

Specific Methyl-CpG Configurations Define Cell Identity through Gene Expression Regulation


 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Specific Methylated Cores Mark the Promoters of Genes Involved in Murine Postnatal Brain Differentiation
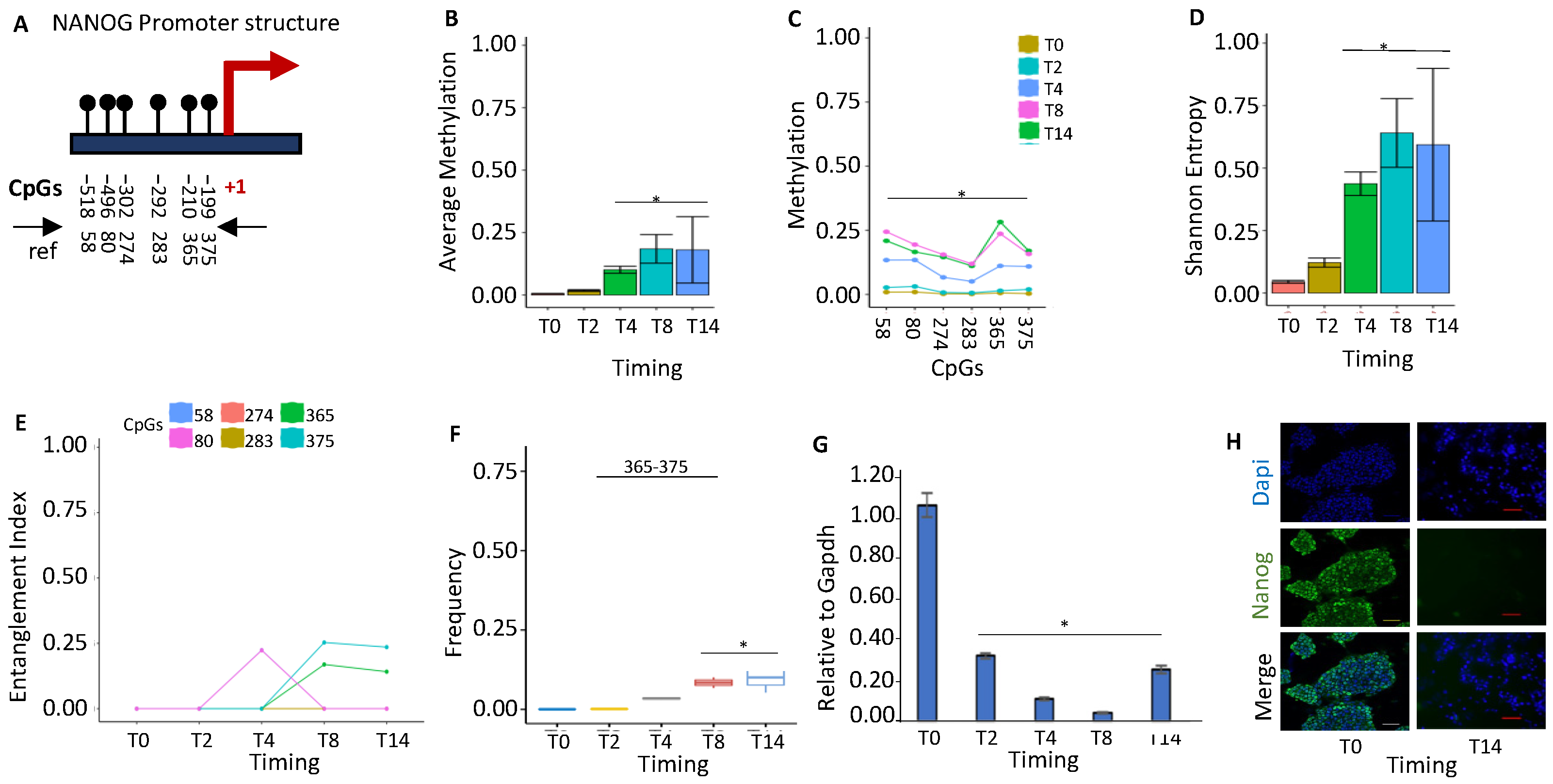
2.1.1. NANOG Repression Is Associated with De Novo Formation of a Methylated Core in the Promoter
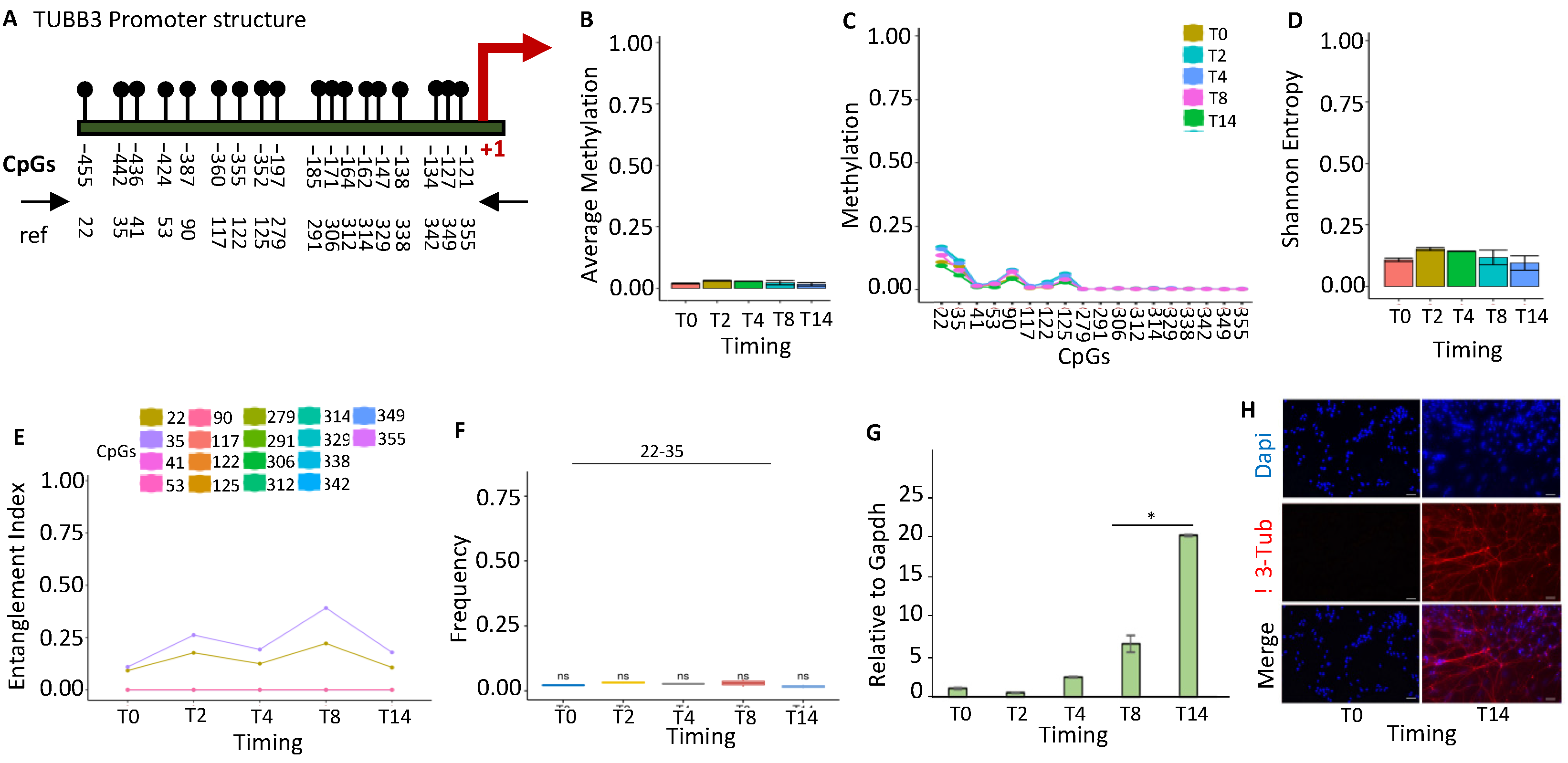
2.1.2. TUBB3 Promoter Methylation Does Not Control the Expression during Neuronal Differentiation
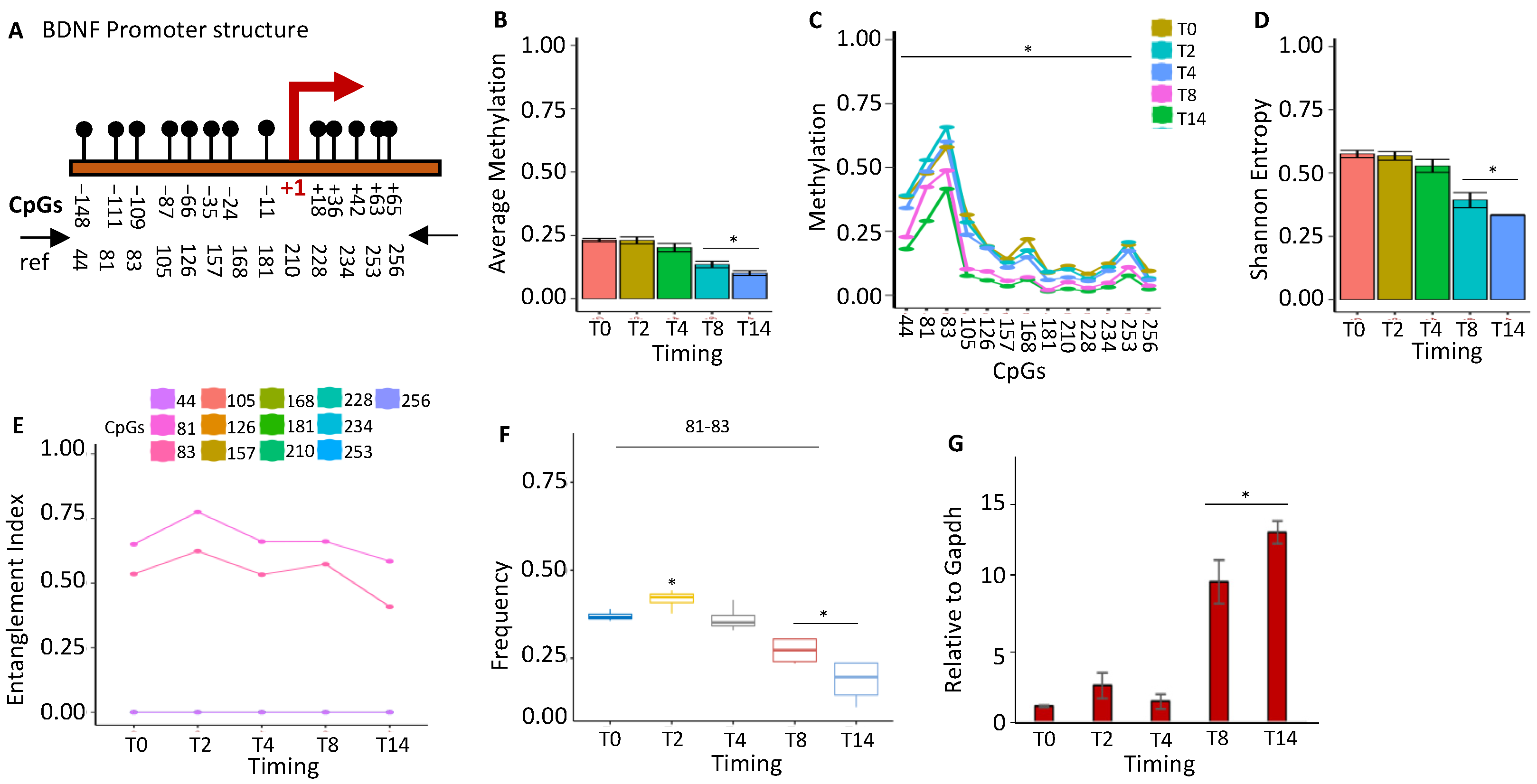
2.1.3. BDNF Gene Activation Parallels the Loss of the Methylated Core at the Promoter during Brain Differentiation
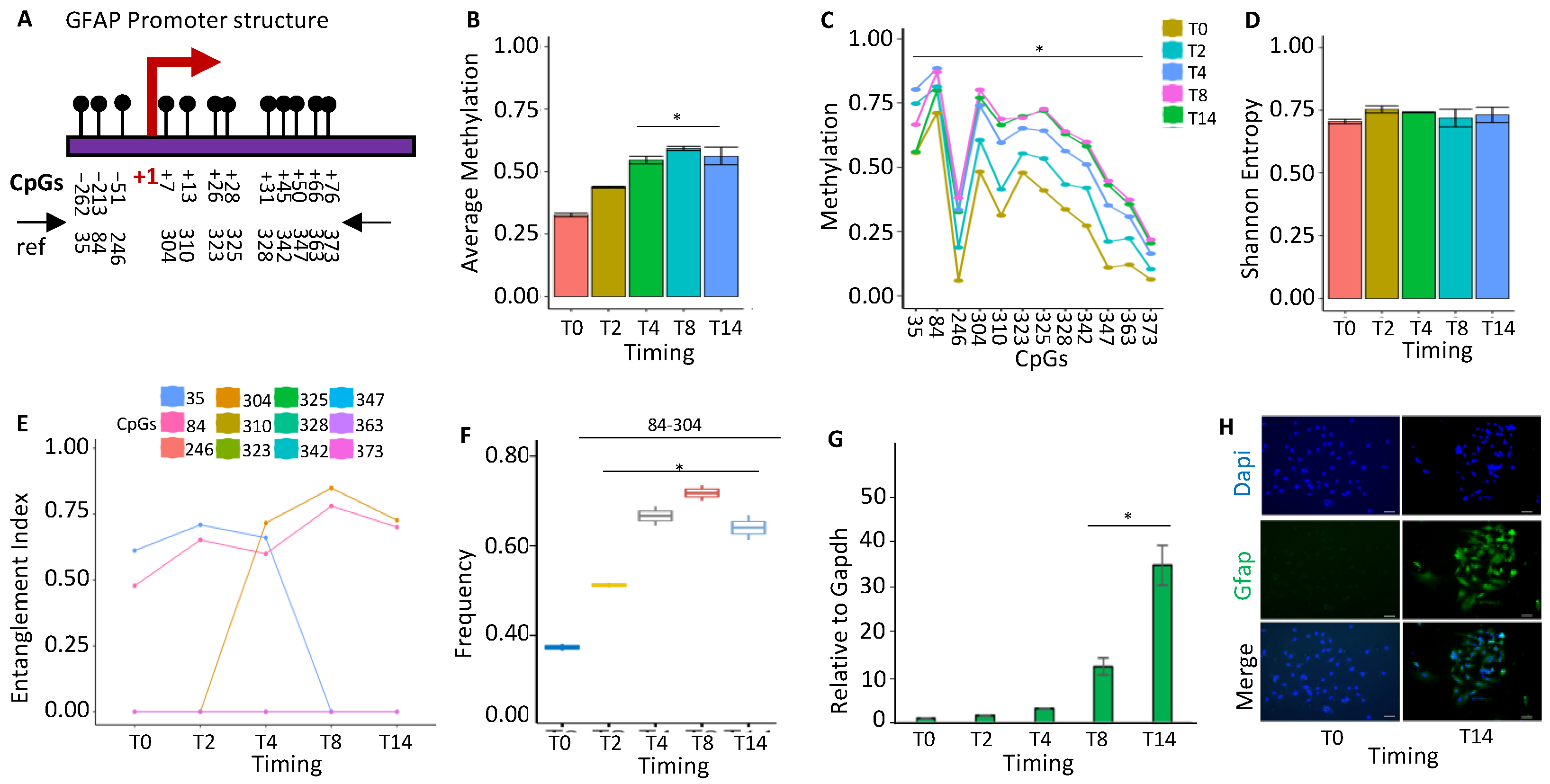
2.1.4. GFAP Gene Activation Changes the Configuration of the Promoter Methylated Core
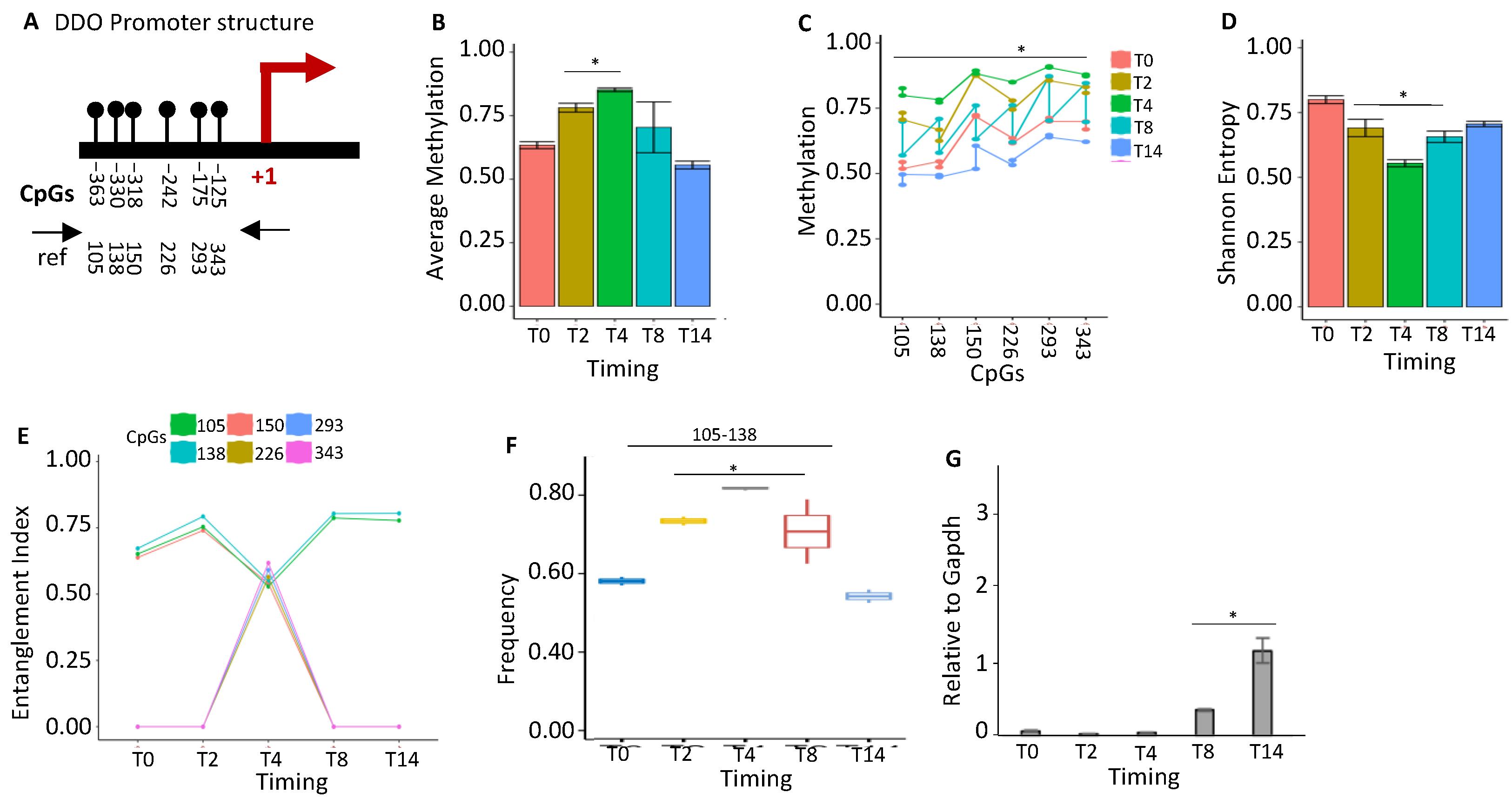
2.1.5. DDO Gene Promoter Methylation Profile and Expression during Postnatal Brain Differentiation
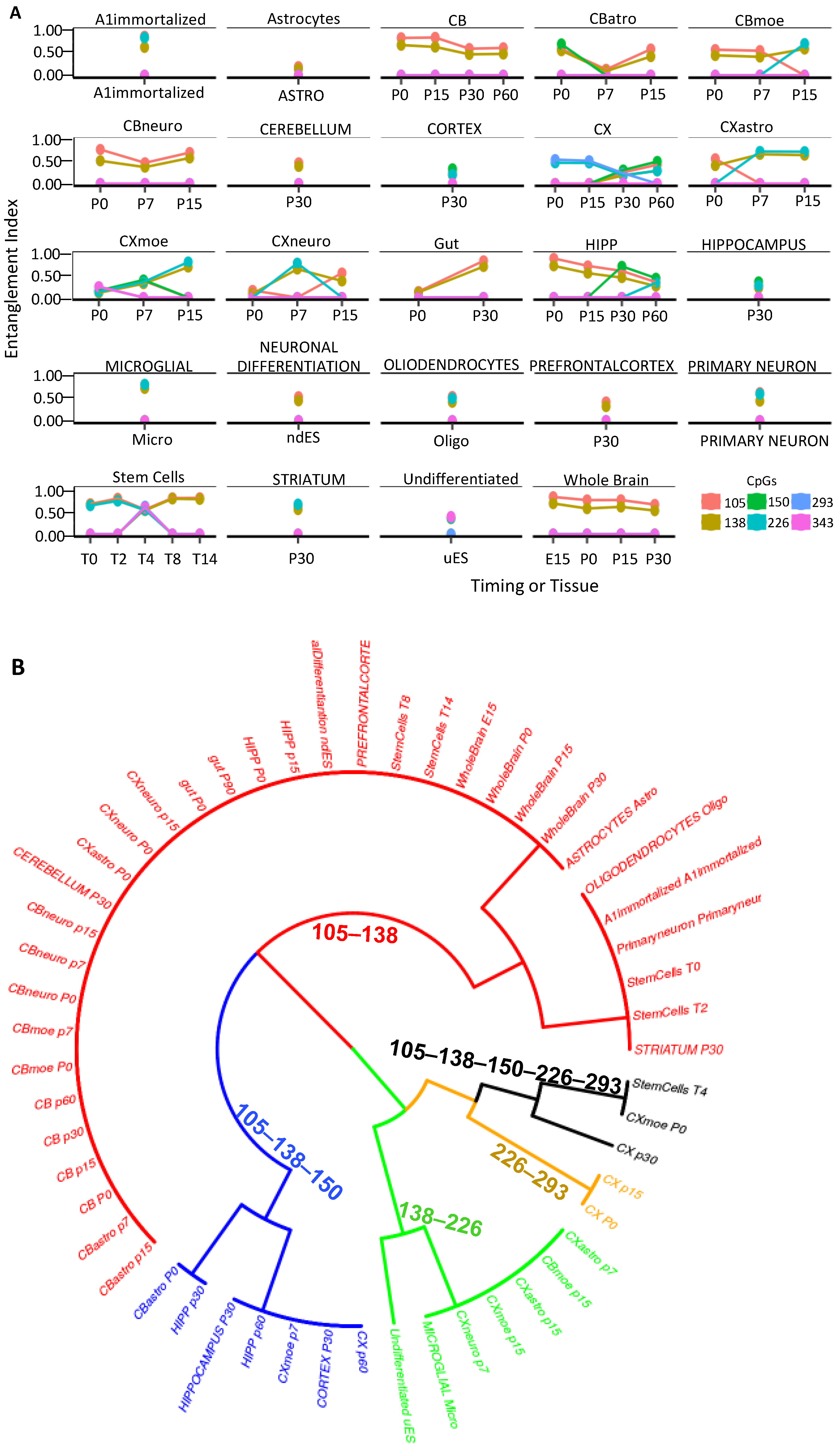
2.2. CpG Methylation Cores Define Cell Identity
3. Discussion
4. Materials and Methods
4.1. ESC culture and Differentiation
4.2. RT–PCR
4.3. Immunofluorescence
4.4. DNA Extraction
4.5. Bisulfite Treatment and Amplicon Library Preparation
4.6. Dataset Description of Figure 6
4.7. Sequence Handling
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moore, L.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Tramontano, A.; Boffo, F.L.; Russo, G.; De Rosa, M.; Iodice, I.; Pezone, A. Methylation of the suppressor gene: Mechanism and consequences. Biomolecules 2020, 10, 446. [Google Scholar] [CrossRef]
- Jin, B.; Ernst, J.; Tiedemann, R.L.; Xu, H.; Sureshchandra, S.; Kellis, M.; Dalton, S.; Liu, C.; Choi, J.-H.; Robertson, K.D. Linking DNA methyltransferases to epigenetic marks and nucleosome structure genome-wide in human tumor cells. Cell Rep. 2012, 2, 1411–1424. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Pastor, W.A.; Shen, Y.; Tahiliani, M.; Liu, D.R.; Rao, A. The behavior of 5-hydroxymethylcytosine in bisulfite sequencing. PLoS ONE 2010, 5, e8888. [Google Scholar]
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Allen, B.; Pezone, A.; Porcellini, A.; Muller, M.T.; Masternak, M.M. Non-homologous end joining induced alterations in DNA methylation: A source of permanent epigenetic change. Oncotarget 2017, 8, 40359–40372. [Google Scholar] [CrossRef] [PubMed]
- Song, C.X.; He, C. Potential functional roles of DNA demethylation intermediates. Trends Biochem Sci. 2013, 38, 480–484. [Google Scholar] [CrossRef]
- Xie, H.; Wang, M.; de Andrade, A.; de Bonaldo, M.F.; Galat, V.; Arndt, K.; Soares, M.B. Genome-wide quantitative assessment of variation in DNA methylation patterns. Nucleic Acids Res. 2011, 39, 4099–4108. [Google Scholar] [CrossRef] [PubMed]
- Barros-Silva, D.; Marques, C.J.; Henrique, R.; Jerónimo, C. Profiling DNA Methylation Based on Next-Generation Sequencing Approaches: New Insights and Clinical Applications. Genes 2018, 9, 429. [Google Scholar] [CrossRef]
- Pezone, A.; Tramontano, A.; Scala, G.; Cuomo, M.; Riccio, P.; De Nicola, S.; Porcellini, A.; Chiariotti, L.; Avvedimento, E.V. Tracing and tracking epiallele families in complex DNA populations. NAR Genom Bioinform. 2020, 2, lqaa096. [Google Scholar] [CrossRef]
- Podobinska, M.; Szablowska-Gadomska, I.; Augustyniak, J.; Sandvig, I.; Sandvig, A.; Buzanska, L. Epigenetic Modulation of Stem Cells in Neurodevelopment: The Role of Methylation and Acetylation. Front Cell Neurosci. 2017, 11, 23. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Costello, J. DNA methylation: An epigenetic mark of cellular memory. Exp. Mol. Med. 2017, 49, e322. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Tiwari, V.K. Epigenetic reprogramming of cell identity: Lessons from development for regenerative medicine. Clin. Epigenet. 2021, 13, 144. [Google Scholar] [CrossRef]
- Yang, Y.; Fan, X.; Yan, J.; Chen, M.; Zhu, M.; Tang, Y.; Liu, S.; Tang, Z. A comprehensive epigenome atlas reveals DNA methylation regulating skeletal muscle development. Nucleic Acids Res. 2021, 49, 1313–1329. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Garrett-Bakelman, F.; Perl, A.E.; Luger, S.M.; Zhang, C.; To, B.L.; Lewis, I.D.; Brown, A.; D’andrea, R.J.; Ross, M.E.; et al. Dynamic evolution of clonal epialleles revealed by methclone. Genome Biol. 2014, 15, 472. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Garrett-Bakelman, F.E.; Chung, S.S.; Sanders, M.A.; Hricik, T.; Rapaport, F.; Patel, J.; Dillon, R.; Vijay, P.; Brown, A.; et al. Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nat. Med. 2016, 22, 792–799. [Google Scholar] [CrossRef] [PubMed]
- Shannon, C.E. A mathematical theory of communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef]
- Verma, R.; Liu, J.; Holland, M.K.; Temple-Smith, P.; Williamson, M.; Verma, P.J. Nanog is an essential factor for induction of pluripotency in somatic cells from endangered felids. Biores. Open Access. 2013, 2, 72–76. [Google Scholar] [CrossRef]
- Radwitz, J.; Hausrat, T.J.; Heisler, F.F.; Janiesch, P.C.; Pechmann, Y.; Rübhausen, M.; Kneussel, M. Tubb3 expression levels are sensitive to neuronal activity changes and determine microtubule growth and kinesin-mediated transport. Cell Mol Life Sci. 2022, 79, 575. [Google Scholar] [CrossRef] [PubMed]
- Errico, F.; Cuomo, M.; Canu, N.; Caputo, V.; Usiello, A. New insights on the influence of free d-aspartate metabolism in the mammalian brain during prenatal and postnatal life. Biochim. Biophys. Acta Proteins Proteom. 2020, 1868, 140471. [Google Scholar] [CrossRef]
- Jurga, A.M.; Paleczna, M.; Kadluczka, J.; Kuter, K.Z. Beyond the GFAP-Astrocyte Protein Markers in the Brain. Biomolecules 2021, 11, 1361. [Google Scholar] [CrossRef]
- Bathina, S.; Das, U.N. Brain-derived neurotrophic factor and its clinical implications. Arch. Med. Sci. 2015, 11, 1164–1178. [Google Scholar] [CrossRef] [PubMed]
- Busskamp, V.; Lewis, N.E.; Guye, P.; Ng, A.H.; Shipman, S.L.; Byrne, S.M.; Sanjana, N.E.; Murn, J.; Li, Y.; Li, S.; et al. Rapid neurogenesis through transcriptional activation in human stem cells. Mol. Syst. Biol. 2014, 10, 760. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.P.; Yang, N.; Vierbuchen, T.; Ostermeier, A.; Fuentes, D.R.; Yang, T.Q.; Citri, A.; Sebastiano, V.; Marro, S.; Südhof, T.C.; et al. Induction of human neuronal cells by defined transcription factors. Nature 2011, 476, 220–223. [Google Scholar] [CrossRef]
- Raspaglio, G.; Petrillo, M.; Martinelli, E.; Puma, D.D.L.; Mariani, M.; De Donato, M.; Filippetti, F.; Mozzetti, S.; Prislei, S.; Zannoni, G.F.; et al. Sox9 and Hif-2α regulate TUBB3 gene expression and affect ovarian cancer aggressiveness. Gene 2014, 542, 173–181. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Wang, J. Decreased serum brain-derived neurotrophic factor in Chinese patients with Type 2 diabetes mellitus. Acta Biochim. Biophys. Sin. 2014, 46, 426–427. [Google Scholar] [CrossRef] [PubMed]
- Mamber, C.; Kamphuis, W.; Haring, N.L.; Peprah, N.; Middeldorp, J.; Hol, E.M. GFAPδ expression in glia of the developmental and adolescent mouse brain. PLoS ONE 2012, 7, e52659. [Google Scholar] [CrossRef]
- Ahmed, A.I.; Shtaya, A.B.; Zaben, M.J.; Owens, E.V.; Kiecker, C.; Gray, W.P. Endogenous GFAP-positive neural stem/progenitor cells in the postnatal mouse cortex are activated following traumatic brain injury. J. Neurotrauma 2012, 29, 828–842. [Google Scholar] [CrossRef]
- Koga, R.; Miyoshi, Y.; Sakaue, H.; Hamase, K.; Konno, R. Mouse D-amino-acid oxidase: Distribution and physiological substrates. Front. Mol. Biosci. 2017, 4, 82. [Google Scholar] [CrossRef]
- Florio, E.; Keller, S.; Coretti, L.; Affinito, O.; Scala, G.; Errico, F.; Fico, A.; Boscia, F.; Sisalli, M.J.; Reccia, M.G.; et al. Tracking the evolution of epialleles during neural differentiation and brain development: D-Aspartate oxidase as a model gene. Epigenetics 2017, 12, 41–54. [Google Scholar] [CrossRef]
- Punzo, D.; Errico, F.; Cristino, L.; Sacchi, S.; Keller, S.; Belardo, C.; Luongo, L.; Nuzzo, T.; Imperatore, R.; Florio, E.; et al. Age-Related Changes in D-Aspartate Oxidase Promoter Methylation Control Extracellular D-Aspartate Levels and Prevent Precocious Cell Death during Brain Aging. J. Neurosci. 2016, 36, 3064–3078. [Google Scholar] [CrossRef] [PubMed]
- Breiling, A.; Lyko, F. Epigenetic regulatory functions of DNA modifications: 5-methylcytosine and beyond. Epigenet. Chromatin 2015, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Hajkova, P.; Ecker, J.R. Dynamic DNA methylation: In the right place at the right time. Science 2018, 361, 1336–1340. [Google Scholar] [CrossRef] [PubMed]
- Fuentealba, L.C.; Rompani, S.B.; Parraguez, J.I.; Obernier, K.; Romero, R.; Cepko, C.L.; Alvarez-Buylla, A. Embryonic origin of postnatal neural stem cells. Cell 2015, 161, 1644–1655. [Google Scholar] [CrossRef]
- Anderson, R.B.; Newgreen, D.F.; Young, H.M. Neural Crest and the Development of the Enteric Nervous System. In Madame Curie Bioscience Database; Landes Bioscience: Austin, TX, USA, 2000–2013. Available online: https://www.ncbi.nlm.nih.gov/books/NBK6273/ (accessed on 5 May 2023).
- Niwa, H.; Miyazaki, J.I.; Smith, A. Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat. Genet. 2000, 24, 372–376. [Google Scholar] [CrossRef]
- Parisi, S.; Passaro, F.; Aloia, L.; Manabe, I.; Nagai, R.; Pastore, L.; Russo, T. Klf5 is involved in self-renewal of mouse embryonic stem cells. J. Cell Sci. 2008, 121 Pt 16, 2629–2634. [Google Scholar] [CrossRef]
- Passaro, F.; De Martino, I.; Zambelli, F.; Di Benedetto, G.; Barbato, M.; D'Erchia, A.M.; Manzari, C.; Pesole, G.; Mutarelli, M.; Cacchiarelli, D.; et al. YAP contributes to DNA methylation remodeling upon mouse embryonic stem cell differentiation. J. Biol. Chem. 2021, 296, 100138. [Google Scholar] [CrossRef]
- Russo, G.; Tramontano, A.; Iodice, I.; Chiariotti, L.; Pezone, A. Epigenome Chaos: Stochastic and Deterministic DNA Methylation Events Drive Cancer Evolution. Cancers 2021, 13, 1800. [Google Scholar] [CrossRef]
- Pezone, A.; Russo, G.; Tramontano, A.; Florio, E.; Scala, G.; Landi, R.; Zuchegna, C.; Romano, A.; Chiariotti, L.; Muller, M.T.; et al. High-coverage methylation data of a gene model before and after DNA damage and homologous repair. Sci. Data 2017, 4, 170043. [Google Scholar] [CrossRef]
- Suelves, M.; Carrió, E.; Núñez-Álvarez, Y.; Peinado, M.A. DNA methylation dynamics in cellular commitment and differentiation. Brief Funct. Genom. 2016, 15, 443–453. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Improda, T.; Morgera, V.; Vitale, M.; Chiariotti, L.; Passaro, F.; Feola, A.; Porcellini, A.; Cuomo, M.; Pezone, A. Specific Methyl-CpG Configurations Define Cell Identity through Gene Expression Regulation. Int. J. Mol. Sci. 2023, 24, 9951. https://doi.org/10.3390/ijms24129951
Improda T, Morgera V, Vitale M, Chiariotti L, Passaro F, Feola A, Porcellini A, Cuomo M, Pezone A. Specific Methyl-CpG Configurations Define Cell Identity through Gene Expression Regulation. International Journal of Molecular Sciences. 2023; 24(12):9951. https://doi.org/10.3390/ijms24129951
Chicago/Turabian StyleImproda, Teresa, Valentina Morgera, Maria Vitale, Lorenzo Chiariotti, Fabiana Passaro, Antonia Feola, Antonio Porcellini, Mariella Cuomo, and Antonio Pezone. 2023. "Specific Methyl-CpG Configurations Define Cell Identity through Gene Expression Regulation" International Journal of Molecular Sciences 24, no. 12: 9951. https://doi.org/10.3390/ijms24129951
APA StyleImproda, T., Morgera, V., Vitale, M., Chiariotti, L., Passaro, F., Feola, A., Porcellini, A., Cuomo, M., & Pezone, A. (2023). Specific Methyl-CpG Configurations Define Cell Identity through Gene Expression Regulation. International Journal of Molecular Sciences, 24(12), 9951. https://doi.org/10.3390/ijms24129951