
COX5A Alleviates Doxorubicin-Induced Cardiotoxicity by Suppressing Oxidative Stress, Mitochondrial Dysfunction and Cardiomyocyte Apoptosis

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. COX5A Is Downregulated in the Heart Tissue of Patients with Dilated Cardiomyopathy
2.2. COX5A Is Decreased by DOX in the Heart of Mice and H9c2 Cardiomyoblasts
2.3. COX5A Upregulation Attenuates DOX-Induced Cardiotoxicity in Mice
2.4. COX5A Overexpression Reduces DOX-Induced Oxidative Stress and Apoptosis in Mice
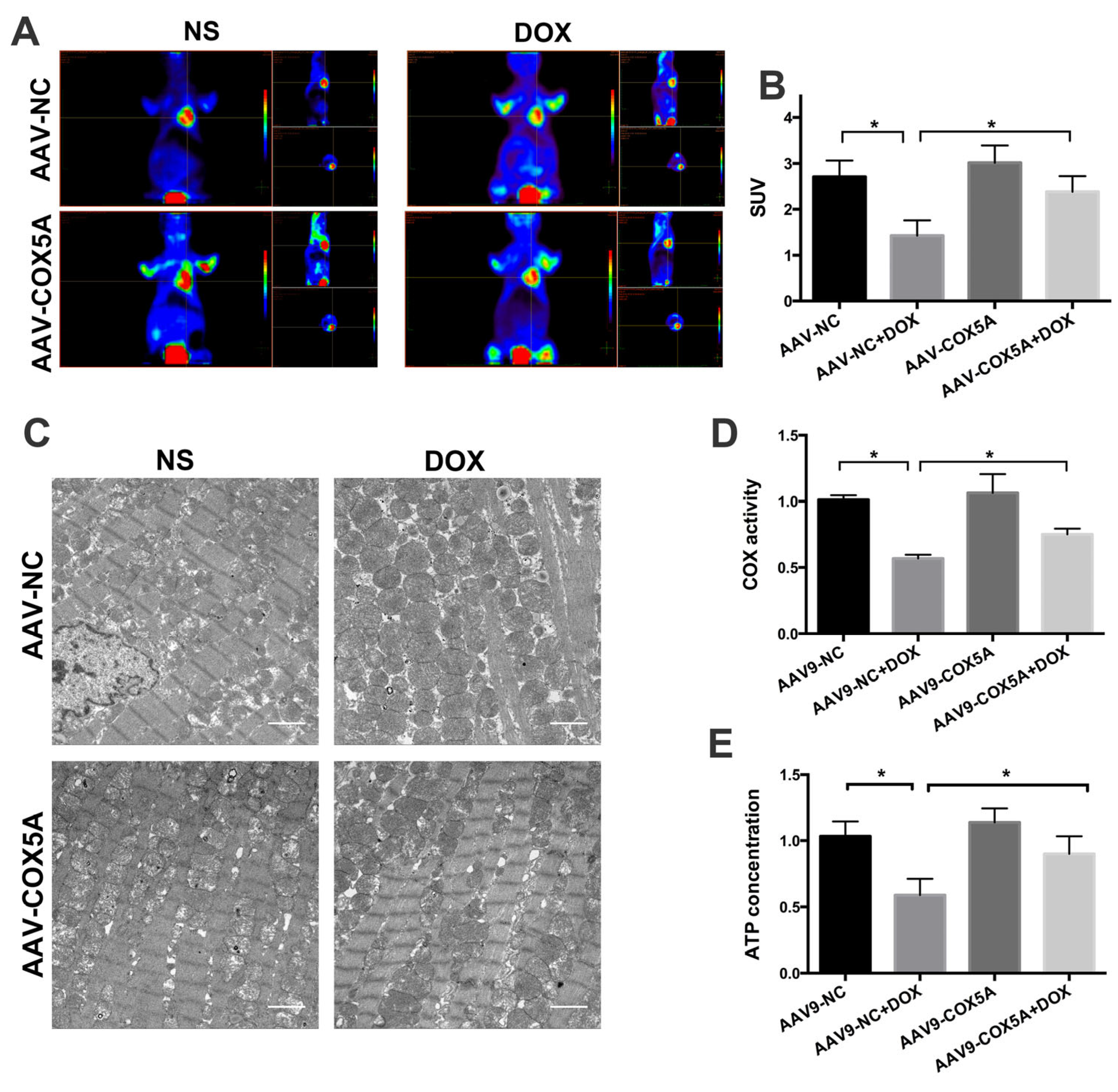
2.5. COX5A Overexpression of Improved Mitochondrial Energy Metabolism in Mice Treated with DOX
2.6. COX5A Upregulation Alleviates DOX-Induced Oxidative Stress, Mitochondrial Dysfunction and Cardiomyocyte Apoptosis In Vitro
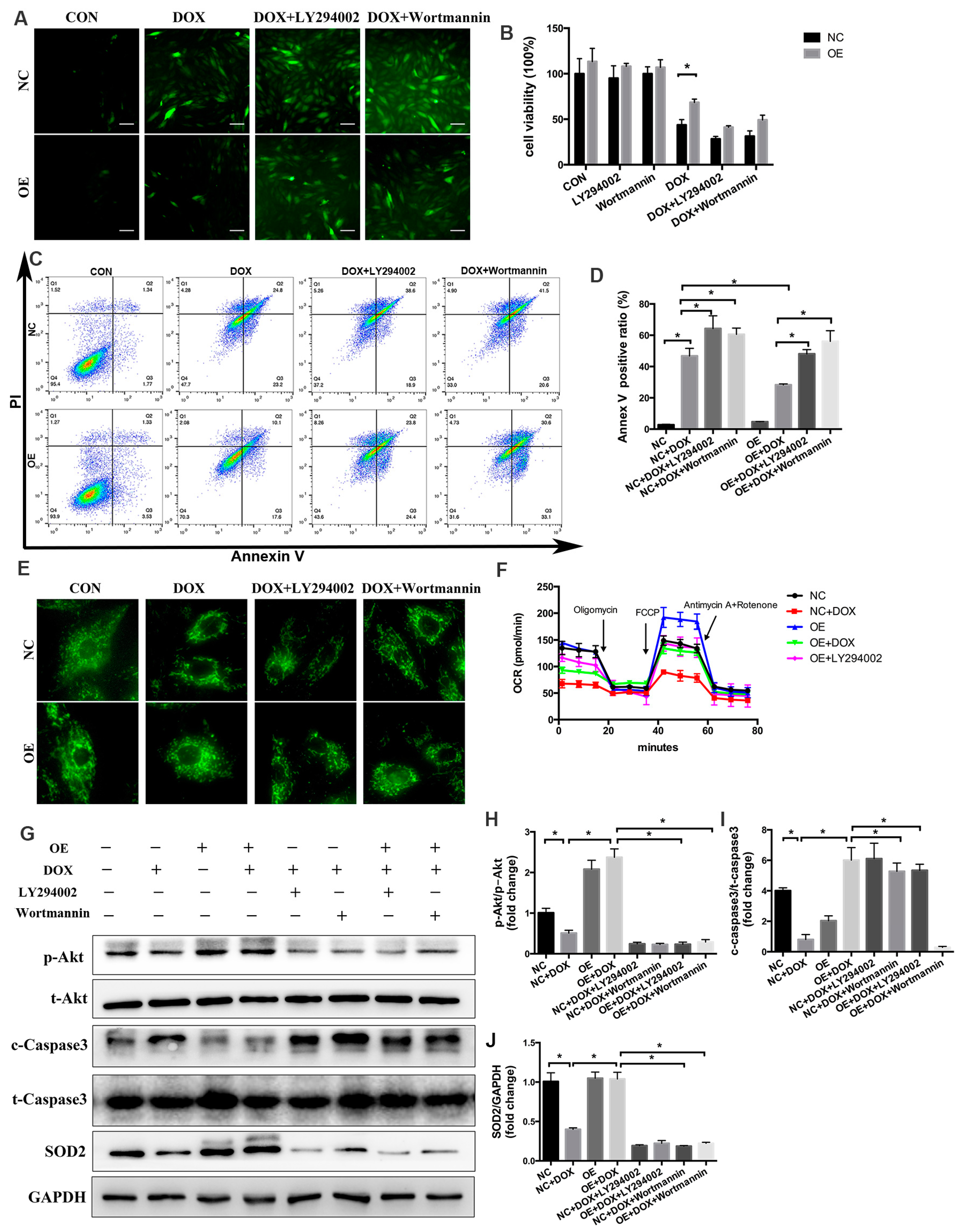
2.7. PI3K/Akt Signaling Is Responsible for the COX5A-Mediated Protective Role against DOX-Induced Cardiotoxicity
3. Discussion
4. Materials and Methods
4.1. Myocardial Samples from Normal and Failing Human Hearts
4.2. Animals and Experimental Protocols
4.3. Echocardiography Evaluation
4.4. Immunohistochemical Staining
4.5. Positron Emission Tomography (PET)
4.6. Transmission Electron Microscopy (TEM)
4.7. Tissue Mitochondria Isolation and COX Activity Assay
4.8. Adenosine Triphosphate (ATP) Assay
4.9. Hematoxylin and Eosin (H &E) Staining and Sirius Red Staining
4.10. Wheat Germ Agglutinin (WGA) Staining
4.11. cTnT, LDH and CK-MB Level Assay
4.12. Dihydroethidium (DHE) Staining
4.13. Cell Culture and Lentivirus Transfection
4.14. Flow-Cytometric Analysis
4.15. DCFH-DA Staining and CCK-8 Assay
4.16. Mitochondrial Respiration Analysis
4.17. Western Blot Analysis
4.18. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Takemura, G.; Fujiwara, H. Doxorubicin-induced cardiomyopathy from the cardiotoxic mechanisms to management. Prog. Cardiovasc. Dis. 2007, 49, 330–352. [Google Scholar] [CrossRef] [PubMed]
- Arinno, A.; Maneechote, C.; Khuanjing, T.; Ongnok, B.; Prathumsap, N.; Chunchai, T.; Arunsak, B.; Kerdphoo, S.; Shinlapawittayatorn, K.; Chattipakorn, S.C.; et al. Cardioprotective effects of melatonin and metformin against doxorubicin-induced cardiotoxicity in rats are through preserving mitochondrial function and dynamics. Biochem. Pharmacol. 2021, 192, 114743. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Sala, A.D.; Tocchetti, C.G.; Porporato, P.E.; Ghigo, A. Metabolic Aspects of Anthracycline Cardiotoxicity. Curr. Treat. Options Oncol. 2021, 22, 18. [Google Scholar] [CrossRef]
- Zhang, S.; Wei, X.; Zhang, H.; Wu, Y.; Jing, J.; Huang, R.; Zhou, T.; Hu, J.; Wu, Y.; Li, Y.; et al. Doxorubicin downregulates autophagy to promote apoptosis-induced dilated cardiomyopathy via regulating the AMPK/mTOR pathway. Biomed. Pharmacother. 2023, 162, 114691. [Google Scholar] [CrossRef] [PubMed]
- Zilinyi, R.; Czompa, A.; Czegledi, A.; Gajtko, A.; Pituk, D.; Lekli, I.; Tosaki, A. The Cardioprotective Effect of Metformin in Doxorubicin-Induced Cardiotoxicity: The Role of Autophagy. Molecules 2018, 23, 1184. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X.; et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680. [Google Scholar] [CrossRef]
- Arola, O.J.; Saraste, A.; Pulkki, K.; Kallajoki, M.; Parvinen, M.; Voipio-Pulkki, L.M. Acute doxorubicin cardiotoxicity involves cardiomyocyte apoptosis. Cancer Res. 2000, 60, 1789–1792. [Google Scholar]
- Satyam, S.M.; Bairy, L.K.; Shetty, P.; Sainath, P.; Bharati, S.; Ahmed, A.Z.; Singh, V.K.; Ashwal, A.J. Metformin and Dapagliflozin Attenuate Doxorubicin-Induced Acute Cardiotoxicity in Wistar Rats: An Electrocardiographic, Biochemical, and Histopathological Approach. Cardiovasc. Toxicol. 2023, 23, 107–119. [Google Scholar] [CrossRef]
- Argun, M.; Uzum, K.; Sonmez, M.F.; Ozyurt, A.; Karabulut, D.; Soyersarica, Z.; Cilenk, K.T.; Unalmis, S.; Pamukcu, O.; Baykan, A.; et al. Cardioprotective effect of metformin against doxorubicin cardiotoxicity in rats. Anatol. J. Cardiol. 2016, 16, 234–241. [Google Scholar] [CrossRef]
- Seifert, C.F.; Nesser, M.E.; Thompson, D.F. Dexrazoxane in the prevention of doxorubicin-induced cardiotoxicity. Ann. Pharmacother. 1994, 28, 1063–1072. [Google Scholar] [CrossRef]
- Yin, J.; Guo, J.; Zhang, Q.; Cui, L.; Zhang, L.; Zhang, T.F.; Zhao, J.; Li, J.; Middleton, A.; Carmichael, P.L.; et al. Doxorubicin-induced mitophagy and mitochondrial damage is associated with dysregulation of the PINK1/parkin pathway. Toxicol. Vitr. 2018, 51, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.P.; Chen, Z.W.; Lu, D.B.; Wu, Y.; Fan, M.K.; Qian, J.Y.; Ge, J.B. Overexpression of COX5A protects H9c2 cells against doxorubicin-induced cardiotoxicity. Biochem. Biophys. Res. Commun. 2020, 524, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Soto, I.C.; Fontanesi, F.; Liu, J.J.; Barrientos, A. Biogenesis and assembly of eukaryotic cytochrome c oxidase catalytic core. BBA-Bioenerg. 2012, 1817, 883–897. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondrial diseases in man and mouse. Science 1999, 283, 1482–1488. [Google Scholar] [CrossRef] [PubMed]
- Giordano, C.; Perli, E.; Orlandi, M.; Pisano, A.; Tuppen, H.A.; He, L.; Ierinò, R.; Petruzziello, L.; Terzi, A.; Autore, C.; et al. Cardiomyopathies due to homoplasmic mitochondrial tRNA mutations: Morphologic and molecular features. Hum. Pathol. 2013, 44, 1262–1270. [Google Scholar] [CrossRef]
- Yu, Q.; Nguyen, T.; Ogbi, M.; Caldwell, R.W.; Johnson, J.A. Differential loss of cytochrome-c oxidase subunits in ischemia-reperfusion injury: Exacerbation of COI subunit loss by PKC-epsilon inhibition. Am. J. Physiol. -Heart Circ. Physiol. 2008, 294, H2637–H2645. [Google Scholar] [CrossRef]
- Arnold, S.; Goglia, F.; Kadenbach, B. 3,5-Diiodothyronine binds to subunit Va of cytochrome-c oxidase and abolishes the allosteric inhibition of respiration by ATP. Eur. J. Biochem. 1998, 252, 325–330. [Google Scholar] [CrossRef]
- Baertling, F.; Al-Murshedi, F.; Sánchez-Caballero, L.; Al-Senaidi, K.; Joshi, N.P.; Venselaar, H.; van den Brand, M.A.; Nijtmans, L.G.; Rodenburg, R. Mutation in mitochondrial complex IV subunit COX5A causes pulmonary arterial hypertension, lactic acidemia, and failure to thrive. Hum. Mutat. 2017, 38, 692–703. [Google Scholar] [CrossRef]
- Torraco, A.; Morlino, S.; Rizza, T.; Nottia, M.D.; Bottaro, G.; Bisceglia, L.; Montanari, A.; Cappa, M.; Castori, M.; Bertini, E.; et al. A novel homozygous variant in COX5A causes an attenuated phenotype with failure to thrive, lactic acidosis, hypoglycemia, and short stature. Clin. Genet. 2022, 102, 56–60. [Google Scholar] [CrossRef]
- Jiang, Y.; Bai, X.; Li, T.-T.; Al-Hawwas, M.; Jin, Y.; Zou, Y.; Hu, Y.; Liu, L.-Y.; Zhang, Y.; Liu, Q.; et al. COX5A over-expression protects cortical neurons from hypoxic ischemic injury in neonatal rats associated with TPI up-regulation. BMC Neurosci. 2020, 21, 18. [Google Scholar] [CrossRef]
- Baden, K.N.; Murray, J.; Capaldi, R.A.; Guillemin, K. Early developmental pathology due to cytochrome c oxidase deficiency is revealed by a new zebrafish model. J. Biol. Chem. 2007, 282, 34839–34849. [Google Scholar] [CrossRef] [PubMed]
- Kaviyarasi, R.; Abilash, V.G.; Tirupathi, P.P.B.; Sankarganesh, A. Molecular mechanism of doxorubicin-induced cardiomyopathy—An update. Eur. J. Pharmacol. 2018, 818, 241–253. [Google Scholar]
- Bourens, M.; Fontanesi, F.; Soto, I.C.; Liu, J.J.; Barrientos, A. Redox and reactive oxygen species regulation of mitochondrial cytochrome C oxidase biogenesis. Antioxid. Redox Signal. 2013, 19, 1940–1952. [Google Scholar] [CrossRef] [PubMed]
- Joshua, A.C.; Sanjeev, A.F.; Douglas, B.S. Anthracycline and Peripartum Cardiomyopathies: Predictably Unpredictable. Circ. Res. 2019, 124, 1633–1646. [Google Scholar]
- Almomani, R.; Herkert, J.C.; Posafalvi, A.; Post, J.G.; Boven, L.G.; Van Der Zwaag, P.A.; Willems, P.H.G.M.; Van Veen-Hof, I.H.; Verhagen, J.M.A.; Wessels, M.W.; et al. Homozygous damaging SOD2 variant causes lethal neonatal dilated cardiomyopathy. J. Med. Genet. 2020, 57, 23–30. [Google Scholar] [CrossRef]
- Li, Y.; Huang, T.-T.; Carlson, E.J.; Melov, S.; Ursell, P.C.; Olson, J.L.; Noble, L.J.; Yoshimura, M.P.; Berger, C.; Chan, P.H.; et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat. Genet. 1995, 11, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Bhattarai, S.; Ara, H.; Sun, G.; Clair, D.K.S.; Bhuiyan, S.; Kevil, C.; Watts, M.N.; Dominic, P.; Shimizu, T.; et al. SOD2 deficiency in cardiomyocytes defines defective mitochondrial bioenergetics as a cause of lethal dilated cardiomyopathy. Redox Biol. 2020, 37, 101740. [Google Scholar] [CrossRef]
- Cunha-Oliveira, T.; Ferreira, L.L.; Coelho, A.R.; Deus, C.M.; Oliveira, P.J. Doxorubicin triggers bioenergetic failure and p53 activation in mouse stem cell-derived cardiomyocytes. Toxicol. Appl. Pharmacol. 2018, 348, 1–13. [Google Scholar] [CrossRef]
- Yen, H.C.; Oberley, T.D.; Gairola, C.G.; Szweda, L.I.; Clair St, D.K. Manganese superoxide dismutase protects mitochondrial complex I against adriamycin-induced cardiomyopathy in transgenic mice. Arch. Biochem. Biophys. 1999, 362, 59–66. [Google Scholar] [CrossRef]
- Chaiswing, L.; Cole, M.P.; Ittarat, W.; Szweda, L.I.; Clair St, D.K.; Oberley, T.D. Manganese superoxide dismutase and inducible nitric oxide synthase modify early oxidative events in acute adriamycin-induced mitochondrial toxicity. Mol. Cancer Ther. 2005, 4, 1056–1064. [Google Scholar] [CrossRef]
- Satish, S.; Narayan, G.A. Cytochrome c oxidase dysfunction in oxidative stress. Free. Radic. Biol. Med. 2012, 53, 1252–1263. [Google Scholar]
- Musatov, A.; Carroll, C.A.; Liu, Y.C.; Henderson, G.I.; Weintraub, S.T.; Robinson, N.C. Identification of bovine heart cytochrome c oxidase subunits modified by the lipid peroxidation product 4-hydroxy-2-nonenal. Biochemistry 2002, 41, 8212–8220. [Google Scholar] [CrossRef] [PubMed]
- Estaquier, J.; Vallette, F.; Vayssiere, J.L.; Mignotte, B. The mitochondrial pathways of apoptosis. Adv. Exp. Med. Biol. 2012, 942, 157–183. [Google Scholar] [PubMed]
- Yajima, N.; Masuda, M.; Miyazaki, M.; Nakajima, N.; Chien, S.; Shyy, J. Oxidative stress is involved in the development of experimental abdominal aortic aneurysm: A study of the transcription profile with complementary DNA microarray. J. Vasc. Surg. 2002, 36, 379–385. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Shi, J.J.; Li, Y.J.; Wei, L. Cardiomyocyte death in doxorubicin-induced cardiotoxicity. Arch. Immuno. Ther. Exp. 2009, 57, 435–445. [Google Scholar] [CrossRef]
- Gong, Y.-Y.; Liu, Y.-Y.; Li, J.; Su, L.; Yu, S.; Zhu, X.-N.; Cao, X.-P.; Xiao, H.-P. Hypermethylation of Cox5a promoter is associated with mitochondrial dysfunction in skeletal muscle of high fat diet-induced insulin resistant rats. PLoS ONE 2014, 9, e113784. [Google Scholar] [CrossRef]
- Varga, E.; Nagy, N.; Lazar, J.; Czifra, G.; Bak, I.; Biro, T.; Tosaki, A. Inhibition of ischemia/reperfusion-induced damage by dexamethasone in isolated working rat hearts: The role of cytochrome c release. Life Sci. 2004, 75, 2411–2423. [Google Scholar] [CrossRef]
- Mocanu, M.M.; Baxter, G.F.; Yellon, D.M. Caspase inhibition and limitation of myocardial infarct size: Protection against lethal reperfusion injury. Br. J. Pharmacol. 2000, 130, 197–200. [Google Scholar] [CrossRef]
- Kovacs, P.; Bak, I.; Szendrei, L.; Vecsernyes, M.; Varga, E.; Blasig, I.E.; Tosaki, A. Non-specific caspase inhibition reduces infarct size and improves post-ischaemic recovery in isolated ischaemic/reperfused rat hearts. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2001, 364, 501–507. [Google Scholar] [CrossRef]
- Tokarska-Schlattner, M.; Zaugg, M.; Zuppinger, C.; Wallimann, T.; Schlattner, U. New insights into doxorubicin-induced cardiotoxicity: The critical role of cellular energetics. J. Mol. Cell. Cardiol. 2006, 41, 389–405. [Google Scholar] [CrossRef]
- McLean, B.A.; Patel, V.B.; Zhabyeyev, P.; Chen, X.; Basu, R.; Wang, F.; Shah, S.; Vanhaesebroeck, B.; Oudit, G.Y. PI3Kα Pathway Inhibition With Doxorubicin Treatment Results in Distinct Biventricular Atrophy and Remodeling With Right Ventricular Dysfunction. J. Am. Heart Assoc. 2019, 8, e010961. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Koide, M.; Akakabe, Y.; Matsuo, K.; Shimoda, Y.; Soma, Y.; Ogata, T.; Ueyama, T.; Matoba, S.; Yamada, H.; et al. Manipulation of cardiac phosphatidylinositol 3-kinase (PI3K)/Akt signaling by apoptosis regulator through modulating IAP expression (ARIA) regulates cardiomyocyte death during doxorubicin-induced cardiomyopathy. J. Biol. Chem. 2014, 289, 2788–2800. [Google Scholar] [CrossRef] [PubMed]
- Curtis, M.J.; Alexander, S.P.H.; Cirino, G.; George, C.H.; Kendall, D.A.; Insel, P.A.; Izzo, A.A.; Ji, Y.; Panettieri, R.A.; Patel, H.H.; et al. Planning experiments: Updated guidance on experimental design and analysis and their reporting III. Br. J. Pharmacol. 2022, 179, 3907–3913. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, P.; Lu, H.; Wu, Y.; Lu, D.; Li, C.; Yang, X.; Chen, Z.; Qian, J.; Ge, J. COX5A Alleviates Doxorubicin-Induced Cardiotoxicity by Suppressing Oxidative Stress, Mitochondrial Dysfunction and Cardiomyocyte Apoptosis. Int. J. Mol. Sci. 2023, 24, 10400. https://doi.org/10.3390/ijms241210400
Zhang P, Lu H, Wu Y, Lu D, Li C, Yang X, Chen Z, Qian J, Ge J. COX5A Alleviates Doxorubicin-Induced Cardiotoxicity by Suppressing Oxidative Stress, Mitochondrial Dysfunction and Cardiomyocyte Apoptosis. International Journal of Molecular Sciences. 2023; 24(12):10400. https://doi.org/10.3390/ijms241210400
Chicago/Turabian StyleZhang, Peipei, Hao Lu, Yuan Wu, Danbo Lu, Chenguang Li, Xiangdong Yang, Zhangwei Chen, Juying Qian, and Junbo Ge. 2023. "COX5A Alleviates Doxorubicin-Induced Cardiotoxicity by Suppressing Oxidative Stress, Mitochondrial Dysfunction and Cardiomyocyte Apoptosis" International Journal of Molecular Sciences 24, no. 12: 10400. https://doi.org/10.3390/ijms241210400
APA StyleZhang, P., Lu, H., Wu, Y., Lu, D., Li, C., Yang, X., Chen, Z., Qian, J., & Ge, J. (2023). COX5A Alleviates Doxorubicin-Induced Cardiotoxicity by Suppressing Oxidative Stress, Mitochondrial Dysfunction and Cardiomyocyte Apoptosis. International Journal of Molecular Sciences, 24(12), 10400. https://doi.org/10.3390/ijms241210400