
New Cases and Mutations in SEC23B Gene Causing Congenital Dyserythropoietic Anemia Type II

 , , , ,
, , , ,
Abstract
1. Introduction
2. Results
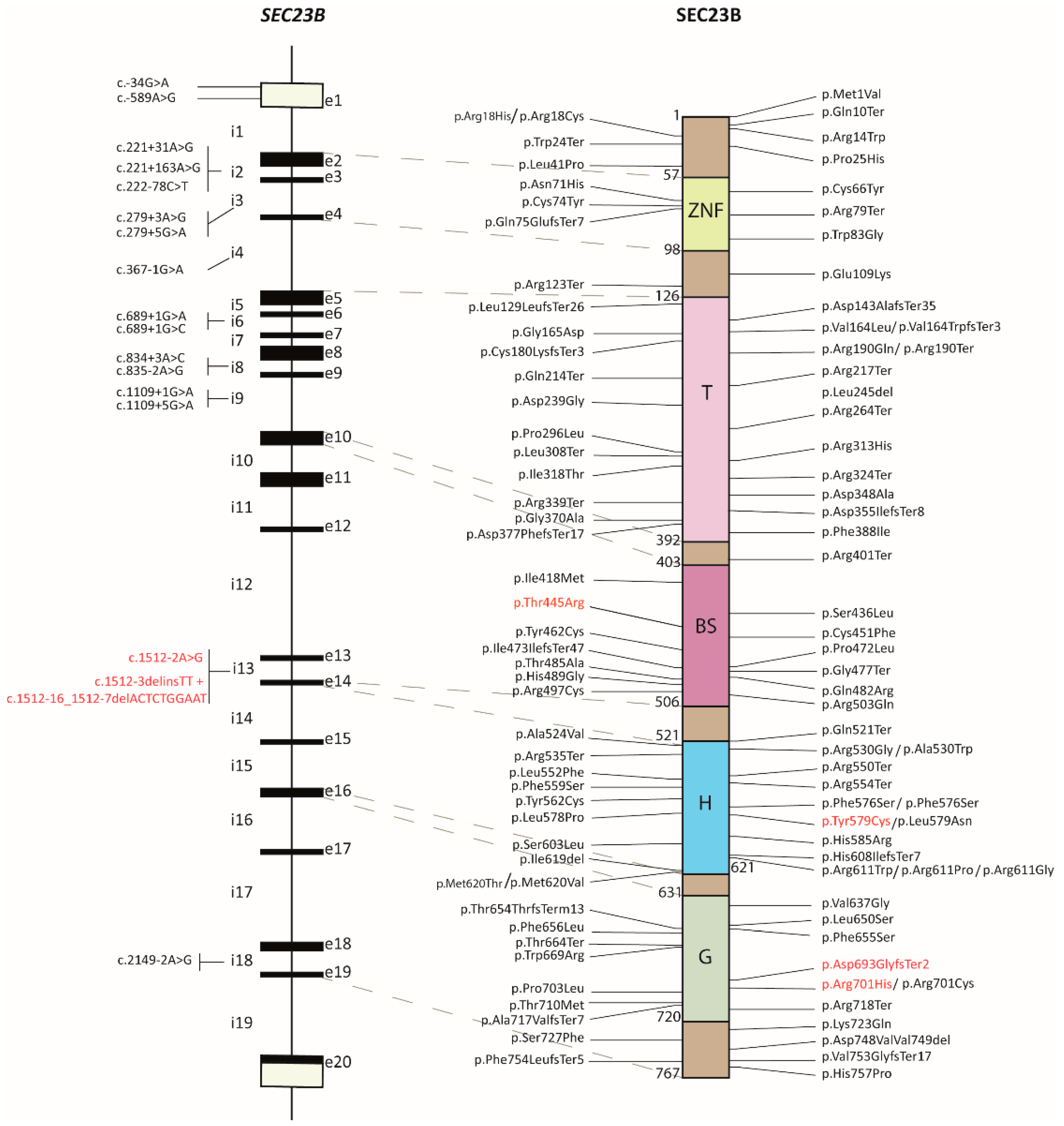
2.1. Review of the SEC23B Mutational Spectrum
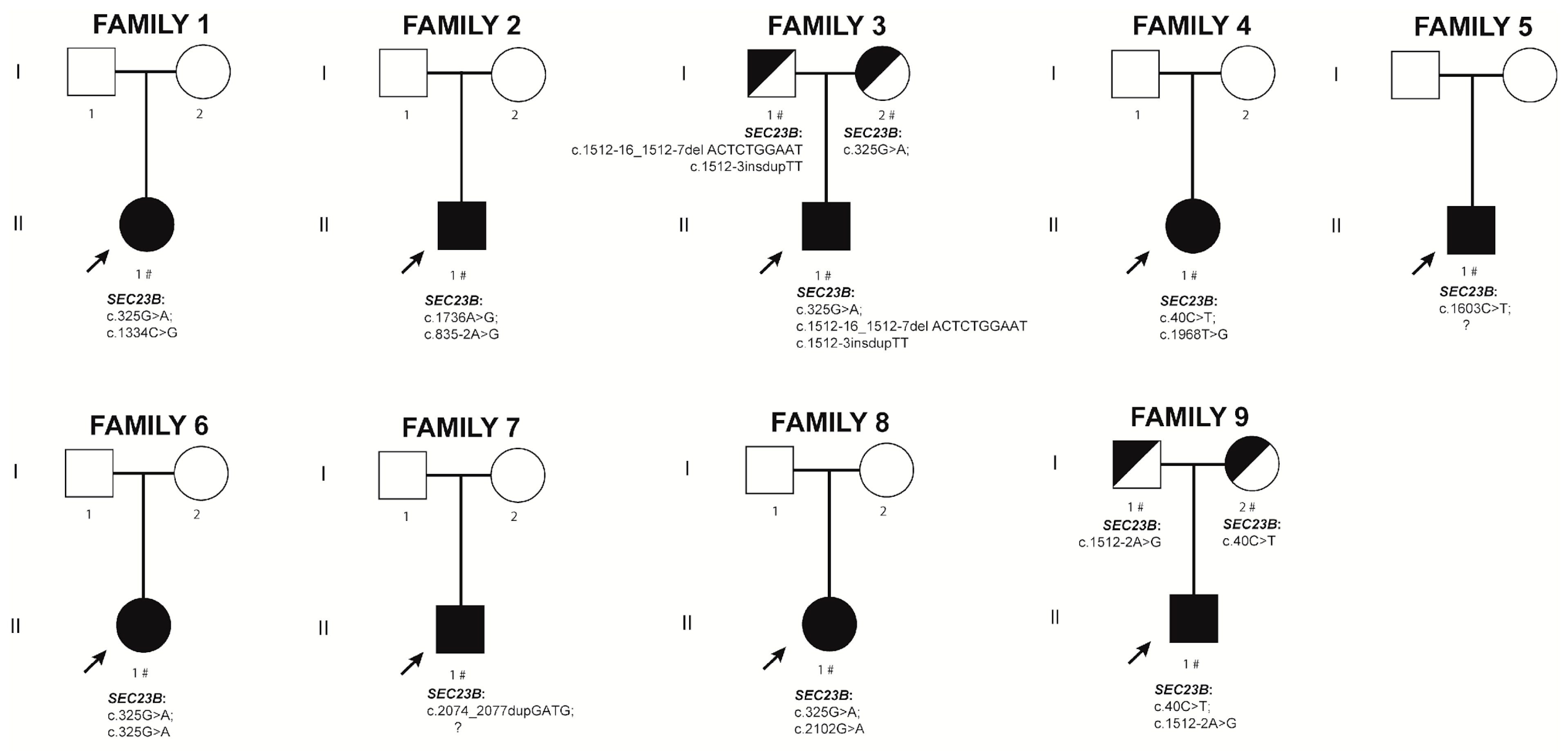
2.2. New CDA II Cases
2.2.1. Family 1. Proband II.1
2.2.2. Family 2. Proband II.1
2.2.3. Family 3. Proband II.1
2.2.4. Family 4. Proband II.1
2.2.5. Family 5. Proband II.1
2.2.6. Family 6. Proband II.1
2.2.7. Family 7. Proband II.1
2.2.8. Family 8. Proband II.1
2.2.9. Family 9. Proband II.1
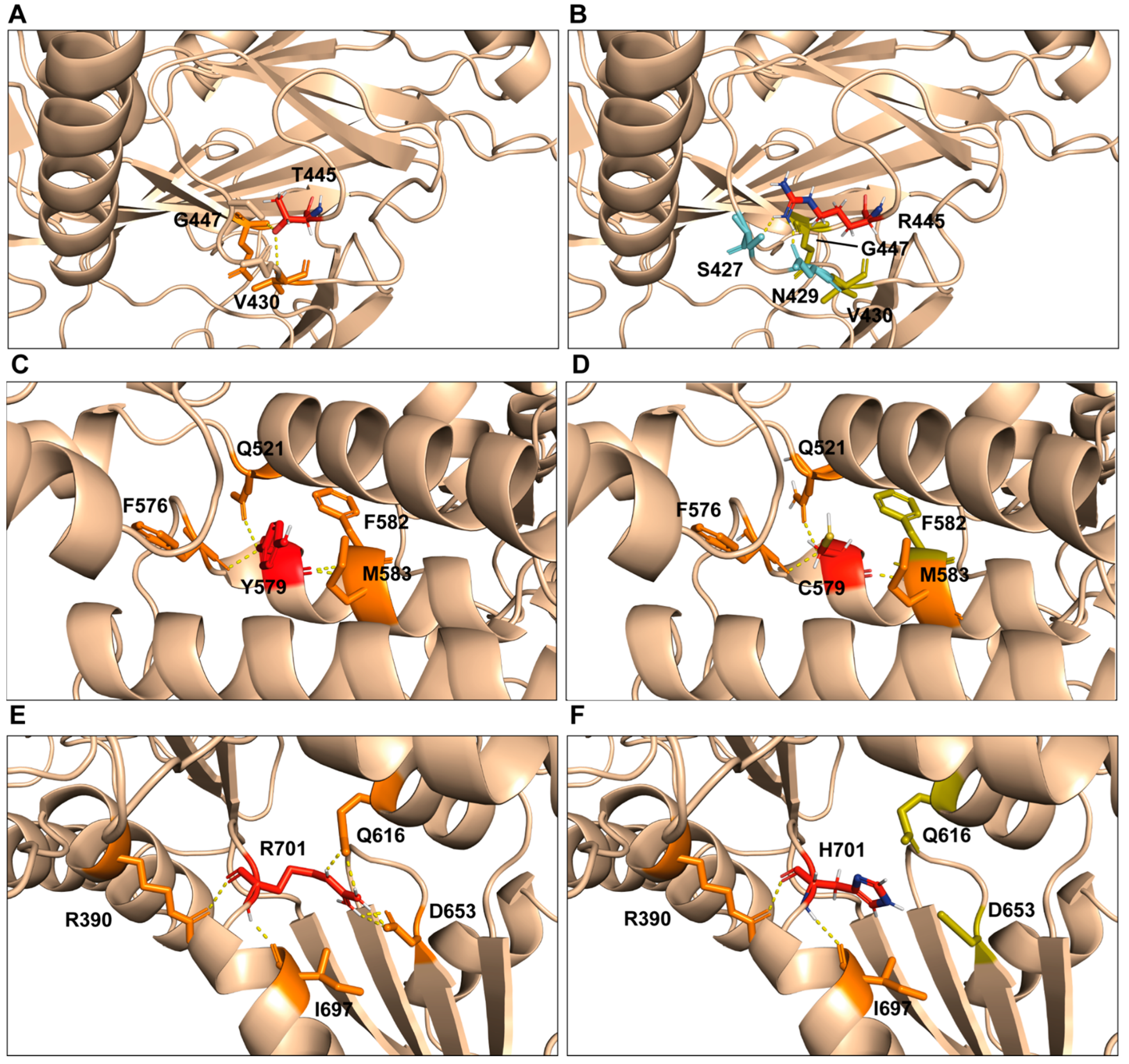
2.3. Computational Model of p.Thr445Arg, p.Tyr579Cys, and p.Arg701His Variants
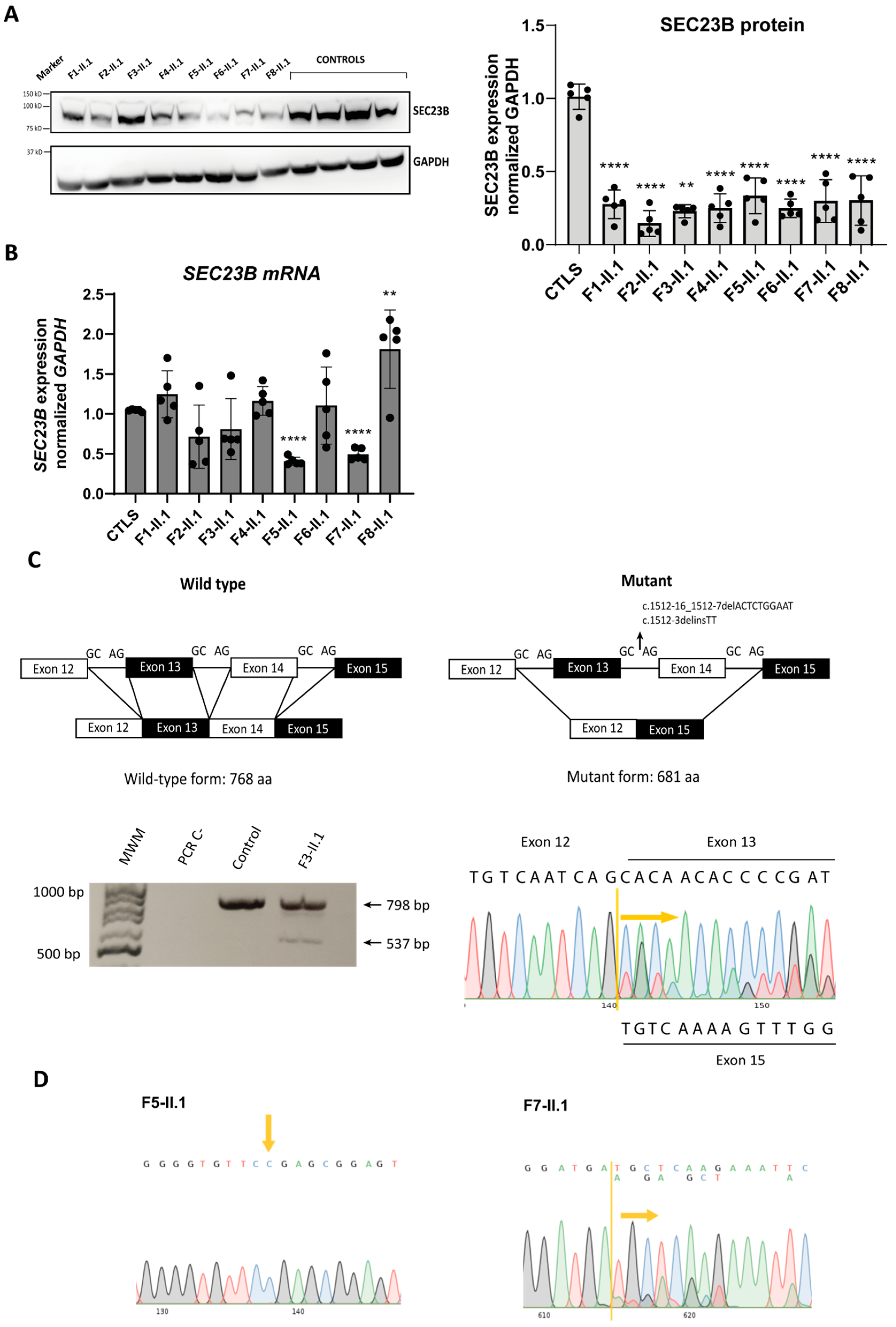
2.4. Analysis of SEC23B mRNA and Protein Levels in Patient-Derived LCLs
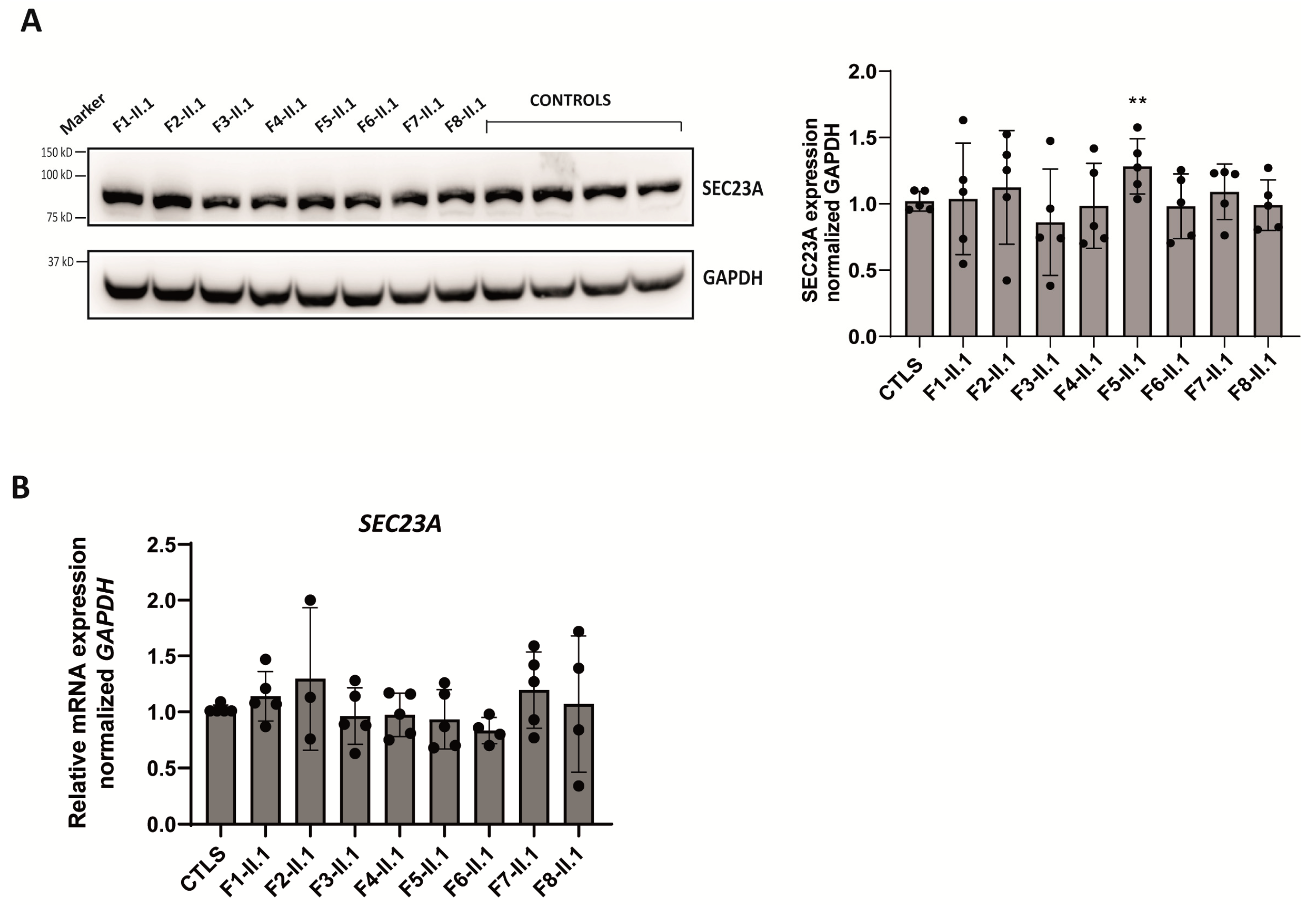
2.5. Expression Analysis of SEC23A Paralog in Patients’ LCLs
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. DNA Sequencing and Analysis
4.3. Generation of Lymphoblastoid Cell Lines (LCLs)
4.4. RNA Extraction, Reverse Transcription, and qPCR
4.5. Immunoblotting
4.6. Computational Studies
4.7. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Iolascon, A.; Andolfo, I.; Russo, R. Congenital dyserythropoietic anemias. Blood 2020, 136, 1274–1283. [Google Scholar] [CrossRef]
- Hernández, G.; Romero-Cortadellas, L.; Ferrer-Cortès, X.; Venturi, V.; Dessy-Rodriguez, M.; Olivella, M.; Husami, A.; De Soto, C.P.; Morales-Camacho, R.M.; Villegas, A.; et al. Mutations in the RACGAP1 gene cause autosomal recessive congenital dyserythropoietic anemia type III. Haematologica 2023, 108, 581–587. [Google Scholar] [CrossRef]
- Cantù-Rajnoldi, A.; Zanella, A.; Conter, U.; Faccini, P.; Soligo, D.; Gornati, G.; Vegni, C.; Nicolini, U. A severe transfusion-dependent congenital dyserythropoietic anaemia presenting as hydrops fetalis. Br. J. Haematol. 1997, 96, 530–533. [Google Scholar] [CrossRef]
- Heimpel, H.; Anselstetter, V.; Chrobak, L.; Denecke, J.; Einsiedler, B.; Gallmeier, K.; Griesshammer, A.; Marquardt, T.; Janka-Schaub, G.; Kron, M.; et al. Congenital dyserythropoietic anemia type II: Epidemiology, clinical appearance, and prognosis based on long-term observation. Blood 2003, 102, 4576–4581. [Google Scholar] [CrossRef]
- Wickramasinghe, S.N. Congenital dyserythropoietic anaemias: Clinical features, haematological morphology and new biochemical data. Blood Rev. 1998, 12, 178–200. [Google Scholar] [CrossRef]
- Heimpel, H.; Wilts, H.; Hirschmann, W.D.; Hofmann, W.K.; Siciliano, R.D.; Steinke, B.; Wechsler, J.G. Aplastic crisis as a complication of congenital dyserythropoietic anemia type II. Acta Haematol. 2007, 117, 115–118. [Google Scholar] [CrossRef]
- Heimpel, H.; Dührsen, U.; Hofbauer, P.; Rigamonti-Wermlinger, V.; Kreuser, E.D.; Schwarz, K.; Solenthaler, M.; Pauls, S. Bulky extramedullary hematopoiesis is not a rare complication of congenital dyserythropoietic anemia. Ann. Hematol. 2009, 88, 937–941. [Google Scholar] [CrossRef]
- Iolascon, A.; Esposito, M.R.; Russo, R. Clinical aspects and pathogenesis of congenital dyserythropoietic anemias: From morphology to molecular approach. Haematologica 2012, 97, 1786–1794. [Google Scholar] [CrossRef]
- Alloisio, N.; Texier, P.; Denoroy, L.; Berger, C.; Miraglia del Giudice, E.; Perrotta, S.; Iolascon, A.; Gilsanz, F.; Berger, G.; Guichard, J. The cisternae decorating the red blood cell membrane in congenital dyserythropoietic anemia (type II) originate from the endoplasmic reticulum. Blood 1996, 87, 4433–4439. [Google Scholar] [CrossRef]
- Heimpel, H.; Kellermann, K.; Neuschwander, N.; Högel, J.; Schwarz, K. The morphological diagnosis of congenital dyserythropoietic anemia: Results of a quantitative analysis of peripheral blood and bone marrow cells. Haematologica 2010, 95, 1034–1036. [Google Scholar] [CrossRef]
- Zdebska, E.; Gołaszewska, E.; Fabijańska-Mitek, J.; Schachter, H.; Shalev, H.; Tamary, H.; Sandström, H.; Wahlin, A.; Kościelak, J. Glycoconjugate abnormalities in patients with congenital dyserythropoietic anaemia type I, II and III. Br. J. Haematol. 2001, 114, 907–913. [Google Scholar] [CrossRef]
- Fukuda, M.N. HEMPAS. Hereditary erythroblastic multinuclearity with positive acidified serum lysis test. Biochim. Biophys. Acta 1999, 1455, 231–239. [Google Scholar] [CrossRef]
- Anselstetter, V.; Horstmann, H.J.; Heimpel, H. Congenital dyserythropoietic anaemia, types I and II: Aberrant pattern of erythrocyte membrane proteins in CDA II, as revealed by two-dimensional polyacrylamide gel electrophoresis. Br. J. Haematol. 1977, 35, 209–215. [Google Scholar] [CrossRef]
- Scartezzini, P.; Forni, G.L.; Baldi, M.; Izzo, C.; Sansone, G. Decreased glycosylation of band 3 and band 4.5 glycoproteins of erythrocyte membrane in congenital dyserythropoietic anaemia type II. Br. J. Haematol. 1982, 51, 569–576. [Google Scholar] [CrossRef]
- Russo, R.; Gambale, A.; Langella, C.; Andolfo, I.; Unal, S.; Iolascon, A. Retrospective cohort study of 205 cases with congenital dyserythropoietic anemia type II: Definition of clinical and molecular spectrum and identification of new diagnostic scores. Am. J. Hematol. 2014, 89, E169–E175. [Google Scholar] [CrossRef]
- Gasparini, P.; Miraglia del Giudice, E.; Delaunay, J.; Totaro, A.; Granatiero, M.; Melchionda, S.; Zelante, L.; Iolascon, A. Localization of the congenital dyserythropoietic anemia II locus to chromosome 20q11.2 by genomewide search. Am. J. Hum. Genet. 1997, 61, 1112–1116. [Google Scholar] [CrossRef]
- Bianchi, P.; Fermo, E.; Vercellati, C.; Boschetti, C.; Barcellini, W.; Iurlo, A.; Marcello, A.P.; Righetti, P.G.; Zanella, A. Congenital dyserythropoietic anemia type II (CDAII) is caused by mutations in the SEC23B gene. Hum. Mutat. 2009, 30, 1292–1298. [Google Scholar] [CrossRef]
- Schwarz, K.; Iolascon, A.; Verissimo, F.; Trede, N.; Horsley, W.; Chen, W.; Paw, B.H.; Hopfner, K.P.; Holzmann, K.; Russo, R.; et al. Mutations affecting the secretory COPII coat component SEC23B cause congenital dyserythropoietic anemia type II. Nat. Genet. 2009, 41, 936–940. [Google Scholar] [CrossRef]
- Iolascon, A.; Delaunay, J. Close to unraveling the secrets of congenital dyserythropoietic anemia types I and II. Haematologica 2009, 94, 599–602. [Google Scholar] [CrossRef]
- Russo, R.; Esposito, M.R.; Asci, R.; Gambale, A.; Perrotta, S.; Ramenghi, U.; Forni, G.L.; Uygun, V.; Delaunay, J.; Iolascon, A. Mutational spectrum in congenital dyserythropoietic anemia type II: Identification of 19 novel variants in SEC23B gene. Am. J. Hematol. 2010, 85, 915–920. [Google Scholar] [CrossRef]
- Rosato, B.E.; Marra, R.; D’Onofrio, V.; Del Giudice, F.; Della Monica, S.; Iolascon, A.; Andolfo, I.; Russo, R. SEC23B Loss-of-Function Suppresses Hepcidin Expression by Impairing Glycosylation Pathway in Human Hepatic Cells. Int. J. Mol. Sci. 2022, 23, 1304. [Google Scholar] [CrossRef] [PubMed]
- Iolascon, A.; Andolfo, I.; Barcellini, W.; Corcione, F.; Garçon, L.; De Franceschi, L.; Pignata, C.; Graziadei, G.; Pospisilova, D.; Rees, D.C.; et al. Recommendations regarding splenectomy in hereditary hemolytic anemias. Haematologica 2017, 102, 1304–1313. [Google Scholar] [CrossRef] [PubMed]
- Shalev, H.; Al-Athamen, K.; Levi, I.; Levitas, A.; Tamary, H. Morbidity and mortality of adult patients with congenital dyserythropoietic anemia type I. Eur. J. Haematol. 2017, 98, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Remacha, A.F.; Badell, I.; Pujol-Moix, N.; Parra, J.; Muñiz-Diaz, E.; Ginovart, G.; Sardà, M.P.; Hernández, A.; Moliner, E.; Torrent, M. Hydrops fetalis-associated congenital dyserythropoietic anemia treated with intrauterine transfusions and bone marrow transplantation. Blood 2002, 100, 356–358. [Google Scholar] [CrossRef]
- Unal, S.; Russo, R.; Gumruk, F.; Kuskonmaz, B.; Cetin, M.; Sayli, T.; Tavil, B.; Langella, C.; Iolascon, A.; Uckan Cetinkaya, D. Successful hematopoietic stem cell transplantation in a patient with congenital dyserythropoietic anemia type II. Pediatr. Transplant. 2014, 18, E130–E133. [Google Scholar] [CrossRef] [PubMed]
- Russo, R.; Langella, C.; Esposito, M.R.; Gambale, A.; Vitiello, F.; Vallefuoco, F.; Ek, T.; Yang, E.; Iolascon, A. Hypomorphic mutations of SEC23B gene account for mild phenotypes of congenital dyserythropoietic anemia type II. Blood Cells Mol. Dis. 2013, 51, 17–21. [Google Scholar] [CrossRef]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef]
- Bianchi, P.; Schwarz, K.; Högel, H.; Fermo, E.; Vercellati, C.; Grosse, R.; van Wijk, R.; van Zwieten, R.; Barcellini, W.; Zanella, A.; et al. Analysis of a cohort of 101 CDAII patients: Description of 24 new molecular variants and genotype-phenotype correlations. Br. J. Haematol. 2016, 175, 696–704. [Google Scholar] [CrossRef]
- Gambale, A.; Iolascon, A.; Andolfo, I.; Russo, R. Diagnosis and management of congenital dyserythropoietic anemias. Expert Rev. Hematol. 2016, 9, 283–296. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Iolascon, A.; Russo, R.; Esposito, M.R.; Asci, R.; Piscopo, C.; Perrotta, S.; Feneant-Thibault, M.; Garcon, L.; Delaunay, J. Molecular analysis of 42 patients with congenital dyserythropoietic anemia type II: New mutations in the SEC23B gene and a search for a genotype-phenotype relationship. Haematologica 2010, 95, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Artimo, P.; Jonnalagedda, M.; Arnold, K.; Baratin, D.; Csardi, G.; de Castro, E.; Duvaud, S.; Flegel, V.; Fortier, A.; Gasteiger, E.; et al. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012, 40, W597–W603. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Khoriaty, R.; Li, L.; McClune, M.; Kalfa, T.A.; Wu, J.; Peltier, D.; Fujiwara, H.; Sun, Y.; Oravecz-Wilson, K.; et al. ER-to-Golgi transport and SEC23-dependent COPII vesicles regulate T cell alloimmunity. J. Clin. Investig. 2021, 131, 136574. [Google Scholar] [CrossRef] [PubMed]
- Iolascon, A.; Heimpel, H.; Wahlin, A.; Tamary, H. Congenital dyserythropoietic anemias: Molecular insights and diagnostic approach. Blood 2013, 122, 2162–2166. [Google Scholar] [CrossRef]
- Tao, J.; Zhu, M.; Wang, H.; Afelik, S.; Vasievich, M.P.; Chen, X.W.; Zhu, G.; Jensen, J.; Ginsburg, D.; Zhang, B. SEC23B is required for the maintenance of murine professional secretory tissues. Proc. Natl. Acad. Sci. USA 2012, 109, E2001–E2009. [Google Scholar] [CrossRef]
- Zhu, M.; Tao, J.; Vasievich, M.P.; Wei, W.; Zhu, G.; Khoriaty, R.N.; Zhang, B. Neural tube opening and abnormal extraembryonic membrane development in SEC23A deficient mice. Sci. Rep. 2015, 5, 15471. [Google Scholar] [CrossRef]
- Boyadjiev, S.A.; Fromme, J.C.; Ben, J.; Chong, S.S.; Nauta, C.; Hur, D.J.; Zhang, G.; Hamamoto, S.; Schekman, R.; Ravazzola, M.; et al. Cranio-lenticulo-sutural dysplasia is caused by a SEC23A mutation leading to abnormal endoplasmic-reticulum-to-Golgi trafficking. Nat. Genet. 2006, 38, 1192–1197. [Google Scholar] [CrossRef]
- Khoriaty, R.; Hesketh, G.G.; Bernard, A.; Weyand, A.C.; Mellacheruvu, D.; Zhu, G.; Hoenerhoff, M.J.; McGee, B.; Everett, L.; Adams, E.J.; et al. Functions of the COPII gene paralogs SEC23A and SEC23B are interchangeable in vivo. Proc. Natl. Acad. Sci. USA 2018, 115, E7748–E7757. [Google Scholar] [CrossRef]
- Roy, N.B.A.; Da Costa, L.; Russo, R.; Bianchi, P.; Del Mar Mañú-Pereira, M.; Fermo, E.; Andolfo, I.; Clark, B.; Proven, M.; Sanchez, M.; et al. The Use of Next-generation Sequencing in the Diagnosis of Rare Inherited Anaemias: A Joint BSH/EHA Good Practice Paper. Hemasphere 2022, 6, e739. [Google Scholar] [CrossRef]
- Fermo, E.; Vercellati, C.; Marcello, A.P.; Keskin, E.Y.; Perrotta, S.; Zaninoni, A.; Brancaleoni, V.; Zanella, A.; Giannotta, J.A.; Barcellini, W.; et al. Targeted Next Generation Sequencing and Diagnosis of Congenital Hemolytic Anemias: A Three Years Experience Monocentric Study. Front. Physiol. 2021, 12, 684569. [Google Scholar] [CrossRef]
- Shefer Averbuch, N.; Steinberg-Shemer, O.; Dgany, O.; Krasnov, T.; Noy-Lotan, S.; Yacobovich, J.; Kuperman, A.A.; Kattamis, A.; Ben Barak, A.; Roth-Jelinek, B.; et al. Targeted next generation sequencing for the diagnosis of patients with rare congenital anemias. Eur. J. Haematol. 2018, 101, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, P.; Kiladjian, J.J.; Platzbecker, U. Luspatercept for the treatment of anemia in myelodysplastic syndromes and primary myelofibrosis. Blood 2019, 133, 790–794. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, G.; Andolfo, I.; Marra, R.; Manna, F.; Rosato, B.E.; Iolascon, A.; Russo, R. RAP-011 Rescues the Disease Phenotype in a Cellular Model of Congenital Dyserythropoietic Anemia Type II by Inhibiting the SMAD2-3 Pathway. Int. J. Mol. Sci. 2020, 21, 5577. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gomez, M.; Calabria, A.; Garcia-Bravo, M.; Benedicenti, F.; Kosinski, P.; López-Manzaneda, S.; Hill, C.; Del Mar Mañu-Pereira, M.; Martín, M.A.; Orman, I.; et al. Safe and Efficient Gene Therapy for Pyruvate Kinase Deficiency. Mol. Ther. 2016, 24, 1187–1198. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, F.; Thompson, A.A.; Kwiatkowski, J.L.; Porter, J.B.; Thrasher, A.J.; Hongeng, S.; Sauer, M.G.; Thuret, I.; Lal, A.; Algeri, M.; et al. Betibeglogene Autotemcel Gene Therapy for Non-β. N. Engl. J. Med. 2022, 386, 415–427. [Google Scholar] [CrossRef]
- Dessy-Rodriguez, M.; Fañanas-Baquero, S.; Venturi, V.; Payan, S.; Tornador, C.; Hernández, G.; Bianchi, P.; Sanchez, M.; Ramirez, J.C.; Segovia, J.C.; et al. Lentiviral Gene Therapy for the Correction of Congenital Dyserythropoietic Anemia Type II. Blood 2021, 138 (Suppl. S1), 1994. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Prob. | S | Age (Years) | Hb (g/L) | MCV (fL) | RBC (106/μL) | Total Bilirubin (mg/dL) | Dir. Bilirubin (mg/dL) | LDH (UI/dL) | Haptoglobin (mg/dL) | Serum Iron (μg/dL) | Serum Ft (μg/L) | Fetal Hb | Nucleotide Change/Protein |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F1-II.1 | F | (a) 15 | (a) N.A. | (a) N/A | (a) N/A | (a) N/A | (a) N/A | (a) N/A | (a) N/A | (a) N/A | (a) N/A | N/A | c.325G>A(p.Glu109Lys); c.1334C>G(p.Thr445Arg) |
| (b) 25 | (b) 90 | (b) 96.1 | (b) 3.06 | (b) 3.16 | (b) 0.62 | (b) 242 | (b) <0.3 | (b) 256 | (b) 647 | N/A | |||
| F2-II.1 | M | (a) 0 | (a) 85 | (a) 85 | (a) N/A | (a) N/A | (a) 0.46 | (a) 431.2 | (a) N/A | (a) 64 | (a) 525 | (d) 1% | c.1736A>G(p.Tyr579Cys); c.835-2A>G |
| (b)12 | (b) N/A | (b) N/A | (b) 3.11 | (b) 4.26 | (b) N/A | (b) N/A | (b) N/A | (b) N/A | (b) N/A | N/A | |||
| F3-II.1 | M | (a) 32 | (a) 130 | (a) 91 | (a) 4.29 | (a) 3.03 | (a) 0.51 | (a) 202 | (a) <6 | (a) 156 | (a) 932 | N/A | c.325G>A(p.Glu109Lys); c.1512-3delinsTT + c.1512-16_1512-7delACTCTGGAAT |
| (b) 42 | (b) 127 | (b) 90 | (b) 4.15 | (b) 2.656 | (b) N/A | (b) 1.04 | (b) <1 | (b) 144 | (c) 297 | N/A | |||
| F4-II.1 | F | (a) 25 | (a) 82 | (a) 90 | (a) 3.50 | (a) 2.92 | (a) 0.87 | (a) N/A | (a) 0.00 | (a) 156.36 | (a) | N/A | c.40C>T(p.Arg14Trp); c.1968T>G(p.Phe656Leu) |
| (b) 60 | (b) 122 | (b) 90.1 | (b) 3.85 | (b) 1.07 | (b) N/A | (b) N/A | (b) N/A | (b) 183.17 | (b) 51 | N/A | |||
| F5-II.1 | M | (a) 21 | (a) N/A | (a) N/A | (a) N/A | (a) N/A | (a) N/A | (a) N/A | (a) N/A | (a) N/A | (a) N/A | N/A | c.1603C>T(p.Arg535Ter) |
| (b) 52 | (b) 127 | (b) 90.6 | (b) 4.20 | (b) 5.9 | (b) 1.2 | (b) 275 | (b) <8 | (b) 158 | (b) 40 | N/A | |||
| F6-II.1 | F | (a) 21 | (a) 110 | (a)103 | (a) 3.46 | (a) 3.34 | (a) 0.67 | (a) 129 | (a) 23 | (a) 217 | (a) 242 | N/A | c.325G>A(p.Glu109Lys); c.325G>A(p.Glu109Lys) |
| (b) 30 | (b) 113 | (b) 104 | (b) 3.45 | (b) 3.18 | (b) 1.12 | (b) 151 | (b) N/A | (b) 196 | (b) 153 | N/A | |||
| F7-II.1 | M | (a) 10 | (a) 833 | (a) 91 | (a) 2.87 | (a) 2.4 | (a) 1.3 | (a) 390 | (a) 38 | (a) 105 | (a) 178 | N/A | c.2074_2077dupGATG(p.Asp693GlyfsTer2) |
| (b) 53 | (b) 105 | (b) 98.5 | (b) 3.34 | (b) 3.9 | (b) 0.9 | (b) 176 | (b) N/A | (b) 227 | (b) 469 | N/A | |||
| F8-II.1 | F | (a) 8 | (a) 95 | (a) 95 | (a) 2.92 | (a) 4.99 | (a) 0.59 | (a) < 500 | (a) 10 | (a) 67 | (a) 74 | (a) 0.5% | c.325G>A(p.Glu109Lys); c.2102G>A (p.Arg701His) |
| (b) 24 | (b) 78 | (b) 106 | (b) 2.13 | (b) 4.82 | (b) 0.45 | (b)167 | (b) | (b) 69 | (b) 659 | N/A | |||
| F9-II.1 | M | (a) 9 | (a) 106 | (a) 83 | (a) 3.45 | (a) 2.0 | (a) 0.5 | (a) 430 | (a) 15.2 | (a) 241 | (a) 37.4 | (a) 1.3 | c.40C>T(p.Arg14Trp); c.1512-2A>G |
| (b) 24 | (b) 99 | (b) 93.8 | (b) 3.29 | (b) 4.3 | (b) 0.4 | (b) 167 | (b) 56 | (b) 228 | (b) 142 | (b) N/A |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Musri, M.M.; Venturi, V.; Ferrer-Cortès, X.; Romero-Cortadellas, L.; Hernández, G.; Leoz, P.; Ricard Andrés, M.P.; Morado, M.; Fernández Valle, M.d.C.; Beneitez Pastor, D.; et al. New Cases and Mutations in SEC23B Gene Causing Congenital Dyserythropoietic Anemia Type II. Int. J. Mol. Sci. 2023, 24, 9935. https://doi.org/10.3390/ijms24129935
Musri MM, Venturi V, Ferrer-Cortès X, Romero-Cortadellas L, Hernández G, Leoz P, Ricard Andrés MP, Morado M, Fernández Valle MdC, Beneitez Pastor D, et al. New Cases and Mutations in SEC23B Gene Causing Congenital Dyserythropoietic Anemia Type II. International Journal of Molecular Sciences. 2023; 24(12):9935. https://doi.org/10.3390/ijms24129935
Chicago/Turabian StyleMusri, Melina Mara, Veronica Venturi, Xènia Ferrer-Cortès, Lídia Romero-Cortadellas, Gonzalo Hernández, Pilar Leoz, María Pilar Ricard Andrés, Marta Morado, María del Carmen Fernández Valle, David Beneitez Pastor, and et al. 2023. "New Cases and Mutations in SEC23B Gene Causing Congenital Dyserythropoietic Anemia Type II" International Journal of Molecular Sciences 24, no. 12: 9935. https://doi.org/10.3390/ijms24129935
APA StyleMusri, M. M., Venturi, V., Ferrer-Cortès, X., Romero-Cortadellas, L., Hernández, G., Leoz, P., Ricard Andrés, M. P., Morado, M., Fernández Valle, M. d. C., Beneitez Pastor, D., Ortuño Cabrero, A., Moreno Gamiz, M., Senent Peris, L., Perez-Valencia, A. I., Pérez-Montero, S., Tornador, C., & Sánchez, M. (2023). New Cases and Mutations in SEC23B Gene Causing Congenital Dyserythropoietic Anemia Type II. International Journal of Molecular Sciences, 24(12), 9935. https://doi.org/10.3390/ijms24129935