
Cholesterol, Amyloid Beta, Fructose, and LPS Influence ROS and ATP Concentrations and the Phagocytic Capacity of HMC3 Human Microglia Cell Line

 ,
,  , , ,
, , ,
Abstract
1. Introduction
2. Results
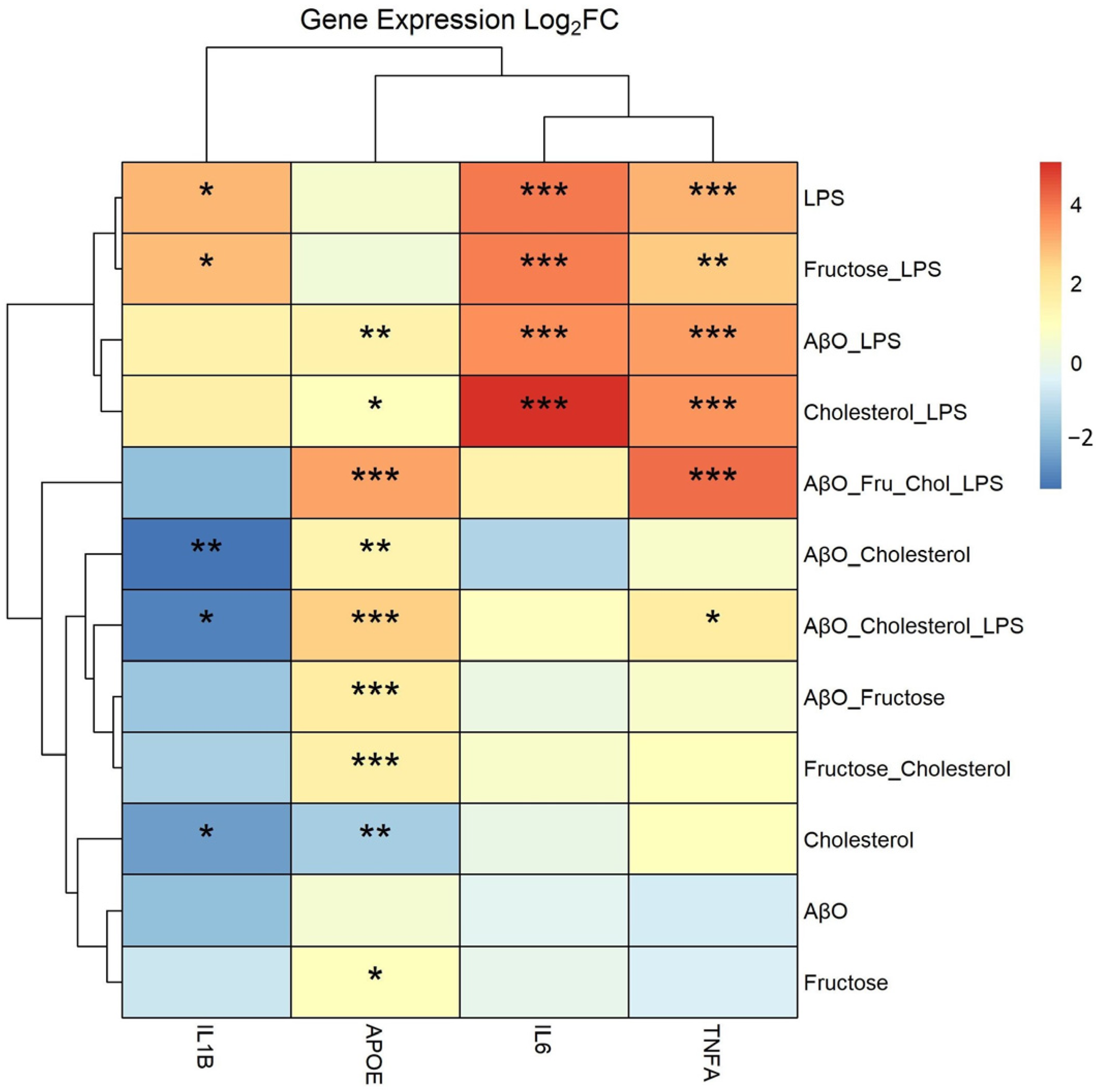
2.1. Effects of Treatments on Expression of Pro-Inflammatory Cytokine Genes and APOE in HMC3 Microglia
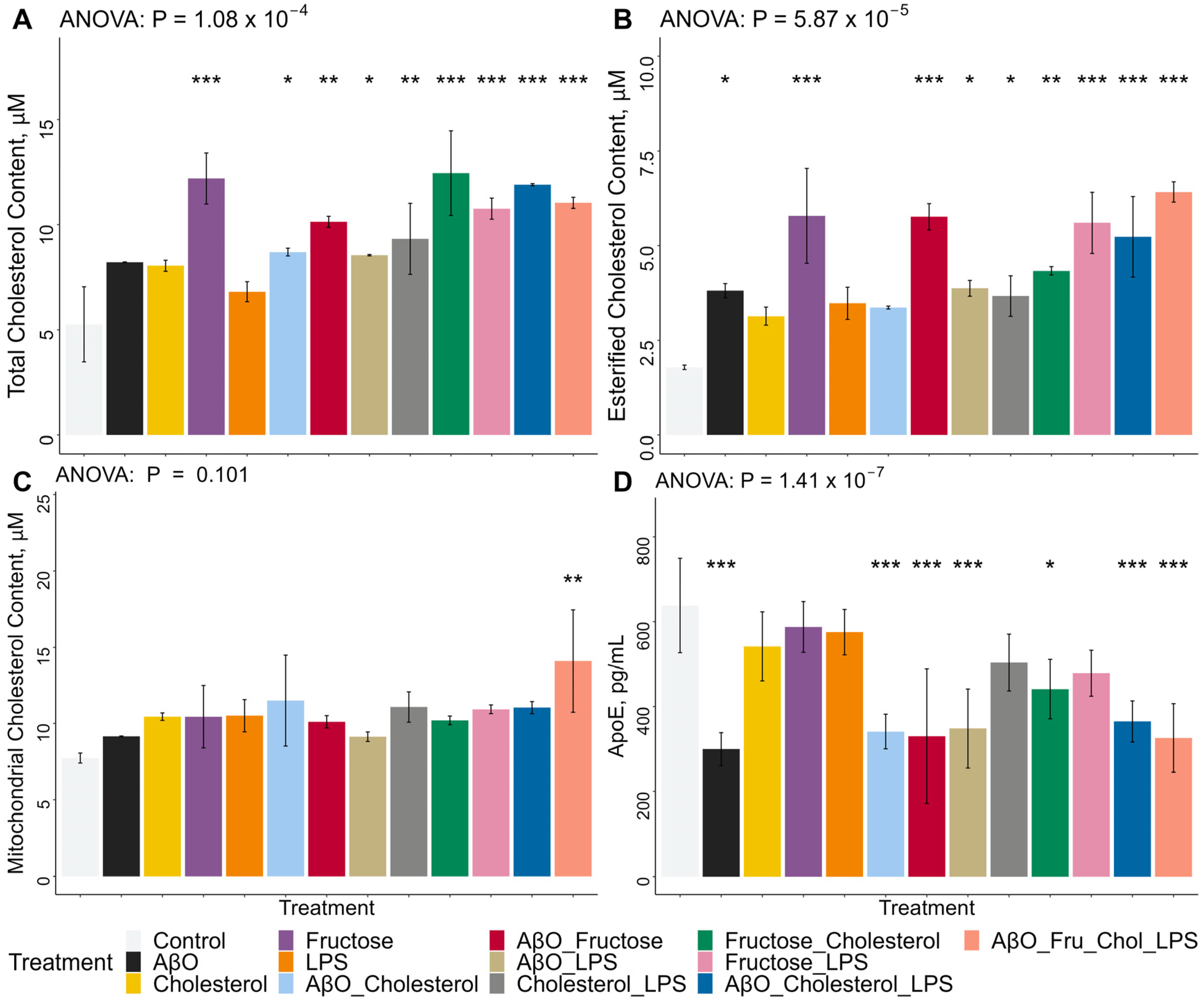
2.2. Effects of Treatments on Whole-Cell Total and Esterified Cholesterol Concentrations, Mitochondrial Total Cholesterol Concentrations and ApoE Secretion in HMC3 Microglia
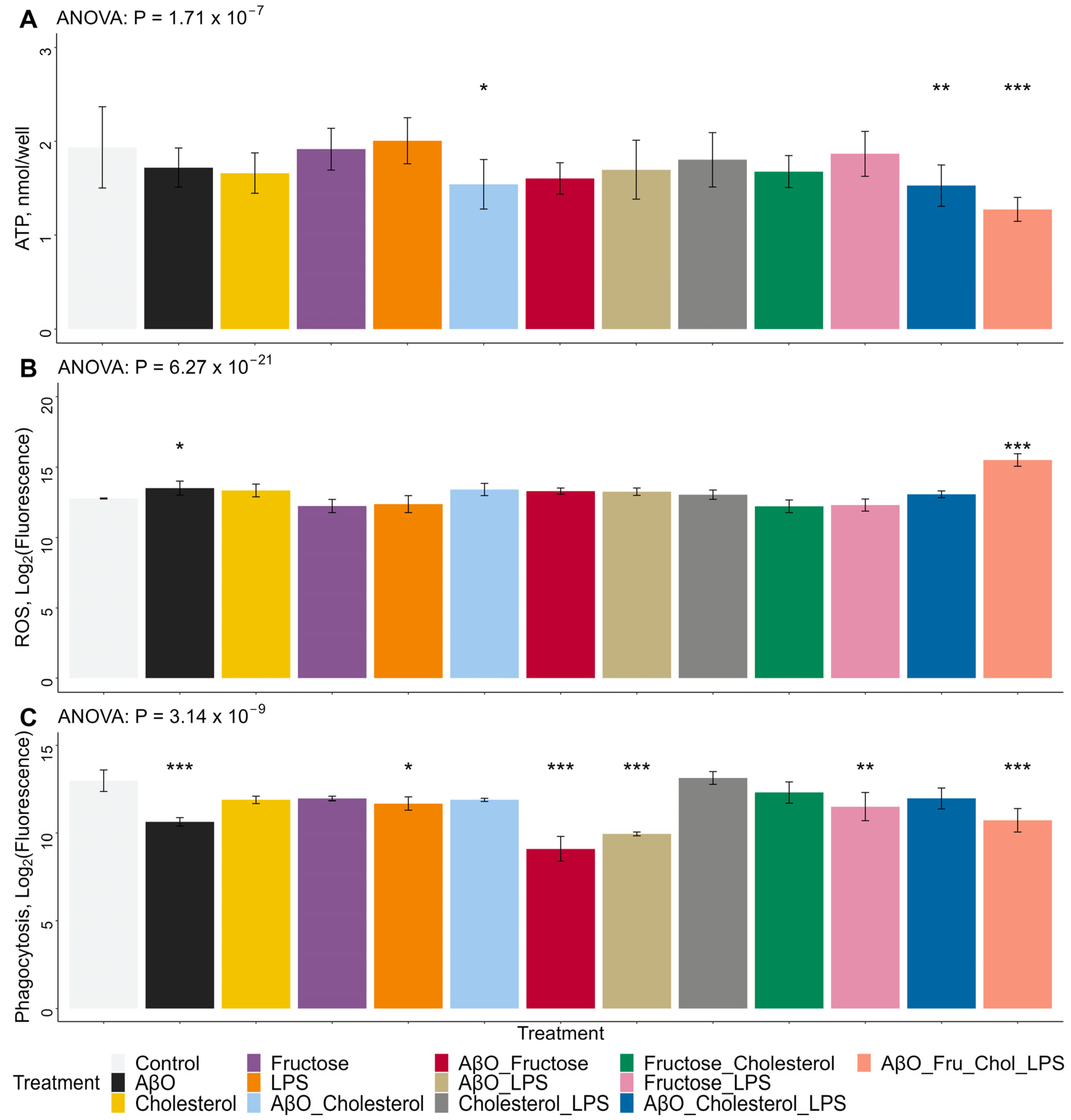
2.3. Effects of Treatments on HMC3 Microglia ROS and ATP Concentrations and Phagocytic Activity
2.4. Effects of Treatments on HMC3 Microglia Morphology
3. Discussion
4. Materials and Methods
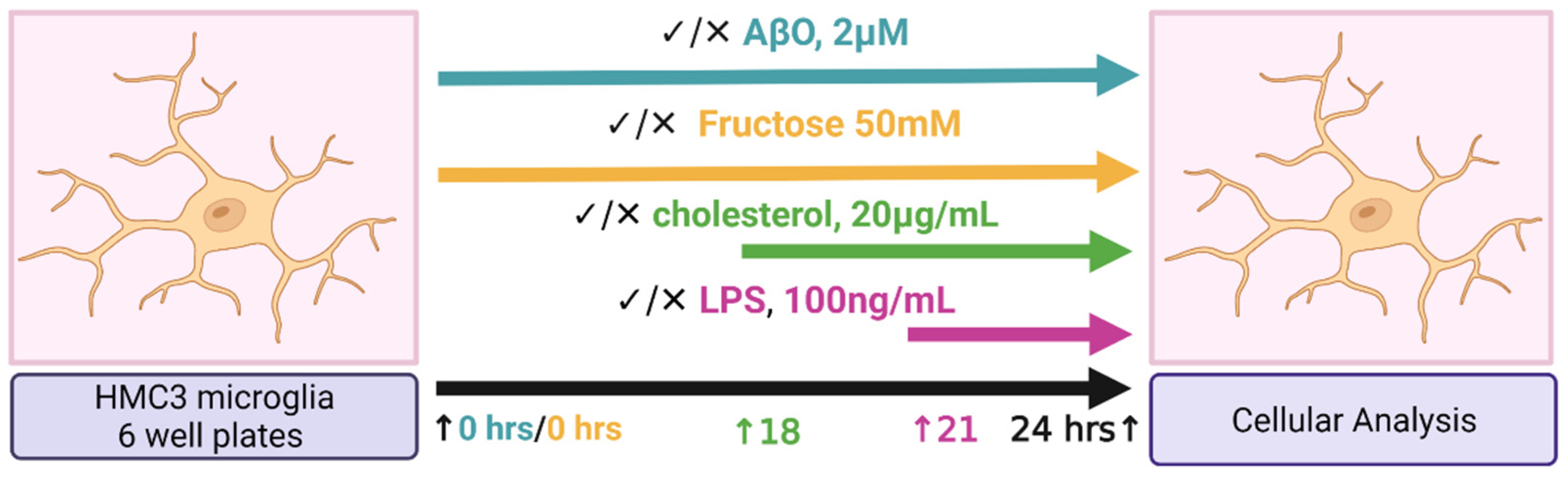
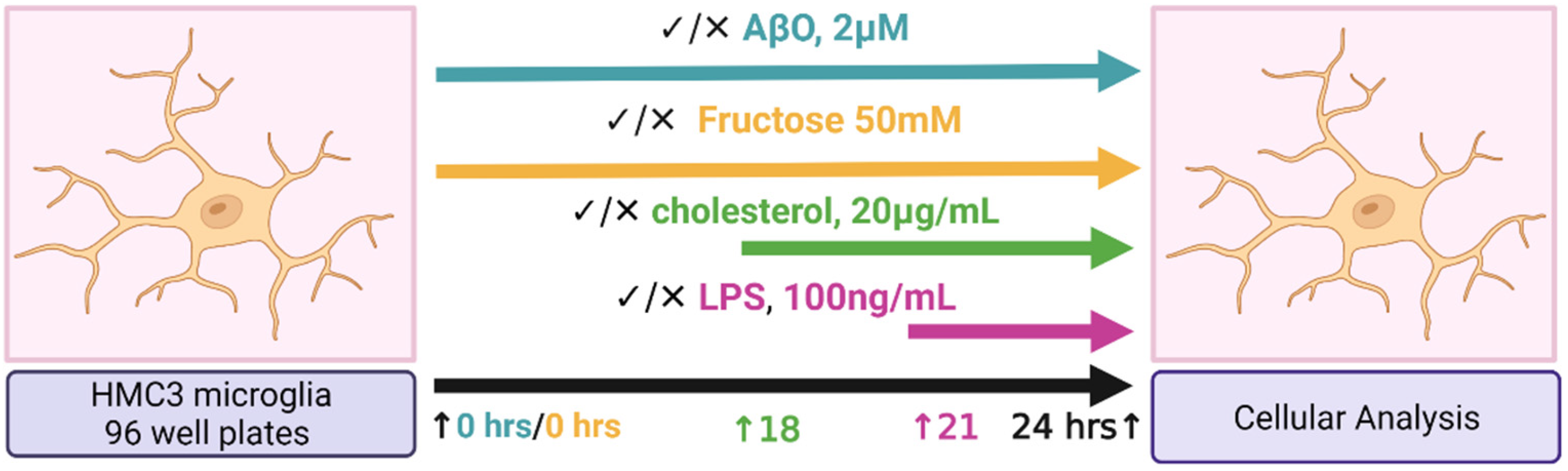
4.1. Study Design
4.2. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain—Implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef]
- Ries, M.; Sastre, M. Mechanisms of Aβ Clearance and Degradation by Glial Cells. Front. Aging Neurosci. 2016, 8, 160. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef]
- Dello Russo, C.; Cappoli, N.; Coletta, I.; Mezzogori, D.; Paciello, F.; Pozzoli, G.; Navarra, P.; Battaglia, A. The human microglial HMC3 cell line: Where do we stand? A systematic literature review. J. Neuroinflamm. 2018, 15, 259. [Google Scholar] [CrossRef]
- Smith, A.M.; Dragunow, M. The human side of microglia. Trends Neurosci. 2014, 37, 125–135. [Google Scholar] [CrossRef]
- Galatro, T.F.; Holtman, I.R.; Lerario, A.M.; Vainchtein, I.D.; Brouwer, N.; Sola, P.R.; Veras, M.M.; Pereira, T.F.; Leite, R.E.P.; Möller, T.; et al. Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat. Neurosci. 2017, 20, 1162–1171. [Google Scholar] [CrossRef]
- Gosselin, D.; Skola, D.; Coufal, N.G.; Holtman, I.R.; Schlachetzki, J.C.M.; Sajti, E.; Jaeger, B.N.; O’Connor, C.; Fitzpatrick, C.; Pasillas, M.P.; et al. An environment-dependent transcriptional network specifies human microglia identity. Science 2017, 356, eaal3222. [Google Scholar] [CrossRef]
- Friedman, B.A.; Srinivasan, K.; Ayalon, G.; Meilandt, W.J.; Lin, H.; Huntley, M.A.; Cao, Y.; Lee, S.-H.; Haddick, P.C.G.; Ngu, H.; et al. Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep. 2018, 22, 832–847. [Google Scholar] [CrossRef] [PubMed]
- Bohlen, C.J.; Bennett, F.C.; Tucker, A.F.; Collins, H.Y.; Mulinyawe, S.B.; Barres, B.A. Diverse Requirements for Microglial Survival, Specification, and Function Revealed by Defined-Medium Cultures. Neuron 2017, 94, 759–773.e8. [Google Scholar] [CrossRef] [PubMed]
- Dawson, T.M.; Golde, T.E.; Lagier-Tourenne, C. Animal models of neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1370–1379. [Google Scholar] [CrossRef] [PubMed]
- Hasselmann, J.; Coburn, M.A.; England, W.; Figueroa Velez, D.X.; Kiani Shabestari, S.; Tu, C.H.; McQuade, A.; Kolahdouzan, M.; Echeverria, K.; Claes, C.; et al. Development of a Chimeric Model to Study and Manipulate Human Microglia In Vivo. Neuron 2019, 103, 1016–1033.e10. [Google Scholar] [CrossRef]
- Ueda, Y.; Gullipalli, D.; Song, W.-C. Modeling complement-driven diseases in transgenic mice: Values and limitations. Immunobiology 2016, 221, 1080–1090. [Google Scholar] [CrossRef]
- Bassil, R.; Shields, K.; Granger, K.; Zein, I.; Ng, S.; Chih, B. Improved modeling of human AD with an automated culturing platform for iPSC neurons, astrocytes and microglia. Nat. Commun. 2021, 12, 5220. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Q. Cholesterol metabolism and homeostasis in the brain. Protein Cell 2015, 6, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-P.; Tang, Y.; Zhou, S.; Toh, B.H.; McLean, C.; Li, H. Cholesterol involvement in the pathogenesis of neurodegenerative diseases. Mol. Cell. Neurosci. 2010, 43, 33–42. [Google Scholar] [CrossRef]
- Puglielli, L.; Tanzi, R.E.; Kovacs, D.M. Alzheimer’s disease: The cholesterol connection. Nat. Neurosci. 2003, 6, 345–351. [Google Scholar] [CrossRef]
- Simons, M.; Keller, P.; De Strooper, B.; Beyreuther, K.; Dotti, C.G.; Simons, K. Cholesterol depletion inhibits the generation of β-amyloid in hippocampal neurons. Proc. Natl. Acad. Sci. USA 1998, 95, 6460–6464. [Google Scholar] [CrossRef] [PubMed]
- Frears, E.R.; Stephens, D.J.; Walters, C.E.; Davies, H.; Austen, B.M. The role of cholesterol in the biosynthesis of β-amyloid. NeuroReport 1999, 10, 1699–1705. [Google Scholar] [CrossRef] [PubMed]
- Fassbender, K.; Simons, M.; Bergmann, C.; Stroick, M.; Lütjohann, D.; Keller, P.; Runz, H.; Kühl, S.; Bertsch, T.; von Bergmann, K.; et al. Simvastatin strongly reduces levels of Alzheimer’s disease β-amyloid peptides Aβ42 and Aβ40 in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2001, 98, 5856–5861. [Google Scholar] [CrossRef]
- Zareba, J.; Peri, F. Microglial ‘fat shaming’ in development and disease. Curr. Opin. Cell Biol. 2021, 73, 105–109. [Google Scholar] [CrossRef]
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K.; Hägg, S.; Athanasiu, L.; et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 2019, 51, 404–413. [Google Scholar] [CrossRef]
- Foley, P. Lipids in Alzheimer’s disease: A century-old story. Biochim. Biophys. Acta BBA—Mol. Cell Biol. Lipids 2010, 1801, 750–753. [Google Scholar] [CrossRef]
- Shibuya, Y.; Chang, C.C.Y.; Huang, L.-H.; Bryleva, E.Y.; Chang, T.-Y. Inhibiting ACAT1/SOAT1 in Microglia Stimulates Autophagy-Mediated Lysosomal Proteolysis and Increases A 1-42 Clearance. J. Neurosci. 2014, 34, 14484–14501. [Google Scholar] [CrossRef]
- Lee, C.Y.D.; Tse, W.; Smith, J.D.; Landreth, G.E. Apolipoprotein E Promotes β-Amyloid Trafficking and Degradation by Modulating Microglial Cholesterol Levels. J. Biol. Chem. 2012, 287, 2032–2044. [Google Scholar] [CrossRef]
- Feringa, F.M.; van der Kant, R. Cholesterol and Alzheimer’s Disease; From Risk Genes to Pathological Effects. Front. Aging Neurosci. 2021, 13, 690372. [Google Scholar] [CrossRef]
- Muñoz Herrera, O.M.; Zivkovic, A.M. Microglia and Cholesterol Handling: Implications for Alzheimer’s Disease. Biomedicines 2022, 10, 3105. [Google Scholar] [CrossRef]
- Block, M.L. NADPH oxidase as a therapeutic target in Alzheimer’s disease. BMC Neurosci. 2008, 9, S8. [Google Scholar] [CrossRef] [PubMed]
- Sumi, N.; Nishioku, T.; Takata, F.; Matsumoto, J.; Watanabe, T.; Shuto, H.; Yamauchi, A.; Dohgu, S.; Kataoka, Y. Lipopolysaccharide-Activated Microglia Induce Dysfunction of the Blood–Brain Barrier in Rat Microvascular Endothelial Cells Co-Cultured with Microglia. Cell. Mol. Neurobiol. 2010, 30, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jin, S.; Sonobe, Y.; Cheng, Y.; Horiuchi, H.; Parajuli, B.; Kawanokuchi, J.; Mizuno, T.; Takeuchi, H.; Suzumura, A. Interleukin-1β Induces Blood–Brain Barrier Disruption by Downregulating Sonic Hedgehog in Astrocytes. PLoS ONE 2014, 9, e110024. [Google Scholar] [CrossRef] [PubMed]
- Zenaro, E.; Piacentino, G.; Constantin, G. The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 2017, 107, 41–56. [Google Scholar] [CrossRef]
- Lukiw, W.J. Gastrointestinal (GI) Tract Microbiome-Derived Neurotoxins—Potent Neuro-Inflammatory Signals From the GI Tract via the Systemic Circulation Into the Brain. Front. Cell. Infect. Microbiol. 2020, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J.; Arceneaux, L.; Li, W.; Bond, T.; Zhao, Y. Gastrointestinal (GI)-Tract Microbiome Derived Neurotoxins and their Potential Contribution to Inflammatory Neurodegeneration in Alzheimer’s Disease (AD). J. Alzheimer’s Dis. Park. 2021, 11, 525. [Google Scholar]
- Zhao, J.; Bi, W.; Xiao, S.; Lan, X.; Cheng, X.; Zhang, J.; Lu, D.; Wei, W.; Wang, Y.; Li, H.; et al. Neuroinflammation induced by lipopolysaccharide causes cognitive impairment in mice. Sci. Rep. 2019, 9, 5790. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Cong, L.; Lukiw, W.J. Lipopolysaccharide (LPS) Accumulates in Neocortical Neurons of Alzheimer’s Disease (AD) Brain and Impairs Transcription in Human Neuronal-Glial Primary Co-cultures. Front. Aging Neurosci. 2017, 9, 407. [Google Scholar] [CrossRef]
- Zhao, Y.; Cong, L.; Jaber, V.; Lukiw, W.J. Microbiome-Derived Lipopolysaccharide Enriched in the Perinuclear Region of Alzheimer’s Disease Brain. Front. Immunol. 2017, 8, 1064. [Google Scholar] [CrossRef]
- Zhao, Y.; Jaber, V.; Lukiw, W.J. Secretory Products of the Human GI Tract Microbiome and Their Potential Impact on Alzheimer’s Disease (AD): Detection of Lipopolysaccharide (LPS) in AD Hippocampus. Front. Cell. Infect. Microbiol. 2017, 7, 318. [Google Scholar] [CrossRef]
- Wang, J.-H.; Wu, Y.-J.; Tee, B.L.; Lo, R.Y. Medical Comorbidity in Alzheimer’s Disease: A Nested Case-Control Study. J. Alzheimers Dis. 2018, 63, 773–781. [Google Scholar] [CrossRef]
- Janson, J.; Laedtke, T.; Parisi, J.E.; O’Brien, P.; Petersen, R.C.; Butler, P.C. Increased Risk of Type 2 Diabetes in Alzheimer Disease. Diabetes 2004, 53, 474–481. [Google Scholar] [CrossRef]
- Kasper Ter Horst; Serlie, M. Fructose Consumption, Lipogenesis, and Non-Alcoholic Fatty Liver Disease. Nutrients 2017, 9, 981. [Google Scholar] [CrossRef]
- Muriel, P.; López-Sánchez, P.; Ramos-Tovar, E. Fructose and the Liver. Int. J. Mol. Sci. 2021, 22, 6969. [Google Scholar] [CrossRef]
- Basciano, H.; Federico, L.; Adeli, K. Fructose, insulin resistance, and metabolic dyslipidemia. Nutr. Metab. 2005, 2, 5. [Google Scholar] [CrossRef]
- Jiménez-Maldonado, A.; Ying, Z.; Byun, H.R.; Gomez-Pinilla, F. Short-term fructose ingestion affects the brain independently from establishment of metabolic syndrome. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 24–33. [Google Scholar] [CrossRef]
- Johnson, R.J.; Gomez-Pinilla, F.; Nagel, M.; Nakagawa, T.; Rodriguez-Iturbe, B.; Sanchez-Lozada, L.G.; Tolan, D.R.; Lanaspa, M.A. Cerebral Fructose Metabolism as a Potential Mechanism Driving Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12, 560865. [Google Scholar] [CrossRef]
- Chiozzi, P.; Sarti, A.C.; Sanz, J.M.; Giuliani, A.L.; Adinolfi, E.; Vultaggio-Poma, V.; Falzoni, S.; Di Virgilio, F. Amyloid β-dependent mitochondrial toxicity in mouse microglia requires P2X7 receptor expression and is prevented by nimodipine. Sci. Rep. 2019, 9, 6475. [Google Scholar] [CrossRef]
- Rahman, M.M.; Lendel, C. Extracellular protein components of amyloid plaques and their roles in Alzheimer’s disease pathology. Mol. Neurodegener. 2021, 16, 59. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Butovsky, O.; Weiner, H.L. Microglial signatures and their role in health and disease. Nat. Rev. Neurosci. 2018, 19, 622–635. [Google Scholar] [CrossRef] [PubMed]
- Arboleda-Velasquez, J.F.; Lopera, F.; O’Hare, M.; Delgado-Tirado, S.; Marino, C.; Chmielewska, N.; Saez-Torres, K.L.; Amarnani, D.; Schultz, A.P.; Sperling, R.A.; et al. Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: A case report. Nat. Med. 2019, 25, 1680–1683. [Google Scholar] [CrossRef] [PubMed]
- Panza, F.; Lozupone, M.; Logroscino, G.; Imbimbo, B.P. A critical appraisal of amyloid-β-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2019, 15, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Pons, V.; Rivest, S. Targeting Systemic Innate Immune Cells as a Therapeutic Avenue for Alzheimer Disease. Pharmacol. Rev. 2022, 74, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.-L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 2018, 98, 1141–1154.e7. [Google Scholar] [CrossRef]
- Olah, M.; Patrick, E.; Villani, A.-C.; Xu, J.; White, C.C.; Ryan, K.J.; Piehowski, P.; Kapasi, A.; Nejad, P.; Cimpean, M.; et al. A transcriptomic atlas of aged human microglia. Nat. Commun. 2018, 9, 539. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Nielsen, M.; Carcione, T.; Li, S.; Shi, J. Apolipoprotein E regulates mitochondrial function through the PGC-1α-sirtuin 3 pathway. Aging 2019, 11, 11148–11156. [Google Scholar] [CrossRef] [PubMed]
- Konttinen, H.; Cabral-da-Silva, M.e.C.; Ohtonen, S.; Wojciechowski, S.; Shakirzyanova, A.; Caligola, S.; Giugno, R.; Ishchenko, Y.; Hernández, D.; Fazaludeen, M.F.; et al. PSEN1ΔE9, APPswe, and APOE4 Confer Disparate Phenotypes in Human iPSC-Derived Microglia. Stem Cell Rep. 2019, 13, 669–683. [Google Scholar] [CrossRef]
- Jena, P.K.; Sheng, L.; Nguyen, M.; Di Lucente, J.; Hu, Y.; Li, Y.; Maezawa, I.; Jin, L.-W.; Wan, Y.-J.Y. Dysregulated bile acid receptor-mediated signaling and IL-17A induction are implicated in diet-associated hepatic health and cognitive function. Biomark. Res. 2020, 8, 59. [Google Scholar] [CrossRef]
- Bernier, L.-P.; York, E.M.; MacVicar, B.A. Immunometabolism in the Brain: How Metabolism Shapes Microglial Function. Trends Neurosci. 2020, 43, 854–869. [Google Scholar] [CrossRef]
- Payne, J.; Maher, F.; Simpson, I.; Mattice, L.; Davies, P. Glucose transporter glut 5 expression in microglial cells. Glia 1997, 21, 327–331. [Google Scholar] [CrossRef]
- Kawasaki, T.; Akanuma, H.; Yamanouchi, T. Increased Fructose Concentrations in Blood and Urine in Patients With Diabetes. Diabetes Care 2002, 25, 353–357. [Google Scholar] [CrossRef]
- Stephan, B.C.M.; Wells, J.C.K.; Brayne, C.; Albanese, E.; Siervo, M. Increased Fructose Intake as a Risk Factor For Dementia. J. Gerontol. A Biol. Sci. Med. Sci. 2010, 65A, 809–814. [Google Scholar] [CrossRef]
- Leng, L.; Yuan, Z.; Pan, R.; Su, X.; Wang, H.; Xue, J.; Zhuang, K.; Gao, J.; Chen, Z.; Lin, H.; et al. Microglial hexokinase 2 deficiency increases ATP generation through lipid metabolism leading to β-amyloid clearance. Nat. Metab. 2022, 4, 1287–1305. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; Yoo, E.; Choi, H.I.; An, G.Y.; Chai, J.C.; Lee, Y.S.; Jung, K.H.; Chai, Y.G. The BET inhibitor attenuates the inflammatory response and cell migration in human microglial HMC3 cell line. Sci. Rep. 2021, 11, 8828. [Google Scholar] [CrossRef]
- Nagai, A.; Nakagawa, E.; Hatori, K.; Choi, H.B.; McLarnon, J.G.; Lee, M.A.; Kim, S.U. Generation and Characterization of Immortalized Human Microglial Cell Lines: Expression of Cytokines and Chemokines. Neurobiol. Dis. 2001, 8, 1057–1068. [Google Scholar] [CrossRef]
- Bhatt, D.P.; Rosenberger, T.A. Acetate treatment increases fatty acid content in LPS-stimulated BV2 microglia. Lipids 2014, 49, 621–631. [Google Scholar] [CrossRef]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.-B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef]
- Karperien, A.; Ahammer, H.; Jelinek, H.F. Quantitating the subtleties of microglial morphology with fractal analysis. Front. Cell. Neurosci. 2013, 7, 3. [Google Scholar] [CrossRef]
- Taylor, S.E.; Morganti-Kossmann, C.; Lifshitz, J.; Ziebell, J.M. Rod Microglia: A Morphological Definition. PLoS ONE 2014, 9, e97096. [Google Scholar] [CrossRef]
- Young, K.; Morrison, H. Quantifying Microglia Morphology from Photomicrographs of Immunohistochemistry Prepared Tissue Using ImageJ. J. Vis. Exp. 2018, 136, 57648. [Google Scholar] [CrossRef]
- Sarode, A.; Fan, Y.; Byrnes, A.E.; Hammel, M.; Hura, G.L.; Fu, Y.; Kou, P.; Hu, C.; Hinz, F.I.; Roberts, J.; et al. Predictive high-throughput screening of PEGylated lipids in oligonucleotide-loaded lipid nanoparticles for neuronal gene silencing. Nanoscale Adv. 2022, 4, 2107–2123. [Google Scholar] [CrossRef] [PubMed]
- Clementino, A.R.; Marchi, C.; Pozzoli, M.; Bernini, F.; Zimetti, F.; Sonvico, F. Anti-Inflammatory Properties of Statin-Loaded Biodegradable Lecithin/Chitosan Nanoparticles: A Step Toward Nose-to-Brain Treatment of Neurodegenerative Diseases. Front. Pharmacol. 2021, 12, 716380. [Google Scholar] [CrossRef]
- Park, D.; Xu, G.; Barboza, M.; Shah, I.M.; Wong, M.; Raybould, H.; Mills, D.A.; Lebrilla, C.B. Enterocyte glycosylation is responsive to changes in extracellular conditions: Implications for membrane functions. Glycobiology 2017, 27, 847–860. [Google Scholar] [CrossRef] [PubMed]
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 2018, 173, 1073–1081. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence (Invitrogen, Waltham, MA, USA) |
|---|---|
| IL-1β (Human) | FW: CCACAGACCTTCCAGGAGAATG RV: GTGCAGTTCAGTGATCGTACAGG |
| IL-6 (Human) | FW: CCAGCTATGAACTCCTTCTC RV: GCTTGTTCCTCACATCTCTC |
| TNF-α (Human) | FW: CTCTTCTGCCTGCTGCACTTTG RV: ATGGGCTACAGGCTTGTCACTC |
| β-Actin (Human) | FW: TCAAGATCATTGCTCCTCCTGAG RV: ACATCTGCTGGAAGGTGGACA |
| Gene | Primer Sequence (Bio-Rad, Hercules, CA, USA) |
| ApoE (Human) | qHsaCED0044297 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muñoz Herrera, O.M.; Hong, B.V.; Ruiz Mendiola, U.; Maezawa, I.; Jin, L.-W.; Lebrilla, C.B.; Harvey, D.J.; Zivkovic, A.M. Cholesterol, Amyloid Beta, Fructose, and LPS Influence ROS and ATP Concentrations and the Phagocytic Capacity of HMC3 Human Microglia Cell Line. Int. J. Mol. Sci. 2023, 24, 10396. https://doi.org/10.3390/ijms241210396
Muñoz Herrera OM, Hong BV, Ruiz Mendiola U, Maezawa I, Jin L-W, Lebrilla CB, Harvey DJ, Zivkovic AM. Cholesterol, Amyloid Beta, Fructose, and LPS Influence ROS and ATP Concentrations and the Phagocytic Capacity of HMC3 Human Microglia Cell Line. International Journal of Molecular Sciences. 2023; 24(12):10396. https://doi.org/10.3390/ijms241210396
Chicago/Turabian StyleMuñoz Herrera, Oscar M., Brian V. Hong, Ulises Ruiz Mendiola, Izumi Maezawa, Lee-Way Jin, Carlito B. Lebrilla, Danielle J. Harvey, and Angela M. Zivkovic. 2023. "Cholesterol, Amyloid Beta, Fructose, and LPS Influence ROS and ATP Concentrations and the Phagocytic Capacity of HMC3 Human Microglia Cell Line" International Journal of Molecular Sciences 24, no. 12: 10396. https://doi.org/10.3390/ijms241210396
APA StyleMuñoz Herrera, O. M., Hong, B. V., Ruiz Mendiola, U., Maezawa, I., Jin, L.-W., Lebrilla, C. B., Harvey, D. J., & Zivkovic, A. M. (2023). Cholesterol, Amyloid Beta, Fructose, and LPS Influence ROS and ATP Concentrations and the Phagocytic Capacity of HMC3 Human Microglia Cell Line. International Journal of Molecular Sciences, 24(12), 10396. https://doi.org/10.3390/ijms241210396