; van den Berg, S.M.; Luque-Martin, R.; Chen, H.-J.; Boshuizen, M.C.S.; Ahmed, M.; et al. Mitochondrial Dysfunction Prevents Repolarization of Inflammatory Macrophages. Cell Rep. 2016, 17, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Peace, C.G.; O’Neill, L.A. The role of itaconate in host defense and inflammation. J. Clin. Investig. 2022, 132, e148548. [Google Scholar] [CrossRef]
- Kelly, B.; Pearce, E.L. Amino Assets: How Amino Acids Support Immunity. Cell Metab. 2020, 32, 154–175. [Google Scholar] [CrossRef]
- O’Neill, L.A.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Shan, X.; Hu, P.; Ni, L.; Shen, L.; Zhang, Y.; Ji, Z.; Cui, Y.; Guo, M.; Wang, H.; Ran, L.; et al. Serine metabolism orchestrates macrophage polarization by regulating the IGF1-p38 axis. Cell. Mol. Immunol. 2022, 19, 1263–1278. [Google Scholar] [CrossRef]
- Gan, Z.; Zhang, M.; Xie, D.; Wu, X.; Hong, C.; Fu, J.; Fan, L.; Wang, S.; Han, S. Glycinergic Signaling in Macrophages and Its Application in Macrophage-Associated Diseases. Front. Immunol. 2021, 12, 762564. [Google Scholar] [CrossRef]
- Rodriguez, A.E.; Ducker, G.S.; Billingham, L.K.; Martinez, C.A.; Mainolfi, N.; Suri, V.; Friedman, A.; Manfredi, M.G.; Weinberg, S.E.; Rabinowitz, J.D.; et al. Serine Metabolism Supports Macrophage IL-1beta Production. Cell Metab. 2019, 29, 1003–1011.e4. [Google Scholar] [CrossRef]
- Yu, W.; Wang, Z.; Zhang, K.; Chi, Z.; Xu, T.; Jiang, D.; Chen, S.; Li, W.; Yang, X.; Zhang, X.; et al. One-Carbon Metabolism Supports S-Adenosylmethionine and Histone Methylation to Drive Inflammatory Macrophages. Mol. Cell 2019, 75, 1147–1160.e5. [Google Scholar] [CrossRef]
- Fang, J.; Ou, Q.; Wu, B.; Li, S.; Wu, M.; Qiu, J.; Cen, N.; Hu, K.; Che, Y.; Ma, Y.; et al. TcpC Inhibits M1 but Promotes M2 Macrophage Polarization via Regulation of the MAPK/NF-κB and Akt/STAT6 Pathways in Urinary Tract Infection. Cells 2022, 11, 2674. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.S.; Wang, H.; Li, X.; Chao, T.; Teav, T.; Christen, S.; Di Conza, G.; Cheng, W.C.; Chou, C.H.; Vavakova, M.; et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol. 2017, 18, 985–994. [Google Scholar] [CrossRef]
- Chiarla, C.; Giovannini, I.; Siegel, J.H. Characterization of alpha-amino-n-butyric acid correlations in sepsis. Transl. Res. J. Lab. Clin. Med. 2011, 158, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Irino, Y.; Toh, R.; Nagao, M.; Mori, T.; Honjo, T.; Shinohara, M.; Tsuda, S.; Nakajima, H.; Satomi-Kobayashi, S.; Shinke, T.; et al. 2-Aminobutyric acid modulates glutathione homeostasis in the myocardium. Sci. Rep. 2016, 6, 36749. [Google Scholar] [CrossRef]
- Adachi, Y.; Toyoshima, K.; Nishimoto, R.; Ueno, S.; Tanaka, T.; Imaizumi, A.; Arashida, N.; Nakamura, M.; Abe, Y.; Hakamada, T.; et al. Association between plasma α-aminobutyric acid and depressive symptoms in older community-dwelling adults in Japan. Geriatr. Gerontol. Int. 2019, 19, 254–258. [Google Scholar] [CrossRef]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef]
- Damas, P.; Ledoux, D.; Nys, M.; Vrindts, Y.; De Groote, D.; Franchimont, P.; Lamy, M. Cytokine serum level during severe sepsis in human IL-6 as a marker of severity. Ann. Surg. 1992, 215, 356–362. [Google Scholar] [CrossRef]
- Symeonides, S.; Balk, R.A. Nitric oxide in the pathogenesis of sepsis. Infect Dis. Clin. N. Am. 1999, 13, 449–463. [Google Scholar] [CrossRef]
- Savi, F.F.; de Oliveira, A.; de Medeiros, G.F.; Bozza, F.A.; Michels, M.; Sharshar, T.; Dal-Pizzol, F.; Ritter, C. What animal models can tell us about long-term cognitive dysfunction following sepsis: A systematic review. Neurosci. Biobehav. Rev. 2021, 124, 386–404. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, R.; Gu, H.; Zhang, E.; Qu, J.; Cao, W.; Huang, X.; Yan, H.; He, J.; Cai, Z. Metabolic reprogramming in macrophage responses. Biomark. Res. 2021, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, G.; Riquelme, S.; Prince, A.; Avraham, R. Immunometabolic crosstalk during bacterial infection. Nat. Microbiol. 2022, 7, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Zhu, Q.; Pan, L.-L.; Sun, J. Macrophage immunometabolism in inflammatory bowel diseases: From pathogenesis to therapy. Pharmacol. Ther. 2022, 238, 108176. [Google Scholar] [CrossRef] [PubMed]
- Boomer, J.S.; Green, J.M.; Hotchkiss, R.S. The changing immune system in sepsis: Is individualized immuno-modulatory therapy the answer? Virulence 2014, 5, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Bouras, M.; Asehnoune, K.; Roquilly, A. Contribution of Dendritic Cell Responses to Sepsis-Induced Immunosuppression and to Susceptibility to Secondary Pneumonia. Front. Immunol. 2018, 9, 2590. [Google Scholar] [CrossRef]
- Wang, F.; Cui, Y.; He, D.; Gong, L.; Liang, H. Natural killer cells in sepsis: Friends or foes? Front. Immunol. 2023, 14, 1101918. [Google Scholar] [CrossRef]
- Kałużna, A.; Olczyk, P.; Komosińska-Vassev, K. The Role of Innate and Adaptive Immune Cells in the Pathogenesis and Development of the Inflammatory Response in Ulcerative Colitis. J. Clin. Med. 2022, 11, 400. [Google Scholar] [CrossRef]
- De Santa, F.; Vitiello, L.; Torcinaro, A.; Ferraro, E. The Role of Metabolic Remodeling in Macrophage Polarization and Its Effect on Skeletal Muscle Regeneration. Antioxid. Redox Signal. 2019, 30, 1553–1598. [Google Scholar] [CrossRef]
- Saha, S.; Shalova, I.N.; Biswas, S.K. Metabolic regulation of macrophage phenotype and function. Immunol. Rev. 2017, 280, 102–111. [Google Scholar] [CrossRef]
- Kieler, M.; Hofmann, M.; Schabbauer, G. More than just protein building blocks: How amino acids and related metabolic pathways fuel macrophage polarization. FEBS J. 2021, 288, 3694–3714. [Google Scholar] [CrossRef]
- Tarditi, A.M.; Klipfel, M.W.; Rodriguez, A.M.; Suvire, F.D.; Chasse, G.A.; Farkas, O.; Perczel, A.; Enriz, R.D. An ab initio exploratory study of side chain conformations for selected backbone conformations of N-acetyl-l-glutamine-N-methylamide. J. Mol. Struct. Theochem. 2001, 545, 29–47. [Google Scholar] [CrossRef]
- van Mierlo, G.; Veenstra, G.J.C.; Vermeulen, M.; Marks, H. The Complexity of PRC2 Subcomplexes. Trends Cell Biol. 2019, 29, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Chi, G.; Pei, J.H.; Li, X.Q. EZH2-mediated H3K27me3 promotes autoimmune hepatitis progression by regulating macrophage polarization. Int. Immunopharmacol. 2022, 106, 108612. [Google Scholar] [CrossRef]
- Kitchen, G.B.; Hopwood, T.; Gali Ramamoorthy, T.; Downton, P.; Begley, N.; Hussell, T.; Dockrell, D.H.; Gibbs, J.E.; Ray, D.W.; Loudon, A.S.I. The histone methyltransferase Ezh2 restrains macrophage inflammatory responses. FASEB J. 2021, 35, e21843. [Google Scholar] [CrossRef] [PubMed]
- Neele, A.E.; de Winther, M.P.J. Repressing the repressor: Ezh2 mediates macrophage activation. J. Exp. Med. 2018, 215, 1269–1271. [Google Scholar] [CrossRef]
- León, I.; Peña, I.; Cabezas, C.; Alonso, E.R.; Alonso, J.L. The last link of the x-aminobutyric acid series: The five conformers of β-aminobutyric acid. Phys. Chem. Chem. Phys. PCCP 2018, 20, 15574–15580. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.W. GABA as a Neurotransmitter in Gastropod Molluscs. Biol. Bull. 2019, 236, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Sun, Q.; Sun, X.; Chen, D.; Wei, C.; Yu, X.; Liu, C.; Li, Y.; Li, J. Activation of GABAA Receptors in Colon Epithelium Exacerbates Acute Colitis. Front. Immunol. 2018, 9, 987. [Google Scholar] [CrossRef] [PubMed]
- Oketch-Rabah, H.A.; Madden, E.F.; Roe, A.L.; Betz, J.M. United States Pharmacopeia (USP) Safety Review of Gamma-Aminobutyric Acid (GABA). Nutrients 2021, 13, 2742. [Google Scholar] [CrossRef]
- Xie, W.; Zou, S.; Dong, C.; Yang, C. SPI1-mediated autophagy of peripheral blood monocyte cells as a mechanism for sepsis based on single-cell RNA sequencing. Int. Immunopharmacol. 2023, 117, 109909. [Google Scholar] [CrossRef] [PubMed]
- Torretta, S.; Scagliola, A.; Ricci, L.; Mainini, F.; Di Marco, S.; Cuccovillo, I.; Kajaste-Rudnitski, A.; Sumpton, D.; Ryan, K.M.; Cardaci, S. D-mannose suppresses macrophage IL-1β production. Nat. Commun. 2020, 11, 6343. [Google Scholar] [CrossRef] [PubMed]
- Xiong, B.; Zhang, W.; Zhang, L.; Huang, X.; Zhou, W.; Zou, Q.; Manyande, A.; Wang, J.; Tian, Y.; Tian, X. Hippocampal glutamatergic synapses impairment mediated novel-object recognition dysfunction in rats with neuropathic pain. Pain 2020, 161, 1824–1836. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Shen, Y.; Wang, M.; Zhang, J. Serum and CSF Metabolites in Stroke-Free Patients Are Associated With Vascular Risk Factors and Cognitive Performance. Front. Aging Neurosci. 2020, 12, 193. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, F.; Xia, Y.; Yuan, S.; Xie, X.; Li, L.; Luo, Y.; Du, Q.; Yuan, Y.; He, R. α-Aminobutyric Acid Constrains Macrophage-Associated Inflammatory Diseases through Metabolic Reprogramming and Epigenetic Modification. Int. J. Mol. Sci. 2023, 24, 10444. https://doi.org/10.3390/ijms241310444
Li F, Xia Y, Yuan S, Xie X, Li L, Luo Y, Du Q, Yuan Y, He R. α-Aminobutyric Acid Constrains Macrophage-Associated Inflammatory Diseases through Metabolic Reprogramming and Epigenetic Modification. International Journal of Molecular Sciences. 2023; 24(13):10444. https://doi.org/10.3390/ijms241310444
Chicago/Turabian StyleLi, Fei, Yuting Xia, Shijie Yuan, Xiaorong Xie, Lin Li, Yuan Luo, Qiuyang Du, Yuqi Yuan, and Ran He. 2023. "α-Aminobutyric Acid Constrains Macrophage-Associated Inflammatory Diseases through Metabolic Reprogramming and Epigenetic Modification" International Journal of Molecular Sciences 24, no. 13: 10444. https://doi.org/10.3390/ijms241310444
APA StyleLi, F., Xia, Y., Yuan, S., Xie, X., Li, L., Luo, Y., Du, Q., Yuan, Y., & He, R. (2023). α-Aminobutyric Acid Constrains Macrophage-Associated Inflammatory Diseases through Metabolic Reprogramming and Epigenetic Modification. International Journal of Molecular Sciences, 24(13), 10444. https://doi.org/10.3390/ijms241310444