
Alzheimer’s Disease: Significant Benefit from the Yeast-Based Models

{kind=link}
{kind=link}
Abstract
1. Introduction
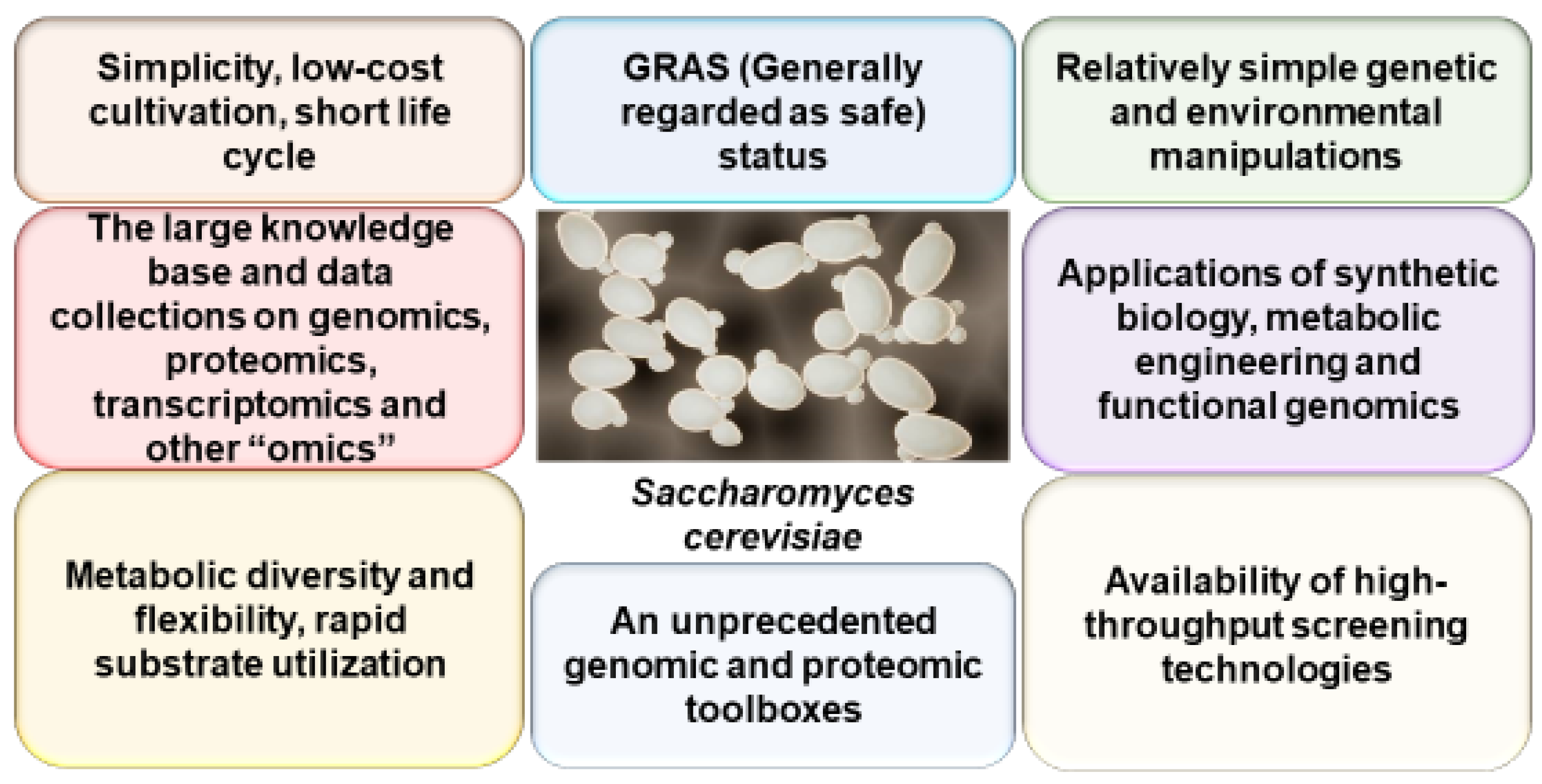
2. Yeast as a Model of AD
2.1. Yeast as a Model of Aβ Generation, Aggregation, Toxicity, and De-Aggregation
2.1.1. Yeast as a Model of APP Processing
2.1.2. Yeast as a Model of Aβ Aggregation
2.1.3. Yeast as a Model for Toxicity of Aβ Aggregates
2.1.4. Yeast as a Model of Aβ De-Aggregation
2.2. Yeast Models of Tau-Protein Aggregation and Toxicity
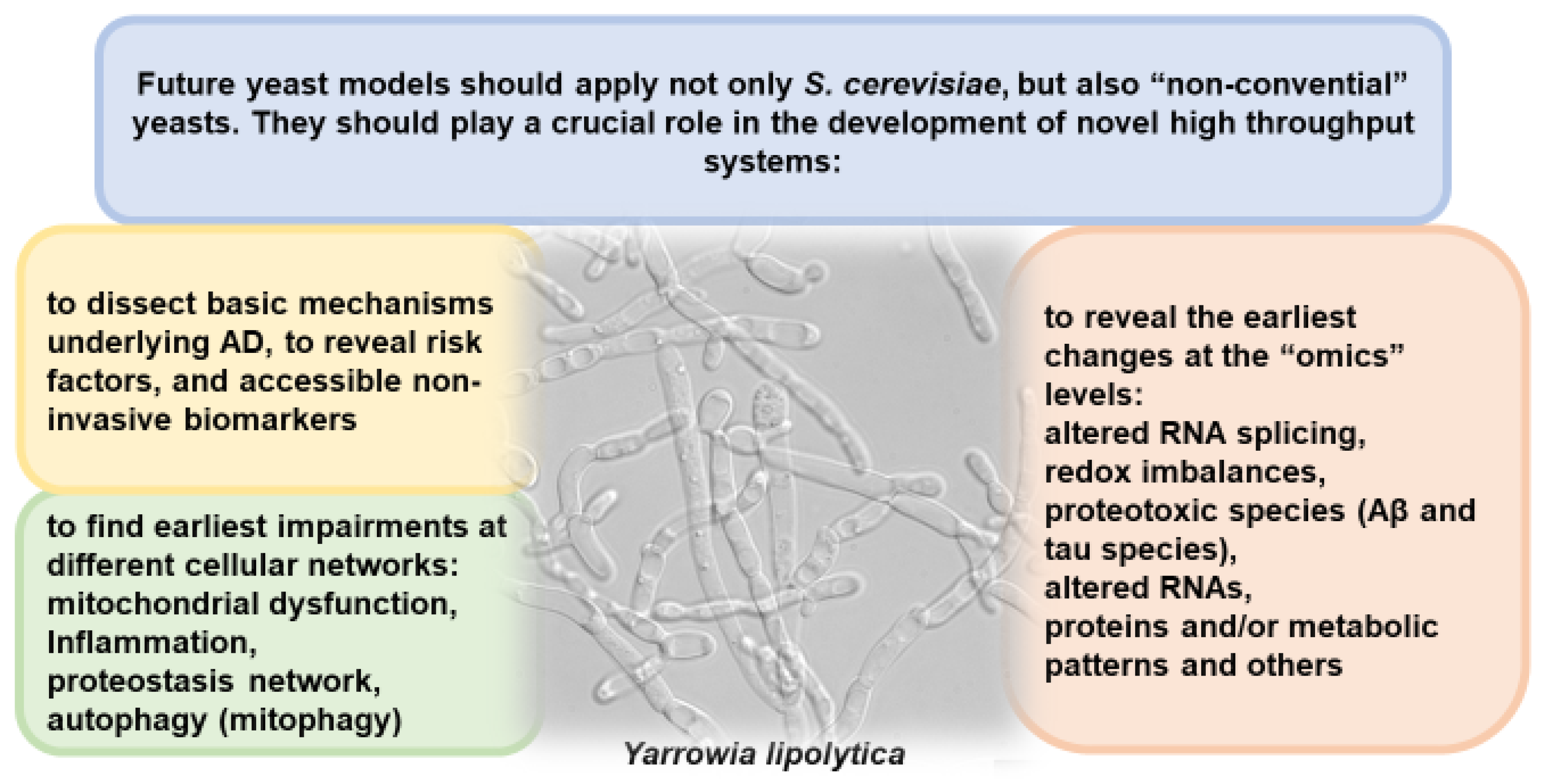
3. Concluding Remarks: The Need to Use Yeast for More In-Depth Study of AD
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alzheimer, A.; Stelzmann, R.A.; Schnitzlein, H.N.; Murtagh, F.R. An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin. Anat. 1995, 8, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Brion, J.P.; Couck, A.M.; Passareiro, E.; Flament-Durand, J. Neurofibrillary tangles of Alzheimer’s disease: An immunohistochemical study. J. Submicrosc. Cytol. 1985, 17, 89–96. [Google Scholar] [PubMed]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Pollock, N.J.; Mirra, S.S.; Binder, L.I.; Hansen, L.A.; Wood, J.G. Filamentous aggregates in Pick’s disease, progressive supranuclear palsy, and Alzheimer’s disease share antigenic determinants with microtubule-associated protein, tau. Lancet 1986, 2, 1211. [Google Scholar] [CrossRef] [PubMed]
- Pritam, P.; Deka, R.; Bhardwaj, A.; Srivastava, R.; Kumar, D.; Jha, A.K.; Jha, N.K.; Villa, C.; Jha, S.K. Antioxidants in Alzheimer’s Disease: Current Therapeutic Significance and Future Prospects. Biology 2022, 11, 212. [Google Scholar] [CrossRef] [PubMed]
- Turkez, H.; Arslan, M.E.; Barboza, J.N.; Kahraman, C.Y.; de Sousa, D.P.; Mardinoğlu, A. Therapeutic Potential of Ferulic Acid in Alzheimer’s Disease. Curr. Drug Deliv. 2022, 19, 860–873. [Google Scholar] [CrossRef]
- Mecca, A.P.; O’Dell, R.S.; Sharp, E.S.; Banks, E.R.; Bartlett, H.H.; Zhao, W.; Lipior, S.; Diepenbrock, N.G.; Chen, M.K.; Naganawa, M.; et al. Synaptic density and cognitive performance in Alzheimer’s disease: A PET imaging study with [(11)C]UCB-J. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2022, 18, 2527–2536. [Google Scholar] [CrossRef]
- Acosta-Baena, N.; Sepulveda-Falla, D.; Lopera-Gómez, C.M.; Jaramillo-Elorza, M.C.; Moreno, S.; Aguirre-Acevedo, D.C.; Saldarriaga, A.; Lopera, F. Pre-dementia clinical stages in presenilin 1 E280A familial early-onset Alzheimer’s disease: A retrospective cohort study. Lancet. Neurol. 2011, 10, 213–220. [Google Scholar] [CrossRef]
- Fernandez, M.A.; Klutkowski, J.A.; Freret, T.; Wolfe, M.S. Alzheimer presenilin-1 mutations dramatically reduce trimming of long amyloid β-peptides (Aβ) by γ-secretase to increase 42-to-40-residue Aβ. J. Biol. Chem. 2014, 289, 31043–31052. [Google Scholar] [CrossRef]
- Fan, L.; Mao, C.; Hu, X.; Zhang, S.; Yang, Z.; Hu, Z.; Sun, H.; Fan, Y.; Dong, Y.; Yang, J.; et al. New Insights into the Pathogenesis of Alzheimer’s Disease. Front. Neurol. 2019, 10, 1312. [Google Scholar] [CrossRef]
- Kaur, D.; Behl, T.; Sehgal, A.; Singh, S.; Sharma, N.; Chigurupati, S.; Alhowail, A.; Abdeen, A.; Ibrahim, S.F.; Vargas-De-La-Cruz, C.; et al. Decrypting the potential role of α-lipoic acid in Alzheimer’s disease. Life Sci. 2021, 284, 119899. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, F. Alzheimer Disease: Controversies in Basic Science Research, Different Theories, and Reasons for Failed Trials. Biomedicines 2021, 9, 254. [Google Scholar] [CrossRef] [PubMed]
- Seynnaeve, D.; Vecchio, M.D.; Fruhmann, G.; Verelst, J.; Cools, M.; Beckers, J.; Mulvihill, D.P.; Winderickx, J.; Franssens, V. Recent Insights on Alzheimer’s Disease Originating from Yeast Models. Int. J. Mol. Sci. 2018, 19, 1947. [Google Scholar] [CrossRef] [PubMed]
- Rajasekhar, K.; Suresh, S.N.; Manjithaya, R.; Govindaraju, T. Rationally designed peptidomimetic modulators of aβ toxicity in Alzheimer’s disease. Sci. Rep. 2015, 5, 8139. [Google Scholar] [CrossRef]
- Kozin, S.A.; Cheglakov, I.B.; Ovsepyan, A.A.; Telegin, G.B.; Tsvetkov, P.O.; Lisitsa, A.V.; Makarov, A.A. Peripherally applied synthetic peptide isoAsp7-Aβ(1-42) triggers cerebral β-amyloidosis. Neurotox. Res. 2013, 24, 370–376. [Google Scholar] [CrossRef]
- Wang, D.; Huang, X.; Yan, L.; Zhou, L.; Yan, C.; Wu, J.; Su, Z.; Huang, Y. The Structure Biology of Tau and Clue for Aggregation Inhibitor Design. Protein J. 2021, 40, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Gabandé-Rodríguez, E.; Keane, L.; Capasso, M. Microglial phagocytosis in aging and Alzheimer’s disease. J. Neurosci. Res. 2020, 98, 284–298. [Google Scholar] [CrossRef]
- Agarwal, M.; Khan, S. Plasma Lipids as Biomarkers for Alzheimer’s Disease: A Systematic Review. Cureus 2020, 12, e12008. [Google Scholar] [CrossRef]
- Montagne, A.; Nation, D.A.; Sagare, A.P.; Barisano, G.; Sweeney, M.D.; Chakhoyan, A.; Pachicano, M.; Joe, E.; Nelson, A.R.; D’Orazio, L.M.; et al. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature 2020, 581, 71–76. [Google Scholar] [CrossRef]
- Narayan, P.; Sienski, G.; Bonner, J.M.; Lin, Y.T.; Seo, J.; Baru, V.; Haque, A.; Milo, B.; Akay, L.A.; Graziosi, A.; et al. PICALM Rescues Endocytic Defects Caused by the Alzheimer’s Disease Risk Factor APOE4. Cell Rep. 2020, 33, 108224. [Google Scholar] [CrossRef]
- Zhang, Q.; Sidorenko, J.; Couvy-Duchesne, B.; Marioni, R.E.; Wright, M.J.; Goate, A.M.; Marcora, E.; Huang, K.L.; Porter, T.; Laws, S.M.; et al. Risk prediction of late-onset Alzheimer’s disease implies an oligogenic architecture. Nat. Commun. 2020, 11, 4799. [Google Scholar] [CrossRef]
- Heath, L.; Earls, J.C.; Magis, A.T.; Kornilov, S.A.; Lovejoy, J.C.; Funk, C.C.; Rappaport, N.; Logsdon, B.A.; Mangravite, L.M.; Kunkle, B.W.; et al. Manifestations of Alzheimer’s disease genetic risk in the blood are evident in a multiomic analysis in healthy adults aged 18 to 90. Sci. Rep. 2022, 12, 6117. [Google Scholar] [CrossRef] [PubMed]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef]
- Rosenberg, A.; Mangialasche, F.; Ngandu, T.; Solomon, A.; Kivipelto, M. Multidomain Interventions to Prevent Cognitive Impairment, Alzheimer’s Disease, and Dementia: From FINGER to World-Wide FINGERS. J. Prev. Alzheimer’s Dis. 2020, 7, 29–36. [Google Scholar] [CrossRef]
- Kao, Y.C.; Ho, P.C.; Tu, Y.K.; Jou, I.M.; Tsai, K.J. Lipids and Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 1505. [Google Scholar] [CrossRef]
- Cohen, A.D.; Landau, S.M.; Snitz, B.E.; Klunk, W.E.; Blennow, K.; Zetterberg, H. Fluid and PET biomarkers for amyloid pathology in Alzheimer’s disease. Mol. Cell. Neurosci. 2019, 97, 3–17. [Google Scholar] [CrossRef]
- Ramesh, M.; Gopinath, P.; Govindaraju, T. Role of Post-translational Modifications in Alzheimer’s Disease. Chembiochem A Eur. J. Chem. Biol. 2020, 21, 1052–1079. [Google Scholar] [CrossRef]
- Zhang, L.; Young, J.I.; Gomez, L.; Silva, T.C.; Schmidt, M.A.; Cai, J.; Chen, X.; Martin, E.R.; Wang, L. Sex-specific DNA methylation differences in Alzheimer’s disease pathology. Acta Neuropathol. Commun. 2021, 9, 77. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Ekundayo, T.C.; Olasehinde, T.A.; Okaiyeto, K.; Okoh, A.I. Microbial Pathogenesis and Pathophysiology of Alzheimer’s Disease: A Systematic Assessment of Microorganisms’ Implications in the Neurodegenerative Disease. Front. Neurosci. 2021, 15, 648484. [Google Scholar] [CrossRef] [PubMed]
- Caruso, A.; Nicoletti, F.; Gaetano, A.; Scaccianoce, S. Risk Factors for Alzheimer’s Disease: Focus on Stress. Front. Pharmacol. 2019, 10, 976. [Google Scholar] [CrossRef] [PubMed]
- Hensley, K.; Carney, J.M.; Mattson, M.P.; Aksenova, M.; Harris, M.; Wu, J.F.; Floyd, R.A.; Butterfield, D.A. A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: Relevance to Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 3270–3274. [Google Scholar] [CrossRef]
- Stoddard-Bennett, T.; Pera, R.R. Stem cell therapy for Parkinson’s disease: Safety and modeling. Neural Regen. Res. 2020, 15, 36–40. [Google Scholar] [CrossRef]
- Tolar, M.; Abushakra, S.; Sabbagh, M. The path forward in Alzheimer’s disease therapeutics: Reevaluating the amyloid cascade hypothesis. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2020, 16, 1553–1560. [Google Scholar] [CrossRef]
- Janowicz, P.W.; Leinenga, G.; Götz, J.; Nisbet, R.M. Ultrasound-mediated blood-brain barrier opening enhances delivery of therapeutically relevant formats of a tau-specific antibody. Sci. Rep. 2019, 9, 9255. [Google Scholar] [CrossRef]
- Pradeepkiran, J.A.; Reddy, P.H. Defective mitophagy in Alzheimer’s disease. Ageing Res. Rev. 2020, 64, 101191. [Google Scholar] [CrossRef] [PubMed]
- Visioli, F.; Rodríguez-Pérez, M.; Gómez-Torres, Ó.; Pintado-Losa, C.; Burgos-Ramos, E. Hydroxytyrosol improves mitochondrial energetics of a cellular model of Alzheimer’s disease. Nutr. Neurosci. 2022, 25, 990–1000. [Google Scholar] [CrossRef]
- Masliah, E.; Terry, R.D.; Alford, M.; DeTeresa, R.; Hansen, L.A. Cortical and subcortical patterns of synaptophysinlike immunoreactivity in Alzheimer’s disease. Am. J. Pathol. 1991, 138, 235–246. [Google Scholar]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef]
- Mucke, L.; Masliah, E.; Yu, G.Q.; Mallory, M.; Rockenstein, E.M.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Johnson-Wood, K.; McConlogue, L. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J. Neurosci. Off. J. Soc. Neurosci. 2000, 20, 4050–4058. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Ta, Q.T.H.; Nguyen, T.K.O.; Nguyen, T.T.D.; Vo, V.G. Role of Body-Fluid Biomarkers in Alzheimer’s Disease Diagnosis. Diagnostics 2020, 10, 326. [Google Scholar] [CrossRef] [PubMed]
- Leuzy, A.; Mattsson-Carlgren, N.; Palmqvist, S.; Janelidze, S.; Dage, J.L.; Hansson, O. Blood-based biomarkers for Alzheimer’s disease. EMBO Mol. Med. 2022, 14, e14408. [Google Scholar] [CrossRef] [PubMed]
- Chrem Mendez, P.; Surace, E.; Bérgamo, Y.; Calandri, I.; Vázquez, S.; Sevlever, G.; Allegri, R.F. Biomarkers for Alzheimer’s disease. Where we stand and where we are headed. Medicina 2019, 79, 546–551. [Google Scholar]
- Dallé, E.; Mabandla, M.V.; Daniels, W.M.U. Dielectric Constant and Conductivity of Blood Plasma: Possible Novel Biomarkers for Alzheimer’s Disease. Oxidative Med. Cell. Longev. 2020, 2020, 5756382. [Google Scholar] [CrossRef] [PubMed]
- Castrillo, J.I.; Oliver, S.G. Alzheimer’s as a Systems-Level Disease Involving the Interplay of Multiple Cellular Networks. Methods Mol. Biol. 2016, 1303, 3–48. [Google Scholar] [CrossRef] [PubMed]
- Matlack, K.E.; Tardiff, D.F.; Narayan, P.; Hamamichi, S.; Caldwell, K.A.; Caldwell, G.A.; Lindquist, S. Clioquinol promotes the degradation of metal-dependent amyloid-β (Aβ) oligomers to restore endocytosis and ameliorate Aβ toxicity. Proc. Natl. Acad. Sci. USA 2014, 111, 4013–4018. [Google Scholar] [CrossRef] [PubMed]
- Goffeau, A.; Barrell, B.G.; Bussey, H.; Davis, R.W.; Dujon, B.; Feldmann, H.; Galibert, F.; Hoheisel, J.D.; Jacq, C.; Johnston, M.; et al. Life with 6000 genes. Science 1996, 274, 546–547. [Google Scholar] [CrossRef]
- Foury, F. Human genetic diseases: A cross-talk between man and yeast. Gene 1997, 195, 1–10. [Google Scholar] [CrossRef]
- Sherman, F. Getting started with yeast. Methods Enzymol. 2002, 350, 3–41. [Google Scholar] [CrossRef]
- Winzeler, E.A.; Shoemaker, D.D.; Astromoff, A.; Liang, H.; Anderson, K.; Andre, B.; Bangham, R.; Benito, R.; Boeke, J.D.; Bussey, H.; et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 1999, 285, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Rolfs, A.; Bhullar, B.; Murthy, T.V.; Zhu, C.; Berger, M.F.; Camargo, A.A.; Kelley, F.; McCarron, S.; Jepson, D.; et al. Approaching a complete repository of sequence-verified protein-encoding clones for Saccharomyces cerevisiae. Genome Res. 2007, 17, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.M.; Stalker, J.; Humphray, S.; West, A.; Cox, T.; Rogers, J.; Dunham, I.; Prelich, G. A systematic library for comprehensive overexpression screens in Saccharomyces cerevisiae. Nat. Methods 2008, 5, 239–241. [Google Scholar] [CrossRef] [PubMed]
- DeRisi, J.L.; Iyer, V.R.; Brown, P.O. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science 1997, 278, 680–686. [Google Scholar] [CrossRef]
- Hoon, S.; St Onge, R.P.; Giaever, G.; Nislow, C. Yeast chemical genomics and drug discovery: An update. Trends Pharmacol. Sci. 2008, 29, 499–504. [Google Scholar] [CrossRef]
- Parsons, A.B.; Lopez, A.; Givoni, I.E.; Williams, D.E.; Gray, C.A.; Porter, J.; Chua, G.; Sopko, R.; Brost, R.L.; Ho, C.H.; et al. Exploring the mode-of-action of bioactive compounds by chemical-genetic profiling in yeast. Cell 2006, 126, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.V.; Schmidt, K.H. Maintenance of Yeast Genome Integrity by RecQ Family DNA Helicases. Genes 2020, 11, 205. [Google Scholar] [CrossRef]
- Rezig, I.M.; Yaduma, W.G.; Gould, G.W.; McInerny, C.J. The role of anillin/Mid1p during medial division and cytokinesis: From fission yeast to cancer cells. Cell Cycle 2023, 22, 633–644. [Google Scholar] [CrossRef]
- Martins Pinto, M.; Paumard, P.; Bouchez, C.; Ransac, S.; Duvezin-Caubet, S.; Mazat, J.P.; Rigoulet, M.; Devin, A. The Warburg effect and mitochondrial oxidative phosphorylation: Friends or foes? Biochim. Biophys. Acta. Bioenerg. 2023, 1864, 148931. [Google Scholar] [CrossRef]
- Sayyed, U.M.H.; Mahalakshmi, R. Mitochondrial protein translocation machinery: From TOM structural biogenesis to functional regulation. J. Biol. Chem. 2022, 298, 101870. [Google Scholar] [CrossRef]
- Brodsky, J.L.; Skach, W.R. Protein folding and quality control in the endoplasmic reticulum: Recent lessons from yeast and mammalian cell systems. Curr. Opin. Cell Biol. 2011, 23, 464–475. [Google Scholar] [CrossRef] [PubMed]
- Dhakal, S.; Macreadie, I. Protein Homeostasis Networks and the Use of Yeast to Guide Interventions in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 8014. [Google Scholar] [CrossRef] [PubMed]
- Kahlhofer, J.; Leon, S.; Teis, D.; Schmidt, O. The α-arrestin family of ubiquitin ligase adaptors links metabolism with selective endocytosis. Biol. Cell 2021, 113, 183–219. [Google Scholar] [CrossRef] [PubMed]
- Kuchitsu, Y.; Mukai, K.; Uematsu, R.; Takaada, Y.; Shinojima, A.; Shindo, R.; Shoji, T.; Hamano, S.; Ogawa, E.; Sato, R.; et al. STING signalling is terminated through ESCRT-dependent microautophagy of vesicles originating from recycling endosomes. Nat. Cell Biol. 2023, 25, 453–466. [Google Scholar] [CrossRef]
- Bonifacino, J.S.; Glick, B.S. The mechanisms of vesicle budding and fusion. Cell 2004, 116, 153–166. [Google Scholar] [CrossRef]
- Kumar, R.; Reichert, A.S. Common Principles and Specific Mechanisms of Mitophagy from Yeast to Humans. Int. J. Mol. Sci. 2021, 22, 4363. [Google Scholar] [CrossRef]
- Manon, S. Yeast as a tool to decipher the molecular mechanisms underlying the functions of Bcl-2 family. Explor. Target. Anti-Tumor Ther. 2022, 3, 128–148. [Google Scholar] [CrossRef]
- Chen, R.E.; Thorner, J. Function and regulation in MAPK signaling pathways: Lessons learned from the yeast Saccharomyces cerevisiae. Biochim. Biophys. Acta 2007, 1773, 1311–1340. [Google Scholar] [CrossRef]
- De Virgilio, C.; Loewith, R. The TOR signalling network from yeast to man. Int. J. Biochem. Cell Biol. 2006, 38, 1476–1481. [Google Scholar] [CrossRef]
- Epremyan, K.K.; Rogov, A.G.; Goleva, T.N.; Lavrushkina, S.V.; Zinovkin, R.A.; Zvyagilskaya, R.A. Altered Mitochondrial Morphology and Bioenergetics in a New Yeast Model Expressing Aβ42. Int. J. Mol. Sci. 2023, 24, 900. [Google Scholar] [CrossRef]
- Fruhmann, G.; Seynnaeve, D.; Zheng, J.; Ven, K.; Molenberghs, S.; Wilms, T.; Liu, B.; Winderickx, J.; Franssens, V. Yeast buddies helping to unravel the complexity of neurodegenerative disorders. Mech. Ageing Dev. 2017, 161, 288–305. [Google Scholar] [CrossRef] [PubMed]
- Mamaev, D.; Zvyagilskaya, R. Yarrowia lipolytica: A multitalented yeast species of ecological significance. FEMS Yeast Res. 2021, 21, foab008. [Google Scholar] [CrossRef]
- Montoliu-Gaya, L.; Esquerda-Canals, G.; Bronsoms, S.; Villegas, S. Production of an anti-Aβ antibody fragment in Pichia pastoris and in vitro and in vivo validation of its therapeutic effect. PLoS ONE 2017, 12, e0181480. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Kachroo, A.H.; Yellman, C.M.; Marcotte, E.M.; Johnson, K.A. Yeast cells expressing the human mitochondrial DNA polymerase reveal correlations between polymerase fidelity and human disease progression. J. Biol. Chem. 2014, 289, 5970–5985. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Petranovic, D. Amyloid-β peptide-induced cytotoxicity and mitochondrial dysfunction in yeast. FEMS Yeast Res. 2015, 15, fov061. [Google Scholar] [CrossRef]
- Heinisch, J.J.; Brandt, R. Signaling pathways and posttranslational modifications of tau in Alzheimer’s disease: The humanization of yeast cells. Microb. Cell 2016, 3, 135–146. [Google Scholar] [CrossRef]
- Rencus-Lazar, S.; DeRowe, Y.; Adsi, H.; Gazit, E.; Laor, D. Yeast Models for the Study of Amyloid-Associated Disorders and Development of Future Therapy. Front. Mol. Biosci. 2019, 6, 15. [Google Scholar] [CrossRef]
- Verduyckt, M.; Vignaud, H.; Bynens, T.; Van den Brande, J.; Franssens, V.; Cullin, C.; Winderickx, J. Yeast as a Model for Alzheimer’s Disease: Latest Studies and Advanced Strategies. Methods Mol. Biol. 2016, 1303, 197–215. [Google Scholar] [CrossRef]
- Andersson, M.J.; Stone, J. Best Medicine for Dementia: The Life-Long Defense of the Brain. J. Alzheimer’s Dis. JAD 2023. [Google Scholar] [CrossRef]
- De Strooper, B.; Annaert, W. Proteolytic processing and cell biological functions of the amyloid precursor protein. J. Cell Sci. 2000, 113 Pt 11, 1857–1870. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Zhang, Y. Animal models of Alzheimer’s disease: Applications, evaluation, and perspectives. Zool. Res. 2022, 43, 1026–1040. [Google Scholar] [CrossRef] [PubMed]
- Futai, E.; Osawa, S.; Cai, T.; Fujisawa, T.; Ishiura, S.; Tomita, T. Suppressor Mutations for Presenilin 1 Familial Alzheimer Disease Mutants Modulate γ-Secretase Activities. J. Biol. Chem. 2016, 291, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Joshi, G.; Wang, Y. Golgi defects enhance APP amyloidogenic processing in Alzheimer’s disease. BioEssays News Rev. Mol. Cell. Dev. Biol. 2015, 37, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Olsson, F.; Schmidt, S.; Althoff, V.; Munter, L.M.; Jin, S.; Rosqvist, S.; Lendahl, U.; Multhaup, G.; Lundkvist, J. Characterization of intermediate steps in amyloid beta (Aβ) production under near-native conditions. J. Biol. Chem. 2014, 289, 1540–1550. [Google Scholar] [CrossRef]
- Furukawa, K.; Sopher, B.L.; Rydel, R.E.; Begley, J.G.; Pham, D.G.; Martin, G.M.; Fox, M.; Mattson, M.P. Increased activity-regulating and neuroprotective efficacy of alpha-secretase-derived secreted amyloid precursor protein conferred by a C-terminal heparin-binding domain. J. Neurochem. 1996, 67, 1882–1896. [Google Scholar] [CrossRef] [PubMed]
- Takasugi, N.; Tomita, T.; Hayashi, I.; Tsuruoka, M.; Niimura, M.; Takahashi, Y.; Thinakaran, G.; Iwatsubo, T. The role of presenilin cofactors in the gamma-secretase complex. Nature 2003, 422, 438–441. [Google Scholar] [CrossRef]
- Laudon, H.; Hansson, E.M.; Melén, K.; Bergman, A.; Farmery, M.R.; Winblad, B.; Lendahl, U.; von Heijne, G.; Näslund, J. A nine-transmembrane domain topology for presenilin 1. J. Biol. Chem. 2005, 280, 35352–35360. [Google Scholar] [CrossRef]
- Tomita, T. Molecular mechanism of intramembrane proteolysis by γ-secretase. J. Biochem. 2014, 156, 195–201. [Google Scholar] [CrossRef]
- Bolduc, D.M.; Montagna, D.R.; Seghers, M.C.; Wolfe, M.S.; Selkoe, D.J. The amyloid-beta forming tripeptide cleavage mechanism of γ-secretase. eLife 2016, 5, e17578. [Google Scholar] [CrossRef]
- Sharma, P.; Srivastava, P.; Seth, A.; Tripathi, P.N.; Banerjee, A.G.; Shrivastava, S.K. Comprehensive review of mechanisms of pathogenesis involved in Alzheimer’s disease and potential therapeutic strategies. Prog. Neurobiol. 2019, 174, 53–89. [Google Scholar] [CrossRef]
- Jarrett, J.T.; Berger, E.P.; Lansbury, P.T., Jr. The C-terminus of the beta protein is critical in amyloidogenesis. Ann. N. Y. Acad. Sci. 1993, 695, 144–148. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Tyler, S.J.; Dawbarn, D.; Wilcock, G.K.; Allen, S.J. alpha- and beta-secretase: Profound changes in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2002, 299, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.C.; Yan, C.; Yang, G.; Lu, P.; Ma, D.; Sun, L.; Zhou, R.; Scheres, S.H.W.; Shi, Y. An atomic structure of human γ-secretase. Nature 2015, 525, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Fukumori, A.; Steiner, H. Substrate recruitment of γ-secretase and mechanism of clinical presenilin mutations revealed by photoaffinity mapping. EMBO J. 2016, 35, 1628–1643. [Google Scholar] [CrossRef]
- Yang, G.; Zhou, R.; Zhou, Q.; Guo, X.; Yan, C.; Ke, M.; Lei, J.; Shi, Y. Structural basis of Notch recognition by human γ-secretase. Nature 2019, 565, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Zhou, R.; Guo, X.; Yan, C.; Lei, J.; Shi, Y. Structural basis of γ-secretase inhibition and modulation by small molecule drugs. Cell 2021, 184, 521.e14–533.e14. [Google Scholar] [CrossRef]
- Suzuki, R.; Takahashi, H.; Yoshida, C.; Hidaka, M.; Ogawa, T.; Futai, E. Specific Mutations near the Amyloid Precursor Protein Cleavage Site Increase γ-Secretase Sensitivity and Modulate Amyloid-β Production. Int. J. Mol. Sci. 2023, 24, 3970. [Google Scholar] [CrossRef]
- De Strooper, B. Loss-of-function presenilin mutations in Alzheimer disease. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 2007, 8, 141–146. [Google Scholar] [CrossRef]
- Cruts, M.; Dermaut, B.; Rademakers, R.; Van den Broeck, M.; Stögbauer, F.; Van Broeckhoven, C. Novel APP mutation V715A associated with presenile Alzheimer’s disease in a German family. J. Neurol. 2003, 250, 1374–1375. [Google Scholar] [CrossRef]
- Kumar-Singh, S.; De Jonghe, C.; Cruts, M.; Kleinert, R.; Wang, R.; Mercken, M.; De Strooper, B.; Vanderstichele, H.; Löfgren, A.; Vanderhoeven, I.; et al. Nonfibrillar diffuse amyloid deposition due to a gamma(42)-secretase site mutation points to an essential role for N-truncated A beta(42) in Alzheimer’s disease. Hum. Mol. Genet. 2000, 9, 2589–2598. [Google Scholar] [CrossRef] [PubMed]
- Lichtenthaler, S.F.; Wang, R.; Grimm, H.; Uljon, S.N.; Masters, C.L.; Beyreuther, K. Mechanism of the cleavage specificity of Alzheimer’s disease gamma-secretase identified by phenylalanine-scanning mutagenesis of the transmembrane domain of the amyloid precursor protein. Proc. Natl. Acad. Sci. USA 1999, 96, 3053–3058. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, T.; Atwal, J.K.; Steinberg, S.; Snaedal, J.; Jonsson, P.V.; Bjornsson, S.; Stefansson, H.; Sulem, P.; Gudbjartsson, D.; Maloney, J.; et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 2012, 488, 96–99. [Google Scholar] [CrossRef]
- Zhang, H.; Komano, H.; Fuller, R.S.; Gandy, S.E.; Frail, D.E. Proteolytic processing and secretion of human beta-amyloid precursor protein in yeast. Evidence for a yeast secretase activity. J. Biol. Chem. 1994, 269, 27799–27802. [Google Scholar] [CrossRef]
- Komano, H.; Seeger, M.; Gandy, S.; Wang, G.T.; Krafft, G.A.; Fuller, R.S. Involvement of cell surface glycosyl-phosphatidylinositol-linked aspartyl proteases in alpha-secretase-type cleavage and ectodomain solubilization of human Alzheimer beta-amyloid precursor protein in yeast. J. Biol. Chem. 1998, 273, 31648–31651. [Google Scholar] [CrossRef] [PubMed]
- Lüthi, U.; Schaerer-Brodbeck, C.; Tanner, S.; Middendorp, O.; Edler, K.; Barberis, A. Human beta-secretase activity in yeast detected by a novel cellular growth selection system. Biochim. Biophys. Acta 2003, 1620, 167–178. [Google Scholar] [CrossRef]
- Sparvero, L.J.; Patz, S.; Brodsky, J.L.; Coughlan, C.M. Proteomic analysis of the amyloid precursor protein fragment C99: Expression in yeast. Anal. Biochem. 2007, 370, 162–170. [Google Scholar] [CrossRef][Green Version]
- Montesinos, J.; Pera, M.; Larrea, D.; Guardia-Laguarta, C.; Agrawal, R.R.; Velasco, K.R.; Yun, T.D.; Stavrovskaya, I.G.; Xu, Y.; Koo, S.Y.; et al. The Alzheimer’s disease-associated C99 fragment of APP regulates cellular cholesterol trafficking. EMBO J. 2020, 39, e103791. [Google Scholar] [CrossRef]
- Galvin, J.; Curran, E.; Arteaga, F.; Goossens, A.; Aubuchon-Endsley, N.; McMurray, M.A.; Moore, J.; Hansen, K.C.; Chial, H.J.; Potter, H.; et al. Proteasome activity modulates amyloid toxicity. FEMS Yeast Res. 2022, 22, foac004. [Google Scholar] [CrossRef] [PubMed]
- Yagishita, S.; Futai, E.; Ishiura, S. In vitro reconstitution of gamma-secretase activity using yeast microsomes. Biochem. Biophys. Res. Commun. 2008, 377, 141–145. [Google Scholar] [CrossRef]
- Futai, E. Advanced Yeast Models of Familial Alzheimer Disease Expressing FAD-Linked Presenilin to Screen Mutations and γ-Secretase Modulators. Methods Mol. Biol. 2019, 2049, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Futai, E.; Yagishita, S.; Ishiura, S. Nicastrin is dispensable for gamma-secretase protease activity in the presence of specific presenilin mutations. J. Biol. Chem. 2009, 284, 13013–13022. [Google Scholar] [CrossRef] [PubMed]
- Paiano, A.; Margiotta, A.; De Luca, M.; Bucci, C. Yeast Two-Hybrid Assay to Identify Interacting Proteins. Curr. Protoc. Protein Sci. 2019, 95, e70. [Google Scholar] [CrossRef]
- Yu, Y.; Li, Y.; Zhang, Y. Yeast Two-Hybrid Screening for Proteins that Interact with the Extracellular Domain of Amyloid Precursor Protein. Neurosci. Bull. 2016, 32, 171–176. [Google Scholar] [CrossRef] [PubMed]
- McLoughlin, D.M.; Miller, C.C. The intracellular cytoplasmic domain of the Alzheimer’s disease amyloid precursor protein interacts with phosphotyrosine-binding domain proteins in the yeast two-hybrid system. FEBS Lett. 1996, 397, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Li, Y.; Zhang, Y. Screening of APP interaction proteins by DUALmembrane yeast two-hybrid system. Int. J. Clin. Exp. Pathol. 2015, 8, 2802–2808. [Google Scholar]
- Lührs, T.; Ritter, C.; Adrian, M.; Riek-Loher, D.; Bohrmann, B.; Döbeli, H.; Schubert, D.; Riek, R. 3D structure of Alzheimer’s amyloid-beta(1-42) fibrils. Proc. Natl. Acad. Sci. USA 2005, 102, 17342–17347. [Google Scholar] [CrossRef] [PubMed]
- Yagi-Utsumi, M.; Kato, K.; Nishimura, K. Membrane-Induced Dichotomous Conformation of Amyloid β with the Disordered N-Terminal Segment Followed by the Stable C-Terminal β Structure. PLoS ONE 2016, 11, e0146405. [Google Scholar] [CrossRef]
- Priller, C.; Bauer, T.; Mitteregger, G.; Krebs, B.; Kretzschmar, H.A.; Herms, J. Synapse formation and function is modulated by the amyloid precursor protein. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 7212–7221. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Moosavi, B.; Mousavi, B.; Macreadie, I.G. Yeast Model of Amyloid-β and Tau Aggregation in Alzheimer’s Disease. J. Alzheimer’s Dis. JAD 2015, 47, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Itoh, S.G.; Okumura, H. Promotion and Inhibition of Amyloid-β Peptide Aggregation: Molecular Dynamics Studies. Int. J. Mol. Sci. 2021, 22, 1859. [Google Scholar] [CrossRef] [PubMed]
- Festa, G.; Mallamace, F.; Sancesario, G.M.; Corsaro, C.; Mallamace, D.; Fazio, E.; Arcidiacono, L.; Garcia Sakai, V.; Senesi, R.; Preziosi, E.; et al. Aggregation States of Aβ(1-40), Aβ(1-42) and Aβp(3-42) Amyloid Beta Peptides: A SANS Study. Int. J. Mol. Sci. 2019, 20, 4126. [Google Scholar] [CrossRef] [PubMed]
- Zhang-Haagen, B.; Biehl, R.; Nagel-Steger, L.; Radulescu, A.; Richter, D.; Willbold, D. Monomeric Amyloid Beta Peptide in Hexafluoroisopropanol Detected by Small Angle Neutron Scattering. PLoS ONE 2016, 11, e0150267. [Google Scholar] [CrossRef]
- Bharadwaj, P.; Martins, R.; Macreadie, I. Yeast as a model for studying Alzheimer’s disease. FEMS Yeast Res. 2010, 10, 961–969. [Google Scholar] [CrossRef][Green Version]
- Porzoor, A.; Macreadie, I. Yeast as a Model for Studies on Aβ Aggregation Toxicity in Alzheimer’s Disease, Autophagic Responses, and Drug Screening. Methods Mol. Biol. 2016, 1303, 217–226. [Google Scholar] [CrossRef]
- Hughes, S.R.; Goyal, S.; Sun, J.E.; Gonzalez-DeWhitt, P.; Fortes, M.A.; Riedel, N.G.; Sahasrabudhe, S.R. Two-hybrid system as a model to study the interaction of beta-amyloid peptide monomers. Proc. Natl. Acad. Sci. USA 1996, 93, 2065–2070. [Google Scholar] [CrossRef]
- Treusch, S.; Hamamichi, S.; Goodman, J.L.; Matlack, K.E.; Chung, C.Y.; Baru, V.; Shulman, J.M.; Parrado, A.; Bevis, B.J.; Valastyan, J.S.; et al. Functional links between Aβ toxicity, endocytic trafficking, and Alzheimer’s disease risk factors in yeast. Science 2011, 334, 1241–1245. [Google Scholar] [CrossRef]
- Chen, X.; Ji, B.; Hao, X.; Li, X.; Eisele, F.; Nyström, T.; Petranovic, D. FMN reduces Amyloid-β toxicity in yeast by regulating redox status and cellular metabolism. Nat. Commun. 2020, 11, 867. [Google Scholar] [CrossRef]
- Chernoff, Y.O.; Grizel, A.V.; Rubel, A.A.; Zelinsky, A.A.; Chandramowlishwaran, P.; Chernova, T.A. Application of yeast to studying amyloid and prion diseases. Adv. Genet. 2020, 105, 293–380. [Google Scholar] [CrossRef]
- Liebman, S.W.; Chernoff, Y.O. Prions in yeast. Genetics 2012, 191, 1041–1072. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B.; Edskes, H.K.; Kryndushkin, D.; McGlinchey, R.; Bateman, D.; Kelly, A. Prion diseases of yeast: Amyloid structure and biology. Semin. Cell Dev. Biol. 2011, 22, 469–475. [Google Scholar] [CrossRef]
- von der Haar, T.; Jossé, L.; Wright, P.; Zenthon, J.; Tuite, M.F. Development of a novel yeast cell-based system for studying the aggregation of Alzheimer’s disease-associated Abeta peptides in vivo. Neuro-Degener. Dis. 2007, 4, 136–147. [Google Scholar] [CrossRef]
- Bagriantsev, S.; Liebman, S. Modulation of Abeta42 low-n oligomerization using a novel yeast reporter system. BMC Biol. 2006, 4, 32. [Google Scholar] [CrossRef]
- Ishikawa, T. Saccharomyces cerevisiae in neuroscience: How unicellular organism helps to better understand prion protein? Neural Regen. Res. 2021, 16, 489–495. [Google Scholar] [CrossRef]
- Rubel, A.A.; Ryzhova, T.A.; Antonets, K.S.; Chernoff, Y.O.; Galkin, A. Identification of PrP sequences essential for the interaction between the PrP polymers and Aβ peptide in a yeast-based assay. Prion 2013, 7, 469–476. [Google Scholar] [CrossRef]
- Apodaca, J.; Kim, I.; Rao, H. Cellular tolerance of prion protein PrP in yeast involves proteolysis and the unfolded protein response. Biochem. Biophys. Res. Commun. 2006, 347, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Caine, J.; Sankovich, S.; Antony, H.; Waddington, L.; Macreadie, P.; Varghese, J.; Macreadie, I. Alzheimer’s Abeta fused to green fluorescent protein induces growth stress and a heat shock response. FEMS Yeast Res. 2007, 7, 1230–1236. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Lindquist, S. De novo generation of a PrPSc-like conformation in living cells. Nat. Cell Biol. 1999, 1, 358–361. [Google Scholar] [CrossRef]
- Yang, W.; Yang, H.; Tien, P. In vitro self-propagation of recombinant PrPSc-like conformation generated in the yeast cytoplasm. FEBS Lett. 2006, 580, 4231–4235. [Google Scholar] [CrossRef][Green Version]
- D’Angelo, F.; Vignaud, H.; Di Martino, J.; Salin, B.; Devin, A.; Cullin, C.; Marchal, C. A yeast model for amyloid-β aggregation exemplifies the role of membrane trafficking and PICALM in cytotoxicity. Dis. Model. Mech. 2013, 6, 206–216. [Google Scholar] [CrossRef]
- Nair, S.; Traini, M.; Dawes, I.W.; Perrone, G.G. Genome-wide analysis of Saccharomyces cerevisiae identifies cellular processes affecting intracellular aggregation of Alzheimer’s amyloid-β42: Importance of lipid homeostasis. Mol. Biol. Cell 2014, 25, 2235–2249. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, P.; Martins, R. A rapid absorbance-based growth assay to screen the toxicity of oligomer Aβ(42) and protect against cell death in yeast. Neural Regen. Res. 2020, 15, 1931–1936. [Google Scholar] [CrossRef]
- Bharadwaj, P.; Waddington, L.; Varghese, J.; Macreadie, I.G. A new method to measure cellular toxicity of non-fibrillar and fibrillar Alzheimer’s Abeta using yeast. J. Alzheimer’s Dis. JAD 2008, 13, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Porzoor, A.; Caine, J.M.; Macreadie, I.G. Pretreatment of chemically-synthesized Aβ42 affects its biological activity in yeast. Prion 2014, 8, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef]
- Lambert, J.C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef]
- França, M.B.; Lima, K.C.; Eleutherio, E.C. Oxidative Stress and Amyloid Toxicity: Insights From Yeast. J. Cell. Biochem. 2017, 118, 1442–1452. [Google Scholar] [CrossRef]
- Chen, X.; Bisschops, M.M.M.; Agarwal, N.R.; Ji, B.; Shanmugavel, K.P.; Petranovic, D. Interplay of Energetics and ER Stress Exacerbates Alzheimer’s Amyloid-β (Aβ) Toxicity in Yeast. Front. Mol. Neurosci. 2017, 10, 232. [Google Scholar] [CrossRef] [PubMed]
- Panaretou, B.; Jones, G.W. Yeast models for amyloid disease. Essays Biochem. 2014, 56, 85–97. [Google Scholar] [CrossRef]
- Mossmann, D.; Vögtle, F.N.; Taskin, A.A.; Teixeira, P.F.; Ring, J.; Burkhart, J.M.; Burger, N.; Pinho, C.M.; Tadic, J.; Loreth, D.; et al. Amyloid-β peptide induces mitochondrial dysfunction by inhibition of preprotein maturation. Cell Metab. 2014, 20, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Dhakal, S.; Macreadie, I. Tyramine and Amyloid Beta 42: A Toxic Synergy. Biomedicines 2020, 8, 145. [Google Scholar] [CrossRef] [PubMed]
- Hamos, J.E.; Oblas, B.; Pulaski-Salo, D.; Welch, W.J.; Bole, D.G.; Drachman, D.A. Expression of heat shock proteins in Alzheimer’s disease. Neurology 1991, 41, 345–350. [Google Scholar] [CrossRef]
- Ring, J.; Tadic, J.; Ristic, S.; Poglitsch, M.; Bergmann, M.; Radic, N.; Mossmann, D.; Liang, Y.; Maglione, M.; Jerkovic, A.; et al. The HSP40 chaperone Ydj1 drives amyloid beta 42 toxicity. EMBO Mol. Med. 2022, 14, e13952. [Google Scholar] [CrossRef]
- Almeida, C.G.; Takahashi, R.H.; Gouras, G.K. Beta-amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 4277–4288. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yan, S.D. Mitochondrial Abeta: A potential cause of metabolic dysfunction in Alzheimer’s disease. IUBMB Life 2006, 58, 686–694. [Google Scholar] [CrossRef]
- Reddy, P.H. Amyloid precursor protein-mediated free radicals and oxidative damage: Implications for the development and progression of Alzheimer’s disease. J. Neurochem. 2006, 96, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Keating, D.J.; Chen, C.; Pritchard, M.A. Alzheimer’s disease and endocytic dysfunction: Clues from the Down syndrome-related proteins, DSCR1 and ITSN1. Ageing Res. Rev. 2006, 5, 388–401. [Google Scholar] [CrossRef]
- Brown, S.T.; Scragg, J.L.; Boyle, J.P.; Hudasek, K.; Peers, C.; Fearon, I.M. Hypoxic augmentation of Ca2+ channel currents requires a functional electron transport chain. J. Biol. Chem. 2005, 280, 21706–21712. [Google Scholar] [CrossRef]
- Andersen, O.M.; Schmidt, V.; Spoelgen, R.; Gliemann, J.; Behlke, J.; Galatis, D.; McKinstry, W.J.; Parker, M.W.; Masters, C.L.; Hyman, B.T.; et al. Molecular dissection of the interaction between amyloid precursor protein and its neuronal trafficking receptor SorLA/LR11. Biochemistry 2006, 45, 2618–2628. [Google Scholar] [CrossRef]
- Panza, F.; Lozupone, M.; Solfrizzi, V.; Watling, M.; Imbimbo, B.P. Time to test antibacterial therapy in Alzheimer’s disease. Brain A J. Neurol. 2019, 142, 2905–2929. [Google Scholar] [CrossRef] [PubMed]
- Park, S.K.; Pegan, S.D.; Mesecar, A.D.; Jungbauer, L.M.; LaDu, M.J.; Liebman, S.W. Development and validation of a yeast high-throughput screen for inhibitors of Aβ42 oligomerization. Dis. Model. Mech. 2011, 4, 822–831. [Google Scholar] [CrossRef]
- Park, S.K.; Ratia, K.; Ba, M.; Valencik, M.; Liebman, S.W. Inhibition of Aβ(42) oligomerization in yeast by a PICALM ortholog and certain FDA approved drugs. Microb. Cell 2016, 3, 53–64. [Google Scholar] [CrossRef]
- Chandramowlishwaran, P.; Sun, M.; Casey, K.L.; Romanyuk, A.V.; Grizel, A.V.; Sopova, J.V.; Rubel, A.A.; Nussbaum-Krammer, C.; Vorberg, I.M.; Chernoff, Y.O. Mammalian amyloidogenic proteins promote prion nucleation in yeast. J. Biol. Chem. 2018, 293, 3436–3450. [Google Scholar] [CrossRef] [PubMed]
- Macreadie, I.; Lotfi-Miri, M.; Mohotti, S.; Shapira, D.; Bennett, L.; Varghese, J. Validation of folate in a convenient yeast assay suited for identification of inhibitors of Alzheimer’s amyloid-beta aggregation. J. Alzheimer’s Dis. JAD 2008, 15, 391–396. [Google Scholar] [CrossRef]
- Son, J.H.; Shim, J.H.; Kim, K.H.; Ha, J.Y.; Han, J.Y. Neuronal autophagy and neurodegenerative diseases. Exp. Mol. Med. 2012, 44, 89–98. [Google Scholar] [CrossRef]
- Dhakal, S.; Subhan, M.; Fraser, J.M.; Gardiner, K.; Macreadie, I. Simvastatin Efficiently Reduces Levels of Alzheimer’s Amyloid Beta in Yeast. Int. J. Mol. Sci. 2019, 20, 3531. [Google Scholar] [CrossRef]
- Bharadwaj, P.R.; Martins, R.N. Autophagy modulates Aβ accumulation and formation of aggregates in yeast. Mol. Cell. Neurosci. 2020, 104, 103466. [Google Scholar] [CrossRef]
- Bharadwaj, P.R.; Verdile, G.; Barr, R.K.; Gupta, V.; Steele, J.W.; Lachenmayer, M.L.; Yue, Z.; Ehrlich, M.E.; Petsko, G.; Ju, S.; et al. Latrepirdine (dimebon) enhances autophagy and reduces intracellular GFP-Aβ42 levels in yeast. J. Alzheimer’s Dis. JAD 2012, 32, 949–967. [Google Scholar] [CrossRef]
- Steele, J.W.; Lachenmayer, M.L.; Ju, S.; Stock, A.; Liken, J.; Kim, S.H.; Delgado, L.M.; Alfaro, I.E.; Bernales, S.; Verdile, G.; et al. Latrepirdine improves cognition and arrests progression of neuropathology in an Alzheimer’s mouse model. Mol. Psychiatry 2013, 18, 889–897. [Google Scholar] [CrossRef]
- Tardiff, D.F.; Brown, L.E.; Yan, X.; Trilles, R.; Jui, N.T.; Barrasa, M.I.; Caldwell, K.A.; Caldwell, G.A.; Schaus, S.E.; Lindquist, S. Dihydropyrimidine-Thiones and Clioquinol Synergize To Target β-Amyloid Cellular Pathologies through a Metal-Dependent Mechanism. ACS Chem. Neurosci. 2017, 8, 2039–2055. [Google Scholar] [CrossRef]
- Cherny, R.A.; Atwood, C.S.; Xilinas, M.E.; Gray, D.N.; Jones, W.D.; McLean, C.A.; Barnham, K.J.; Volitakis, I.; Fraser, F.W.; Kim, Y.; et al. Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron 2001, 30, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Lannfelt, L.; Blennow, K.; Zetterberg, H.; Batsman, S.; Ames, D.; Harrison, J.; Masters, C.L.; Targum, S.; Bush, A.I.; Murdoch, R.; et al. Safety, efficacy, and biomarker findings of PBT2 in targeting Abeta as a modifying therapy for Alzheimer’s disease: A phase IIa, double-blind, randomised, placebo-controlled trial. Lancet. Neurol. 2008, 7, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Rafii, M.S. Tau PET Imaging for Staging of Alzheimer’s Disease in Down Syndrome. Dev. Neurobiol. 2019, 79, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Rösler, T.W.; Höglinger, G.U. Tau links developmental to neurodegenerative diseases. Neurosci. Biobehav. Rev. 2019, 104, 26–27. [Google Scholar] [CrossRef] [PubMed]
- Fichou, Y.; Lin, Y.; Rauch, J.N.; Vigers, M.; Zeng, Z.; Srivastava, M.; Keller, T.J.; Freed, J.H.; Kosik, K.S.; Han, S. Cofactors are essential constituents of stable and seeding-active tau fibrils. Proc. Natl. Acad. Sci. USA 2018, 115, 13234–13239. [Google Scholar] [CrossRef] [PubMed]
- Kubo, A.; Ueda, S.; Yamane, A.; Wada-Kakuda, S.; Narita, M.; Matsuyama, M.; Nomori, A.; Takashima, A.; Kato, T.; Onodera, O.; et al. Ectopic Expression Induces Abnormal Somatodendritic Distribution of Tau in the Mouse Brain. J. Neurosci. Off. J. Soc. Neurosci. 2019, 39, 6781–6797. [Google Scholar] [CrossRef]
- Reznik, E.; Miller, M.L.; Şenbabaoğlu, Y.; Riaz, N.; Sarungbam, J.; Tickoo, S.K.; Al-Ahmadie, H.A.; Lee, W.; Seshan, V.E.; Hakimi, A.A.; et al. Mitochondrial DNA copy number variation across human cancers. eLife 2016, 5, e10769. [Google Scholar] [CrossRef] [PubMed]
- Stoccoro, A.; Siciliano, G.; Migliore, L.; Coppedè, F. Decreased Methylation of the Mitochondrial D-Loop Region in Late-Onset Alzheimer’s Disease. J. Alzheimer’s Dis. JAD 2017, 59, 559–564. [Google Scholar] [CrossRef]
- De Vos, A.; Anandhakumar, J.; Van den Brande, J.; Verduyckt, M.; Franssens, V.; Winderickx, J.; Swinnen, E. Yeast as a model system to study tau biology. Int. J. Alzheimer’s Dis. 2011, 2011, 428970. [Google Scholar] [CrossRef]
- Vandebroek, T.; Vanhelmont, T.; Terwel, D.; Borghgraef, P.; Lemaire, K.; Snauwaert, J.; Wera, S.; Van Leuven, F.; Winderickx, J. Identification and isolation of a hyperphosphorylated, conformationally changed intermediate of human protein tau expressed in yeast. Biochemistry 2005, 44, 11466–11475. [Google Scholar] [CrossRef] [PubMed]
- Rosseels, J.; Van den Brande, J.; Violet, M.; Jacobs, D.; Grognet, P.; Lopez, J.; Huvent, I.; Caldara, M.; Swinnen, E.; Papegaey, A.; et al. Tau monoclonal antibody generation based on humanized yeast models: Impact on Tau oligomerization and diagnostics. J. Biol. Chem. 2015, 290, 4059–4074. [Google Scholar] [CrossRef] [PubMed]
- Carmel, G.; Mager, E.M.; Binder, L.I.; Kuret, J. The structural basis of monoclonal antibody Alz50’s selectivity for Alzheimer’s disease pathology. J. Biol. Chem. 1996, 271, 32789–32795. [Google Scholar] [CrossRef] [PubMed]
- Weaver, C.L.; Espinoza, M.; Kress, Y.; Davies, P. Conformational change as one of the earliest alterations of tau in Alzheimer’s disease. Neurobiol. Aging 2000, 21, 719–727. [Google Scholar] [CrossRef]
- Hallows, J.L.; Chen, K.; DePinho, R.A.; Vincent, I. Decreased cyclin-dependent kinase 5 (cdk5) activity is accompanied by redistribution of cdk5 and cytoskeletal proteins and increased cytoskeletal protein phosphorylation in p35 null mice. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 10633–10644. [Google Scholar] [CrossRef]
- Spittaels, K.; Van den Haute, C.; Van Dorpe, J.; Bruynseels, K.; Vandezande, K.; Laenen, I.; Geerts, H.; Mercken, M.; Sciot, R.; Van Lommel, A.; et al. Prominent axonopathy in the brain and spinal cord of transgenic mice overexpressing four-repeat human tau protein. Am. J. Pathol. 1999, 155, 2153–2165. [Google Scholar] [CrossRef]
- Jicha, G.A.; Weaver, C.; Lane, E.; Vianna, C.; Kress, Y.; Rockwood, J.; Davies, P. cAMP-dependent protein kinase phosphorylations on tau in Alzheimer’s disease. J. Neurosci. Off. J. Soc. Neurosci. 1999, 19, 7486–7494. [Google Scholar] [CrossRef]
- Zheng-Fischhöfer, Q.; Biernat, J.; Mandelkow, E.M.; Illenberger, S.; Godemann, R.; Mandelkow, E. Sequential phosphorylation of Tau by glycogen synthase kinase-3beta and protein kinase A at Thr212 and Ser214 generates the Alzheimer-specific epitope of antibody AT100 and requires a paired-helical-filament-like conformation. Eur. J. Biochem. 1998, 252, 542–552. [Google Scholar] [CrossRef]
- Wen, Y.; Planel, E.; Herman, M.; Figueroa, H.Y.; Wang, L.; Liu, L.; Lau, L.F.; Yu, W.H.; Duff, K.E. Interplay between cyclin-dependent kinase 5 and glycogen synthase kinase 3 beta mediated by neuregulin signaling leads to differential effects on tau phosphorylation and amyloid precursor protein processing. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 2624–2632. [Google Scholar] [CrossRef]
- Vandebroek, T.; Terwel, D.; Vanhelmont, T.; Gysemans, M.; Van Haesendonck, C.; Engelborghs, Y.; Winderickx, J.; Van Leuven, F. Microtubule binding and clustering of human Tau-4R and Tau-P301L proteins isolated from yeast deficient in orthologues of glycogen synthase kinase-3beta or cdk5. J. Biol. Chem. 2006, 281, 25388–25397. [Google Scholar] [CrossRef]
- Vanhelmont, T.; Vandebroek, T.; De Vos, A.; Terwel, D.; Lemaire, K.; Anandhakumar, J.; Franssens, V.; Swinnen, E.; Van Leuven, F.; Winderickx, J. Serine-409 phosphorylation and oxidative damage define aggregation of human protein tau in yeast. FEMS Yeast Res. 2010, 10, 992–1005. [Google Scholar] [CrossRef] [PubMed]
- Gamblin, T.C.; Berry, R.W.; Binder, L.I. Tau polymerization: Role of the amino terminus. Biochemistry 2003, 42, 2252–2257. [Google Scholar] [CrossRef] [PubMed]
- Sergeant, N.; Delacourte, A.; Buée, L. Tau protein as a differential biomarker of tauopathies. Biochim. Biophys. Acta 2005, 1739, 179–197. [Google Scholar] [CrossRef] [PubMed]
- Zambrano, C.A.; Egaña, J.T.; Núñez, M.T.; Maccioni, R.B.; González-Billault, C. Oxidative stress promotes tau dephosphorylation in neuronal cells: The roles of cdk5 and PP1. Free Radic. Biol. Med. 2004, 36, 1393–1402. [Google Scholar] [CrossRef]
- De Vos, A.; Bynens, T.; Rosseels, J.; Coun, C.; Ring, J.; Madeo, F.; Galas, M.-C.; Winderickx, J.; Franssens, V. The peptidyl prolyl cis/trans isomerase Pin1/Ess1 inhibits phosphorylation and toxicity of tau in a yeast model for Alzheimer’s disease. AIMS Mol. Sci. 2015, 2, 144–160. [Google Scholar] [CrossRef]
- Ciaccioli, G.; Martins, A.; Rodrigues, C.; Vieira, H.; Calado, P. A powerful yeast model to investigate the synergistic interaction of α-synuclein and tau in neurodegeneration. PLoS ONE 2013, 8, e55848. [Google Scholar] [CrossRef]
- Zabrocki, P.; Pellens, K.; Vanhelmont, T.; Vandebroek, T.; Griffioen, G.; Wera, S.; Van Leuven, F.; Winderickx, J. Characterization of alpha-synuclein aggregation and synergistic toxicity with protein tau in yeast. FEBS J. 2005, 272, 1386–1400. [Google Scholar] [CrossRef]
- Verelst, J.; Geukens, N.; Eddarkaoui, S.; Vliegen, D.; De Smidt, E.; Rosseels, J.; Franssens, V.; Molenberghs, S.; Francois, C.; Stoops, E.; et al. A Novel Tau Antibody Detecting the First Amino-Terminal Insert Reveals Conformational Differences Among Tau Isoforms. Front. Mol. Biosci. 2020, 7, 48. [Google Scholar] [CrossRef]
- Chen, X.Q.; Mobley, W.C. Alzheimer Disease Pathogenesis: Insights From Molecular and Cellular Biology Studies of Oligomeric Aβ and Tau Species. Front. Neurosci. 2019, 13, 659. [Google Scholar] [CrossRef]
- Chételat, G.; La Joie, R.; Villain, N.; Perrotin, A.; de La Sayette, V.; Eustache, F.; Vandenberghe, R. Amyloid imaging in cognitively normal individuals, at-risk populations and preclinical Alzheimer’s disease. NeuroImage Clin. 2013, 2, 356–365. [Google Scholar] [CrossRef]
- Baek, S.H.; Park, S.J.; Jeong, J.I.; Kim, S.H.; Han, J.; Kyung, J.W.; Baik, S.H.; Choi, Y.; Choi, B.Y.; Park, J.S.; et al. Inhibition of Drp1 Ameliorates Synaptic Depression, Aβ Deposition, and Cognitive Impairment in an Alzheimer’s Disease Model. J. Neurosci. Off. J. Soc. Neurosci. 2017, 37, 5099–5110. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Manczak, M.; Yin, X. Mitochondria-Division Inhibitor 1 Protects Against Amyloid-β induced Mitochondrial Fragmentation and Synaptic Damage in Alzheimer’s Disease. J. Alzheimer’s Dis. JAD 2017, 58, 147–162. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Epremyan, K.K.; Mamaev, D.V.; Zvyagilskaya, R.A. Alzheimer’s Disease: Significant Benefit from the Yeast-Based Models. Int. J. Mol. Sci. 2023, 24, 9791. https://doi.org/10.3390/ijms24129791
Epremyan KK, Mamaev DV, Zvyagilskaya RA. Alzheimer’s Disease: Significant Benefit from the Yeast-Based Models. International Journal of Molecular Sciences. 2023; 24(12):9791. https://doi.org/10.3390/ijms24129791
Chicago/Turabian StyleEpremyan, Khoren K., Dmitry V. Mamaev, and Renata A. Zvyagilskaya. 2023. "Alzheimer’s Disease: Significant Benefit from the Yeast-Based Models" International Journal of Molecular Sciences 24, no. 12: 9791. https://doi.org/10.3390/ijms24129791
APA StyleEpremyan, K. K., Mamaev, D. V., & Zvyagilskaya, R. A. (2023). Alzheimer’s Disease: Significant Benefit from the Yeast-Based Models. International Journal of Molecular Sciences, 24(12), 9791. https://doi.org/10.3390/ijms24129791