
Mitochondrial Regulation of Ferroptosis in Cancer Therapy


Abstract
1. Introduction
2. Hallmarks of Ferroptosis
2.1. Morphological Hallmarks
2.2. Biochemical Hallmarks
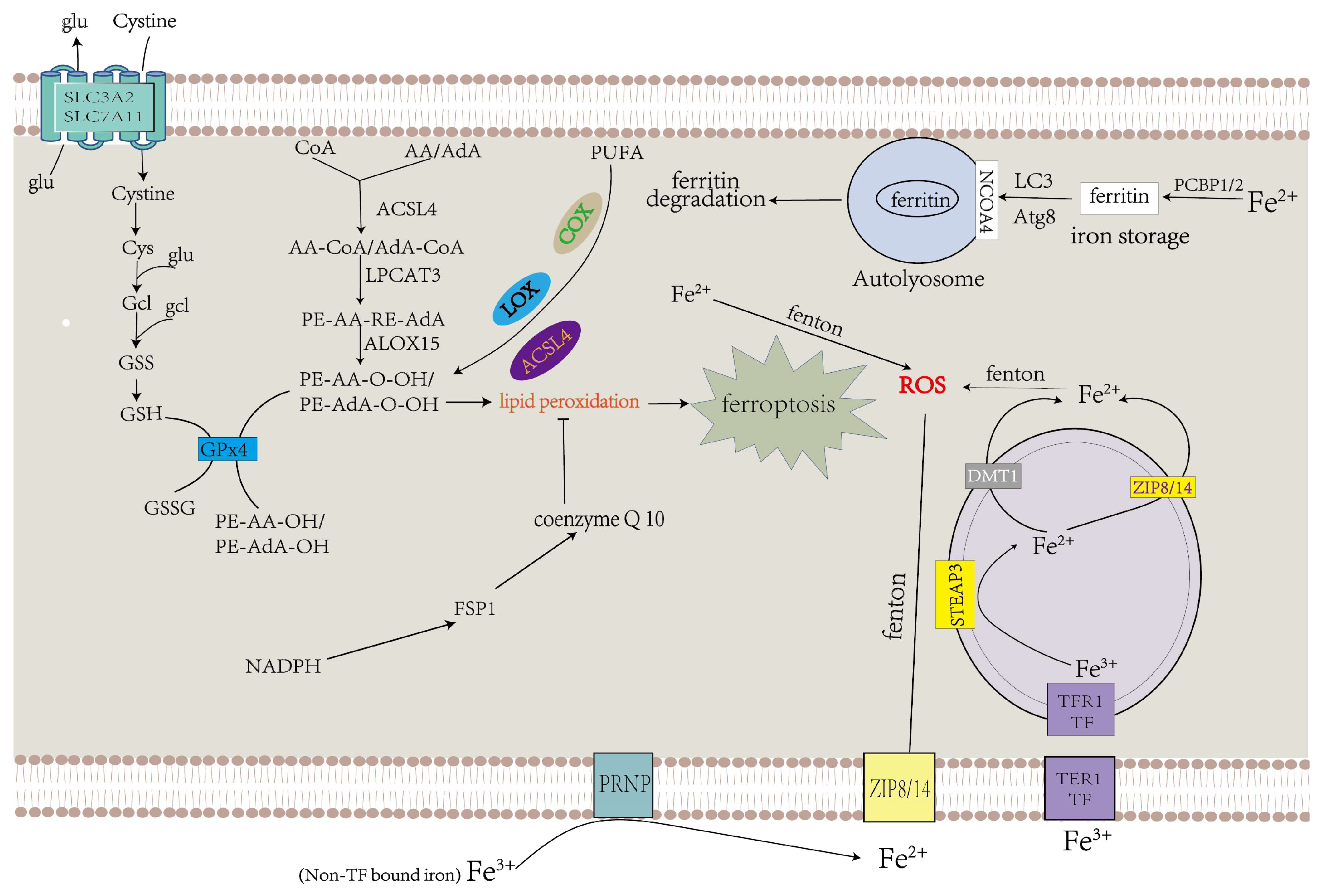
2.2.1. Iron Accumulation
2.2.2. Lipid Peroxidation
2.2.3. Inhibition of Antioxidant Systems
2.3. Protein Concentration Changes
2.3.1. GPX4
2.3.2. P53
2.3.3. ACSL4
3. Mechanisms of Ferroptosis
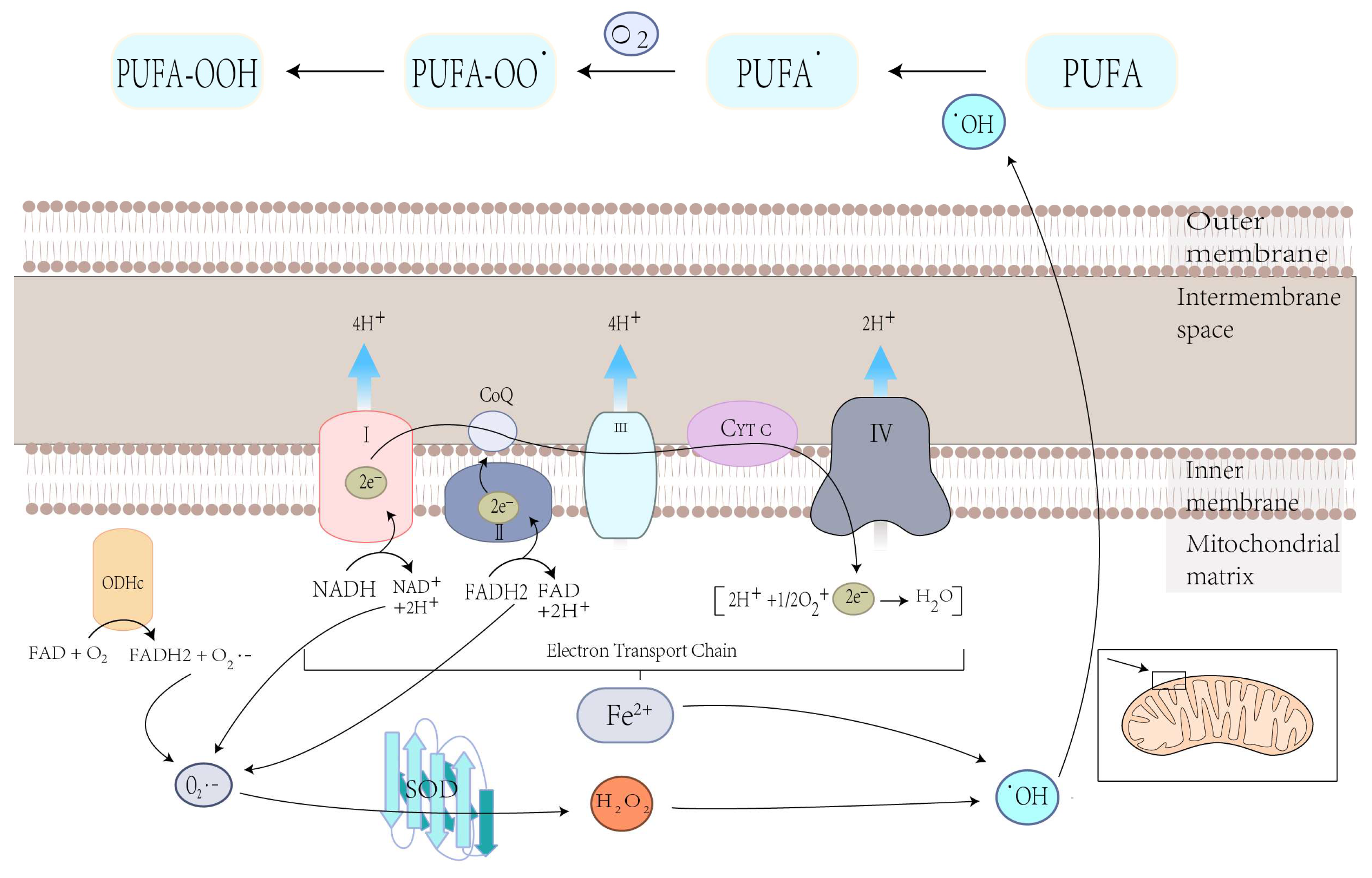
3.1. Mitochondrial Mechanisms of Ferroptosis
3.2. GPX4 and Lipid Metabolism Mechanisms in Ferroptosis

3.3. Iron Metabolism in Ferroptosis

{kind=link}
{kind=link}
{kind=link}
| Name | Function | Reference |
|---|---|---|
| TFR1 | A membrane receptor that internalizes iron-bound transferrin through receptor-mediated endocytosis | [78] |
| FtH | Primary iron-storage protein that degrades to release iron under the effect of lysosomes | [79] |
| ACSF2 | Increases the content of intracellular lipids to promote ferroptosis | [27] |
| ZIP8/14 | Divalent metal transporter that transports non-transferrin-bound iron across the cell membrane | [80] |
| DMT1 | Involved in transport of iron (Fe2+) from endosomes to the cytoplasm | [81] |
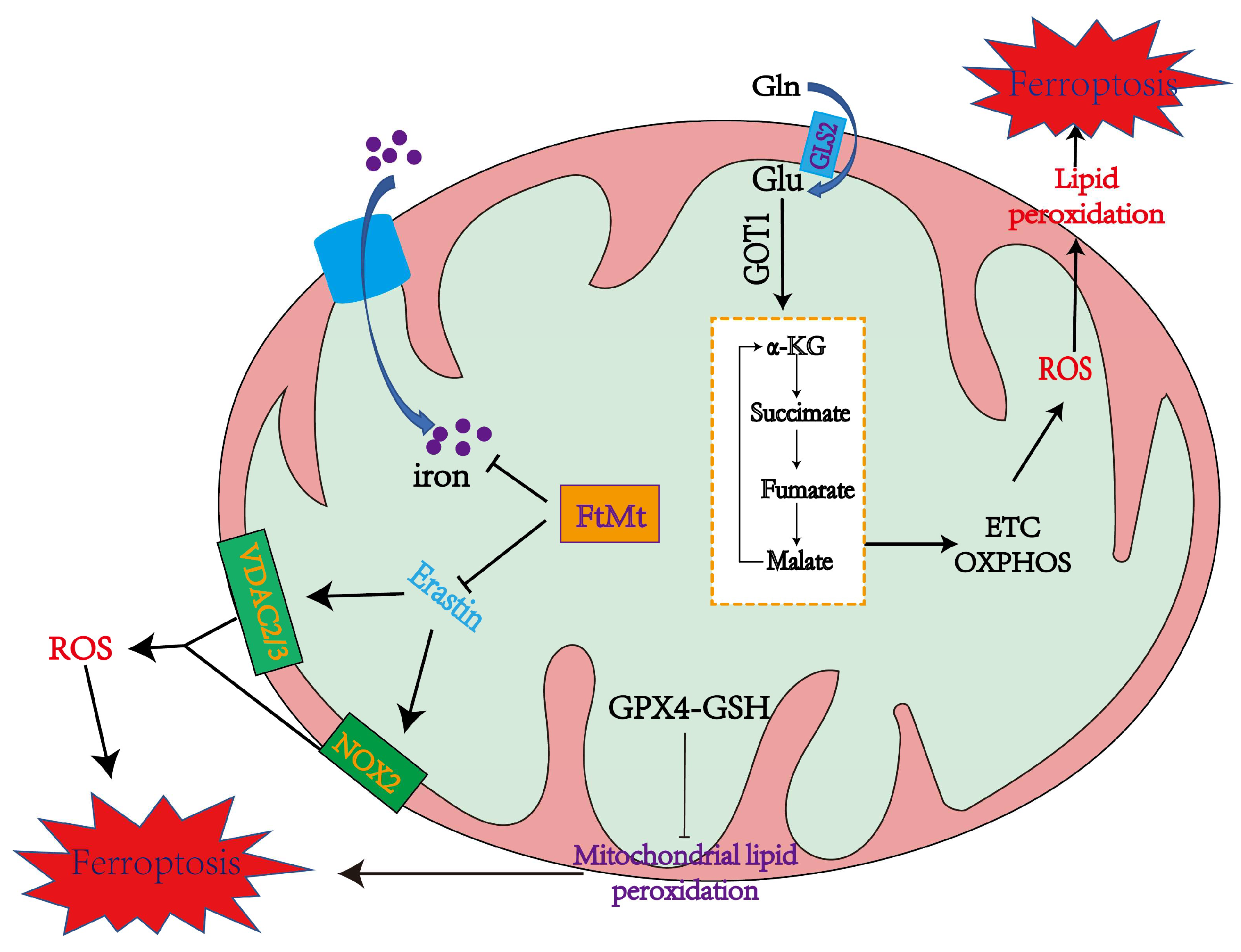
4. Mitochondrial Function in Ferroptosis
4.1. The Role of Mitochondria in Inducing Ferroptosis
4.1.1. Mitochondrial ROS Production and Lipid Peroxidation in Ferroptosis
4.1.2. The Mitochondrial TCA Cycle May Cause Ferroptosis
4.1.3. Iron Overload in Mitochondria Promotes Ferroptosis
4.2. The Role of Mitochondria in Inhibiting Ferroptosis
5. Ferroptosis Inducer
5.1. Ferroptosis Inducers Targeting Different Mechanisms
5.1.1. Ferroptosis Inducers Targeting the Production of ROS via Iron Accumulation
5.1.2. Ferroptosis Inducers Targeting System Xc−
5.1.3. Ferroptosis Inducers Targeting the Consumption of GSH
5.1.4. Ferroptosis Inducers Targeting GPX4
5.1.5. Ferroptosis Inducers Targeting DHODH
5.1.6. Multiple Targeted Ferroptosis Inducers
5.2. Ferroptosis-Based Combinational Cancer Therapy
5.3. Nanomedicines Treat Tumors via Inducing Ferroptosis
| Names | Targets | Mechanisms | Cancers | References |
|---|---|---|---|---|
| Erastin | System Xc− | Inhibition of the cystine–glutamate antiporter | Meningioma, Diffuse large B-cell lymphoma (DLBCL) and Renal cell carcinoma | [29,105,106] |
| Sulfasalazine | System Xc− | Inhibition of the cystine–glutamate antiporter | Endometrial cancer | [107,108] |
| Bavachin | STAT3/p53/SLC7A11 axis | Upregulating the expression of P53 and downregulating the expression of SLC7A11 | Osteosarcoma | [109] |
| Metformin | SLC7A11 | Inhibiting SLC7A11 acylation from reducing its stability | Breast cancer cells | [110] |
| Artesunate | GSH | Decreasing cellular GSH levels and increasing lipid ROS levels | Head and neck cancer | [112] |
| ML-162 | GPX4 | Reducing the activity of GPX4 | Thyroid cancer | [113] |
| RSL3 | GPX4 | Decreasing the expression of GPX4 | DLBCL and renal cell carcinoma | [29] |
| Brequinar | DHODH | Damaging the ferroptosis defense systems by inhibiting mitochondrial DHODH | GPX4low tumor | [96] |
| DHA | AMPK/ mTOR/p70S6k signaling pathway | Degradation of ferritin and accumulation of ROS | Acute myeloid leukemia (AML) | [102] |
| Auriculasin | Fe2+ | Accumulation of Fe2+ to increase the production of ROS | Colorectal cancer | [103] |
| Alloimperatorin | SLC7A11 and GPX4 | Accumulation of Fe2+, ROS, and MDA | Breast cancer cell | [114] |
| Bupivacaine | xCT and GPX4 | Restraining the expression of xCT and GPX4 | Bladder cancer | [115] |
| ipGdIO-Dox | Fe2+ | Releasing abundant Fe(II) ions and then catalyzing H2O2 into highly toxic OH• | [118] | |
| ZVI-NP | AMPK/mTOR signaling pathway | Enhanced GSK3 β/β-Trcp-dependent Nrf2 degradation | Lung cancer | [119] |
6. Ferroptosis Inhibitor
7. Conclusions and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Soerjomataram, I.; Bray, F. Planning for tomorrow: Global cancer incidence and the role of prevention 2020–2070. Nat. Rev. Clin. Oncol. 2021, 18, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Malik, D.; Mahendiratta, S.; Kaur, H.; Medhi, B. Futuristic approach to cancer treatment. Gene 2021, 805, 145906. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Gu, T.; Patel, S.; Bode, A.M.; Lee, M.H.; Dong, Z. CRISPR/Cas9—An evolving biological tool kit for cancer biology and oncology. NPJ Precis. Oncol. 2019, 3, 8. [Google Scholar] [CrossRef]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Broadening horizons: The role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296. [Google Scholar] [CrossRef]
- Eagle, H. The specific amino acid requirements of a human carcinoma cell (Stain HeLa) in tissue culture. J. Exp. Med. 1955, 102, 37–48. [Google Scholar] [CrossRef]
- Bannai, S.; Tsukeda, H.; Okumura, H. Effect of antioxidants on cultured human diploid fibroblasts exposed to cystine-free medium. Biochem. Biophys. Res. Commun. 1977, 74, 1582–1588. [Google Scholar] [CrossRef] [PubMed]
- Murphy, T.H.; Miyamoto, M.; Sastre, A.; Schnaar, R.L.; Coyle, J.T. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron 1989, 2, 1547–1558. [Google Scholar] [CrossRef]
- Tan, S.; Schubert, D.; Maher, P. Oxytosis: A novel form of programmed cell death. Curr. Top. Med. Chem. 2001, 1, 497–506. [Google Scholar]
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003, 3, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Stockwell, B.R. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 2018, 16, e2006203. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.L.; Goncalves, R.L.; Hey-Mogensen, M.; Yadava, N.; Bunik, V.I.; Brand, M.D. The 2-oxoacid dehydrogenase complexes in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex I. J. Biol. Chem. 2014, 289, 8312–8325. [Google Scholar] [CrossRef]
- Wu, H.; Wang, F.; Ta, N.; Zhang, T.; Gao, W. The Multifaceted Regulation of Mitochondria in Ferroptosis. Life 2021, 11, 222. [Google Scholar] [CrossRef]
- Conrad, M.; Angeli, J.P.; Vandenabeele, P.; Stockwell, B.R. Regulated necrosis: Disease relevance and therapeutic opportunities. Nat. Rev. Drug. Discov. 2016, 15, 348–366. [Google Scholar] [CrossRef] [PubMed]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef]
- Liu, J.; Kuang, F.; Kroemer, G.; Klionsky, D.J.; Kang, R.; Tang, D. Autophagy-Dependent Ferroptosis: Machinery and Regulation. Cell. Chem. Biol. 2020, 27, 420–435. [Google Scholar] [CrossRef]
- Zhou, B.; Liu, J.; Kang, R.; Klionsky, D.J.; Kroemer, G.; Tang, D. Ferroptosis is a type of autophagy-dependent cell death. Semin. Cancer Biol. 2020, 66, 89–100. [Google Scholar] [CrossRef]
- Wang, Y.; Kanneganti, T.D. From pyroptosis, apoptosis and necroptosis to PANoptosis: A mechanistic compendium of programmed cell death pathways. Comput. Struct. Biotechnol. J. 2021, 19, 4641–4657. [Google Scholar] [CrossRef]
- Chen, X.; Yu, C.; Kang, R.; Tang, D. Iron Metabolism in Ferroptosis. Front. Cell. Dev. Biol. 2020, 8, 590226. [Google Scholar] [CrossRef]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., 3rd; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef] [PubMed]
- Kuang, F.; Liu, J.; Tang, D.; Kang, R. Oxidative Damage and Antioxidant Defense in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 586578. [Google Scholar] [CrossRef]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef]
- Xie, Y.; Zhu, S.; Song, X.; Sun, X.; Fan, Y.; Liu, J.; Zhong, M.; Yuan, H.; Zhang, L.; Billiar, T.R.; et al. The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep. 2017, 20, 1692–1704. [Google Scholar] [CrossRef] [PubMed]
- Chu, B.; Kon, N.; Chen, D.; Li, T.; Liu, T.; Jiang, L.; Song, S.; Tavana, O.; Gu, W. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat. Cell Biol. 2019, 21, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem. Biophys. Res. Commun. 2016, 478, 1338–1343. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Kraft, V.A.N.; Bezjian, C.T.; Pfeiffer, S.; Ringelstetter, L.; Muller, C.; Zandkarimi, F.; Merl-Pham, J.; Bao, X.; Anastasov, N.; Kossl, J.; et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent. Sci. 2020, 6, 41–53. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Soula, M.; Weber, R.A.; Zilka, O.; Alwaseem, H.; La, K.; Yen, F.; Molina, H.; Garcia-Bermudez, J.; Pratt, D.A.; Birsoy, K. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat. Chem. Biol. 2020, 16, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kroemer, G. Ferroptosis. Curr. Biol. 2020, 30, R1292–R1297. [Google Scholar] [CrossRef] [PubMed]
- Bentinger, M.; Brismar, K.; Dallner, G. The antioxidant role of coenzyme Q. Mitochondrion 2007, 7, S41–S50. [Google Scholar] [CrossRef]
- Cao, J.Y.; Dixon, S.J. Mechanisms of ferroptosis. Cell. Mol. Life Sci. 2016, 73, 2195–2209. [Google Scholar] [CrossRef]
- Sharbeen, G.; McCarroll, J.A.; Akerman, A.; Kopecky, C.; Youkhana, J.; Kokkinos, J.; Holst, J.; Boyer, C.; Erkan, M.; Goldstein, D.; et al. Cancer-Associated Fibroblasts in Pancreatic Ductal Adenocarcinoma Determine Response to SLC7A11 Inhibition. Cancer Res. 2021, 81, 3461–3479. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef]
- Song, X.; Xie, Y.; Kang, R.; Hou, W.; Sun, X.; Epperly, M.W.; Greenberger, J.S.; Tang, D. FANCD2 protects against bone marrow injury from ferroptosis. Biochem. Biophys. Res. Commun. 2016, 480, 443–449. [Google Scholar] [CrossRef]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef]
- Ooko, E.; Saeed, M.E.; Kadioglu, O.; Sarvi, S.; Colak, M.; Elmasaoudi, K.; Janah, R.; Greten, H.J.; Efferth, T. Artemisinin derivatives induce iron-dependent cell death (ferroptosis) in tumor cells. Phytomedicine 2015, 22, 1045–1054. [Google Scholar] [CrossRef] [PubMed]
- Refaat, B.; Abdelghany, A.H.; BaSalamah, M.A.; El-Boshy, M.; Ahmad, J.; Idris, S. Acute and Chronic Iron Overloading Differentially Modulates the Expression of Cellular Iron-homeostatic Molecules in Normal Rat Kidney. J. Histochem. Cytochem. 2018, 66, 825–839. [Google Scholar] [CrossRef]
- Jennis, M.; Kung, C.P.; Basu, S.; Budina-Kolomets, A.; Leu, J.I.; Khaku, S.; Scott, J.P.; Cai, K.Q.; Campbell, M.R.; Porter, D.K.; et al. An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes. Dev. 2016, 30, 918–930. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Kroemer, G.; Tang, D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic. Biol. Med. 2019, 133, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; He, S.; Cai, Q.; Li, F.; Wang, S.; Tao, K.; Xi, Y.; Qin, H.; Gao, G.; Feng, D. Polydatin alleviates traumatic brain injury: Role of inhibiting ferroptosis. Biochem. Biophys. Res. Commun. 2021, 556, 149–155. [Google Scholar] [CrossRef]
- Ma, C.; Li, F.; Luo, H. Prognostic and immune implications of a novel ferroptosis-related ten-gene signature in lung adenocarcinoma. Ann. Transl. Med. 2021, 9, 1058. [Google Scholar] [CrossRef]
- Bogdan, A.R.; Miyazawa, M.; Hashimoto, K.; Tsuji, Y. Regulators of Iron Homeostasis: New Players in Metabolism, Cell Death, and Disease. Trends Biochem. Sci. 2016, 41, 274–286. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Badgley, M.A.; Kremer, D.M.; Maurer, H.C.; DelGiorno, K.E.; Lee, H.J.; Purohit, V.; Sagalovskiy, I.R.; Ma, A.; Kapilian, J.; Firl, C.E.M.; et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 2020, 368, 85–89. [Google Scholar] [CrossRef]
- Qi, F.; Pradhan, R.K.; Dash, R.K.; Beard, D.A. Detailed kinetics and regulation of mammalian 2-oxoglutarate dehydrogenase. BMC Biochem. 2011, 12, 53. [Google Scholar] [CrossRef]
- Levi, S.; Corsi, B.; Bosisio, M.; Invernizzi, R.; Volz, A.; Sanford, D.; Arosio, P.; Drysdale, J. A human mitochondrial ferritin encoded by an intronless gene. J. Biol. Chem. 2001, 276, 24437–24440. [Google Scholar] [CrossRef] [PubMed]
- Santambrogio, P.; Biasiotto, G.; Sanvito, F.; Olivieri, S.; Arosio, P.; Levi, S. Mitochondrial ferritin expression in adult mouse tissues. J. Histochem. Cytochem. 2007, 55, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, A.M.; Sacco, A.; Perrotta, I.D.; Faniello, M.C.; Scalise, M.; Torella, D.; Levi, S.; Costanzo, F.; Biamonte, F. Iron Administration Overcomes Resistance to Erastin-Mediated Ferroptosis in Ovarian Cancer Cells. Front. Oncol. 2022, 12, 868351. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Conrad, M. The Metabolic Underpinnings of Ferroptosis. Cell Metab. 2020, 32, 920–937. [Google Scholar] [CrossRef] [PubMed]
- Bachhawat, A.K.; Yadav, S. The glutathione cycle: Glutathione metabolism beyond the gamma-glutamyl cycle. IUBMB Life 2018, 70, 585–592. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef]
- Chen, Q.; Wang, B.; Ding, H.; Zhang, J.; Li, S. Review: The role of NADP-malic enzyme in plants under stress. Plant Sci. 2019, 281, 206–212. [Google Scholar] [CrossRef]
- Drincovich, M.F.; Casati, P.; Andreo, C.S. NADP-malic enzyme from plants: A ubiquitous enzyme involved in different metabolic pathways. FEBS Lett. 2001, 490, 1–6. [Google Scholar] [CrossRef]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef]
- Zhu, J.; Xiong, Y.; Zhang, Y.; Wen, J.; Cai, N.; Cheng, K.; Liang, H.; Zhang, W. The Molecular Mechanisms of Regulating Oxidative Stress-Induced Ferroptosis and Therapeutic Strategy in Tumors. Oxid. Med. Cell. Longev. 2020, 2020, 8810785. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Yan, J.; Hu, X.; Li, H.; Li, H.; Liu, Q.; Chen, Y.; Zou, Z. Targeting lipid metabolism for ferroptotic cancer therapy. Apoptosis 2023, 28, 81–107. [Google Scholar] [CrossRef] [PubMed]
- Gkouvatsos, K.; Papanikolaou, G.; Pantopoulos, K. Regulation of iron transport and the role of transferrin. Biochim. Biophys. Acta 2012, 1820, 188–202. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.M.; Chua, A.C.; Herbison, C.E.; Olynyk, J.K.; Trinder, D. Liver iron transport. World J. Gastroenterol. 2007, 13, 4725–4736. [Google Scholar] [CrossRef] [PubMed]
- Turcu, A.L.; Versini, A.; Khene, N.; Gaillet, C.; Caneque, T.; Muller, S.; Rodriguez, R. DMT1 Inhibitors Kill Cancer Stem Cells by Blocking Lysosomal Iron Translocation. Chemistry 2020, 26, 7369–7373. [Google Scholar] [CrossRef] [PubMed]
- Pinilla-Tenas, J.J.; Sparkman, B.K.; Shawki, A.; Illing, A.C.; Mitchell, C.J.; Zhao, N.; Liuzzi, J.P.; Cousins, R.J.; Knutson, M.D.; Mackenzie, B. Zip14 is a complex broad-scope metal-ion transporter whose functional properties support roles in the cellular uptake of zinc and nontransferrin-bound iron. Am. J. Physiol. Cell Physiol. 2011, 301, C862–C871. [Google Scholar] [CrossRef]
- Tripathi, A.K.; Haldar, S.; Qian, J.; Beserra, A.; Suda, S.; Singh, A.; Hopfer, U.; Chen, S.G.; Garrick, M.D.; Turner, J.R.; et al. Prion protein functions as a ferrireductase partner for ZIP14 and DMT1. Free Radic. Biol. Med. 2015, 84, 322–330. [Google Scholar] [CrossRef]
- Bagyinszky, E.; Giau, V.V.; Youn, Y.C.; An, S.S.A.; Kim, S. Characterization of mutations in PRNP (prion) gene and their possible roles in neurodegenerative diseases. Neuropsychiatr. Dis. Treat. 2018, 14, 2067–2085. [Google Scholar] [CrossRef]
- Ryskalin, L.; Busceti, C.L.; Biagioni, F.; Limanaqi, F.; Familiari, P.; Frati, A.; Fornai, F. Prion Protein in Glioblastoma Multiforme. Int. J. Mol. Sci. 2019, 20, 5107. [Google Scholar] [CrossRef] [PubMed]
- Wulf, M.A.; Senatore, A.; Aguzzi, A. The biological function of the cellular prion protein: An update. BMC Biol. 2017, 15, 34. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Haldar, S.; Horback, K.; Tom, C.; Zhou, L.; Meyerson, H.; Singh, N. Prion protein regulates iron transport by functioning as a ferrireductase. J. Alzheimers Dis. 2013, 35, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Kajarabille, N.; Latunde-Dada, G.O. Programmed Cell-Death by Ferroptosis: Antioxidants as Mitigators. Int. J. Mol. Sci. 2019, 20, 4968. [Google Scholar] [CrossRef] [PubMed]
- Frey, A.G.; Nandal, A.; Park, J.H.; Smith, P.M.; Yabe, T.; Ryu, M.S.; Ghosh, M.C.; Lee, J.; Rouault, T.A.; Park, M.H.; et al. Iron chaperones PCBP1 and PCBP2 mediate the metallation of the dinuclear iron enzyme deoxyhypusine hydroxylase. Proc. Natl. Acad. Sci. USA 2014, 111, 8031–8036. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Yamamoto, A.; Oshitani-Okamoto, S.; Ohsumi, Y.; Yoshimori, T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 2004, 117 Pt 13, 2805–2812. [Google Scholar] [CrossRef] [PubMed]
- Santana-Codina, N.; Gikandi, A.; Mancias, J.D. The Role of NCOA4-Mediated Ferritinophagy in Ferroptosis. Adv. Exp. Med. Biol. 2021, 1301, 41–57. [Google Scholar] [PubMed]
- Jin, J.; Schorpp, K.; Samaga, D.; Unger, K.; Hadian, K.; Stockwell, B.R. Machine Learning Classifies Ferroptosis and Apoptosis Cell Death Modalities with TfR1 Immunostaining. ACS Chem. Biol. 2022, 17, 654–660. [Google Scholar] [CrossRef]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef]
- van Raaij, S.E.G.; Srai, S.K.S.; Swinkels, D.W.; van Swelm, R.P.L. Iron uptake by ZIP8 and ZIP14 in human proximal tubular epithelial cells. Biometals 2019, 32, 211–226. [Google Scholar] [CrossRef]
- Masaldan, S.; Clatworthy, S.A.S.; Gamell, C.; Meggyesy, P.M.; Rigopoulos, A.T.; Haupt, S.; Haupt, Y.; Denoyer, D.; Adlard, P.A.; Bush, A.I.; et al. Iron accumulation in senescent cells is coupled with impaired ferritinophagy and inhibition of ferroptosis. Redox Biol. 2018, 14, 100–115. [Google Scholar] [CrossRef]
- Neitemeier, S.; Jelinek, A.; Laino, V.; Hoffmann, L.; Eisenbach, I.; Eying, R.; Ganjam, G.K.; Dolga, A.M.; Oppermann, S.; Culmsee, C. BID links ferroptosis to mitochondrial cell death pathways. Redox Biol. 2017, 12, 558–570. [Google Scholar] [CrossRef]
- Lennicke, C.; Cocheme, H.M. Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Mol. Cell 2021, 81, 3691–3707. [Google Scholar] [CrossRef]
- Prieto-Bermejo, R.; Hernandez-Hernandez, A. The Importance of NADPH Oxidases and Redox Signaling in Angiogenesis. Antioxidants 2017, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Stoyanovsky, D.A.; Tyurina, Y.Y.; Shrivastava, I.; Bahar, I.; Tyurin, V.A.; Protchenko, O.; Jadhav, S.; Bolevich, S.B.; Kozlov, A.V.; Vladimirov, Y.A.; et al. Iron catalysis of lipid peroxidation in ferroptosis: Regulated enzymatic or random free radical reaction? Free Radic. Biol. Med. 2019, 133, 153–161. [Google Scholar] [CrossRef]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Wu, L.; Zhang, K.; Wang, H.; Zhang, T.; Gutierrez, L.; O’Connell, D.; Zhang, P.; Li, Y.; Gao, T.; et al. miR-137 regulates ferroptosis by targeting glutamine transporter SLC1A5 in melanoma. Cell Death Differ. 2018, 25, 1457–1472. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Zandkarimi, F.; Zhang, Y.; Meena, J.K.; Kim, J.; Zhuang, L.; Tyagi, S.; Ma, L.; Westbrook, T.F.; Steinberg, G.R.; et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat. Cell Biol. 2020, 22, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Sheftel, A.D.; Zhang, A.S.; Brown, C.; Shirihai, O.S.; Ponka, P. Direct interorganellar transfer of iron from endosome to mitochondrion. Blood 2007, 110, 125–132. [Google Scholar] [CrossRef]
- Das, A.; Nag, S.; Mason, A.B.; Barroso, M.M. Endosome-mitochondria interactions are modulated by iron release from transferrin. J. Cell Biol. 2016, 214, 831–845. [Google Scholar] [CrossRef]
- Paradkar, P.N.; Zumbrennen, K.B.; Paw, B.H.; Ward, D.M.; Kaplan, J. Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2. Mol. Cell. Biol. 2009, 29, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Zhou, Y.; Li, Y.; Xia, J.; Chen, Y.; Chen, S.; Wang, X.; Sun, W.; Wang, T.; Ren, X.; et al. Identification of Frataxin as a regulator of ferroptosis. Redox Biol. 2020, 32, 101483. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X.; et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; An, P.; Xie, E.; Wu, Q.; Fang, X.; Gao, H.; Zhang, Z.; Li, Y.; Wang, X.; Zhang, J.; et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology 2017, 66, 449–465. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Chang, S.Y.; Wu, Q.; Gou, Y.J.; Jia, L.; Cui, Y.M.; Yu, P.; Shi, Z.H.; Wu, W.S.; Gao, G.; et al. The Protective Role of Mitochondrial Ferritin on Erastin-Induced Ferroptosis. Front. Aging Neurosci. 2016, 8, 308. [Google Scholar] [CrossRef]
- Mao, C.; Liu, X.; Zhang, Y.; Lei, G.; Yan, Y.; Lee, H.; Koppula, P.; Wu, S.; Zhuang, L.; Fang, B.; et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 2021, 593, 586–590. [Google Scholar] [CrossRef]
- Tadokoro, T.; Ikeda, M.; Ide, T.; Deguchi, H.; Ikeda, S.; Okabe, K.; Ishikita, A.; Matsushima, S.; Koumura, T.; Yamada, K.I.; et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight 2020, 5, e132747. [Google Scholar] [CrossRef]
- Madak, J.T.; Bankhead, A., 3rd; Cuthbertson, C.R.; Showalter, H.D.; Neamati, N. Revisiting the role of dihydroorotate dehydrogenase as a therapeutic target for cancer. Pharmacol. Ther. 2019, 195, 111–131. [Google Scholar] [CrossRef]
- Ribas, V.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Glutathione and mitochondria. Front. Pharm. 2014, 5, 151. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Henning, Y.; Blind, U.S.; Larafa, S.; Matschke, J.; Fandrey, J. Hypoxia aggravates ferroptosis in RPE cells by promoting the Fenton reaction. Cell Death Dis. 2022, 13, 662. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Wang, T.; Li, Y.; Zhou, Y.; Wang, X.; Yu, X.; Ren, X.; An, Y.; Wu, Y.; Sun, W.; et al. DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radic. Biol. Med. 2019, 131, 356–369. [Google Scholar] [CrossRef]
- Wang, C.X.; Chen, L.H.; Zhuang, H.B.; Shi, Z.S.; Chen, Z.C.; Pan, J.P.; Hong, Z.S. Auriculasin enhances ROS generation to regulate colorectal cancer cell apoptosis, ferroptosis, oxeiptosis, invasion and colony formation. Biochem. Biophys. Res. Commun. 2022, 587, 99–106. [Google Scholar] [CrossRef]
- Liu, M.R.; Zhu, W.T.; Pei, D.S. System Xc−: A key regulatory target of ferroptosis in cancer. Investig. New Drugs 2021, 39, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Bao, Z.; Hua, L.; Ye, Y.; Wang, D.; Li, C.; Xie, Q.; Wakimoto, H.; Gong, Y.; Ji, J. MEF2C silencing downregulates NF2 and E-cadherin and enhances Erastin-induced ferroptosis in meningioma. Neuro Oncol. 2021, 23, 2014–2027. [Google Scholar] [CrossRef]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014, 3, e02523. [Google Scholar] [CrossRef] [PubMed]
- Gout, P.W.; Buckley, A.R.; Simms, C.R.; Bruchovsky, N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the Xc− cystine transporter: A new action for an old drug. Leukemia 2001, 15, 1633–1640. [Google Scholar] [CrossRef] [PubMed]
- Sendo, K.; Seino, M.; Ohta, T.; Nagase, S. Impact of the glutathione synthesis pathway on sulfasalazine-treated endometrial cancer. Oncotarget 2022, 13, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Gao, X.; Zou, L.; Lei, M.; Feng, J.; Hu, Z. Bavachin Induces Ferroptosis through the STAT3/P53/SLC7A11 Axis in Osteosarcoma Cells. Oxid. Med. Cell. Longev. 2021, 2021, 1783485. [Google Scholar] [CrossRef]
- Dias Lopes, N.M.; Marinello, P.C.; Sanches, L.J.; da Silva Brito, W.A.; Lovo-Martins, M.I.; Pinge-Filho, P.; Luiz, R.C.; Cecchini, R.; Cecchini, A.L. Patterns of cell death induced by metformin in human MCF-7 breast cancer cells. Pathol. Res. Pract. 2020, 216, 153199. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Roh, J.L.; Kim, E.H.; Jang, H.; Shin, D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol. 2017, 11, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Sekhar, K.R.; Cyr, S.; Baregamian, N. Ferroptosis Inducers in Thyroid Cancer. World J. Surg. 2023, 47, 371–381. [Google Scholar] [CrossRef]
- Zhang, J.; Gao, R.F.; Li, J.; Yu, K.D.; Bi, K.X. Alloimperatorin activates apoptosis, ferroptosis and oxeiptosis to inhibit the growth and invasion of breast cancer cells in vitro. Biochem. Cell Biol. 2022, 100, 213–222. [Google Scholar] [CrossRef]
- Hao, J.; Zhang, W.; Huang, Z. Bupivacaine modulates the apoptosis and ferroptosis in bladder cancer via phosphatidylinositol 3-kinase (PI3K)/AKT pathway. Bioengineered 2022, 13, 6794–6806. [Google Scholar] [CrossRef]
- Ackermann, A.; Capci, A.; Buchfelder, M.; Tsogoeva, S.B.; Savaskan, N. Chemical hybridization of sulfasalazine and dihydroartemisinin promotes brain tumor cell death. Sci. Rep. 2021, 11, 20766. [Google Scholar] [CrossRef]
- Gao, W.; Huang, Z.; Duan, J.; Nice, E.C.; Lin, J.; Huang, C. Elesclomol induces copper-dependent ferroptosis in colorectal cancer cells via degradation of ATP7A. Mol. Oncol. 2021, 15, 3527–3544. [Google Scholar] [CrossRef]
- Zhu, L.; Wang, J.; Tang, X.; Zhang, C.; Wang, P.; Wu, L.; Gao, W.; Ding, W.; Zhang, G.; Tao, X. Efficient Magnetic Nanocatalyst-Induced Chemo- and Ferroptosis Synergistic Cancer Therapy in Combination with T1-T2 Dual-Mode Magnetic Resonance Imaging Through Doxorubicin Delivery. ACS Appl. Mater. Interfaces 2022, 14, 3621–3632. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.H.; Hsieh, H.C.; Shih, F.S.; Wang, P.W.; Yang, L.X.; Shieh, D.B.; Wang, Y.C. An innovative NRF2 nano-modulator induces lung cancer ferroptosis and elicits an immunostimulatory tumor microenvironment. Theranostics 2021, 11, 7072–7091. [Google Scholar] [CrossRef] [PubMed]
- Costa, I.; Barbosa, D.J.; Benfeito, S.; Silva, V.; Chavarria, D.; Borges, F.; Remiao, F.; Silva, R. Molecular mechanisms of ferroptosis and their involvement in brain diseases. Pharmacol. Ther. 2023, 244, 108373. [Google Scholar] [CrossRef]
- Martin-Sanchez, D.; Fontecha-Barriuso, M.; Martinez-Moreno, J.M.; Ramos, A.M.; Sanchez-Nino, M.D.; Guerrero-Hue, M.; Moreno, J.A.; Ortiz, A.; Sanz, A.B. Ferroptosis and kidney disease. Nefrologia 2020, 40, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Ratan, R.R. The Chemical Biology of Ferroptosis in the Central Nervous System. Cell Chem. Biol. 2020, 27, 479–498. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Jiang, X.; Gu, W. Emerging Mechanisms and Disease Relevance of Ferroptosis. Trends Cell Biol. 2020, 30, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.H.; Fefelova, N.; Pamarthi, S.H.; Gwathmey, J.K. Molecular Mechanisms of Ferroptosis and Relevance to Cardiovascular Disease. Cells 2022, 11, 2726. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Guo, Z. Recent progress in ferroptosis: Inducers and inhibitors. Cell Death Discov. 2022, 8, 501. [Google Scholar] [CrossRef]
- Yeang, C.; Hasanally, D.; Que, X.; Hung, M.Y.; Stamenkovic, A.; Chan, D.; Chaudhary, R.; Margulets, V.; Edel, A.L.; Hoshijima, M.; et al. Reduction of myocardial ischaemia-reperfusion injury by inactivating oxidized phospholipids. Cardiovasc. Res. 2019, 115, 179–189. [Google Scholar] [CrossRef]
- Bai, T.; Li, M.; Liu, Y.; Qiao, Z.; Wang, Z. Inhibition of ferroptosis alleviates atherosclerosis through attenuating lipid peroxidation and endothelial dysfunction in mouse aortic endothelial cell. Free Radic. Biol. Med. 2020, 160, 92–102. [Google Scholar] [CrossRef]
- Skouta, R.; Dixon, S.J.; Wang, J.; Dunn, D.E.; Orman, M.; Shimada, K.; Rosenberg, P.A.; Lo, D.C.; Weinberg, J.M.; Linkermann, A.; et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 2014, 136, 4551–4556. [Google Scholar] [CrossRef]
- Wang, F.; Li, J.; Zhao, Y.; Guo, D.; Liu, D.; Chang, S.; Qiao, H.; Li, J.; Yang, Y.; Zhang, C.; et al. miR-672-3p Promotes Functional Recovery in Rats with Contusive Spinal Cord Injury by Inhibiting Ferroptosis Suppressor Protein 1. Oxid. Med. Cell. Longev. 2022, 2022, 6041612. [Google Scholar] [CrossRef]
- Liu, Q.; Jiang, J.; Fu, Y.; Liu, T.; Yu, Y.; Zhang, X. MiR-129-5p functions as a tumor suppressor in gastric cancer progression through targeting ADAM9. Biomed. Pharm. 2018, 105, 420–427. [Google Scholar] [CrossRef]
- Yadav, P.; Sharma, P.; Sundaram, S.; Venkatraman, G.; Bera, A.K.; Karunagaran, D. SLC7A11/xCT is a target of miR-5096 and its restoration partially rescues miR-5096-mediated ferroptosis and anti-tumor effects in human breast cancer cells. Cancer Lett. 2021, 522, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Luo, M.; Cui, X.; O’Connell, D.; Yang, Y. Long noncoding RNA NEAT1 promotes ferroptosis by modulating the miR-362-3p/MIOX axis as a ceRNA. Cell Death Differ. 2022, 29, 1850–1863. [Google Scholar] [CrossRef]
- Fuhrmann, D.C.; Brune, B. A graphical journey through iron metabolism, microRNAs, and hypoxia in ferroptosis. Redox Biol. 2022, 54, 102365. [Google Scholar] [CrossRef]
- Zhang, H.; Deng, T.; Liu, R.; Ning, T.; Yang, H.; Liu, D.; Zhang, Q.; Lin, D.; Ge, S.; Bai, M.; et al. CAF secreted miR-522 suppresses ferroptosis and promotes acquired chemo-resistance in gastric cancer. Mol. Cancer 2020, 19, 43. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Wang, J.; Xu, W.; Ma, C.; Wan, F.; Huang, Y.; Yao, M.; Zhang, H.; Qu, Y.; Ye, D.; et al. LncRNA RP11-89 facilitates tumorigenesis and ferroptosis resistance through PROM2-activated iron export by sponging miR-129-5p in bladder cancer. Cell Death Dis. 2021, 12, 1043. [Google Scholar] [CrossRef] [PubMed]


| Type | Morphological Hallmarks |
|---|---|
| Ferroptosis | Mitochondrial membrane density increases, mitochondrial volume decreases, mitochondrial crest disappearance, the outer membrane of the mitochondria ruptures, and the nucleus is normal. |
| Apoptosis | Cells shrink, apoptotic bodies form, and surrounding tissues are normal. |
| Necrosis | The chromatin concentration changes, nucleus volume shrinkage, nuclear membrane rupture, and nuclear contours disappear. |
| Pyroptosis | Cells swell and cell membranes rupture, causing an inflammatory response. |
| Autophagy | A double-membrane autophagosome forms, cell membranes rupture, and organelles are engulfed. |
| PANoptosis | Cells swell, cell membranes rupture, and inflammasome formation occurs. |
| Protein | Function | Reference |
|---|---|---|
| FANCD2 | Reduces GSH consumption and LPO production to inhibit iron accumulation and lipid peroxidation during ferroptosis through transcriptional and non-transcriptional-dependent mechanisms | [39] |
| NFE2L2 | Encodes NRF2 and acts as an antioxidant and has anti-ferroptosis effects | [40] |
| Protein | Function | References |
|---|---|---|
| TFRC | Inputs iron into cells to promote ferroptosis | [41,42] |
| GLS2 | It involves glutamine decomposition and promotes ferroptosis, regulated by P53 | [43,44] |
| ATP5G3 | Inhibition of its expression alleviates erastin-type ferroptosis | [45,46] |
| NCOA4 | Participates in the control of ferritinophagy and free iron abundance and promotes ferroptosis | [47] |
| Names | Type | Mechanisms | Targets | Reference |
|---|---|---|---|---|
| miR-672-3p | microRNA, inducer | Differentially expressed after spinal cord injury (SCI) and induced ferroptosis by inhibiting FSP1 | FSP1 and System Xc− | [129] |
| miR-129-5p | microRNA, inducer | Inhibits the expression of ACSL4 | ACSL4 | [130] |
| miR-5096 | microRNA, inducer | Directly targets the SLC7A11 system and facilitates lipid peroxidation | SLC7A11 | [131] |
| miR-541-3p | microRNA, inducer | Directly targets the GPX4 system and facilitates lipid peroxidation | GPX4 | [131] |
| nuclear enriched transcript 1 (NEAT1) | incRNA, inducer | Inhibits the expression of ACSL4 and the occurrence of ferroptosis | ACSL4 | [132] |
| miR-7-5p | microRNA, inhibitor | Indirectly reduces the labile iron pool and Fenton reactions | Transferrin | [133] |
| miR-522 | microRNA, inhibitor | Reduces iron levels via another enzyme called arachidonate lipoxygenase 15 (ALOX15) to inhibit ferroptosis | ALOX15 | [134] |
| miR-200a | microRNA, inhibitor | Targeting Keap1 and activating Nrf2 to inhibit ferroptosis | Keap1 | [131] |
| RP11-89 | IncRNA, inhibitor | Reducing iron level via regulating miR-129-5p to inhibit ferroptosis | miR-129-5p | [135] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, X.; Zhang, J.; Xiao, Y.; Wang, Z.; He, J.; Ke, M.; Liu, S.; Wang, Q.; Zhang, L. Mitochondrial Regulation of Ferroptosis in Cancer Therapy. Int. J. Mol. Sci. 2023, 24, 10037. https://doi.org/10.3390/ijms241210037
Cheng X, Zhang J, Xiao Y, Wang Z, He J, Ke M, Liu S, Wang Q, Zhang L. Mitochondrial Regulation of Ferroptosis in Cancer Therapy. International Journal of Molecular Sciences. 2023; 24(12):10037. https://doi.org/10.3390/ijms241210037
Chicago/Turabian StyleCheng, Xiaoxia, Jiale Zhang, Yichen Xiao, Zhihang Wang, Jin He, Mengquan Ke, Sijie Liu, Qun Wang, and Lei Zhang. 2023. "Mitochondrial Regulation of Ferroptosis in Cancer Therapy" International Journal of Molecular Sciences 24, no. 12: 10037. https://doi.org/10.3390/ijms241210037
APA StyleCheng, X., Zhang, J., Xiao, Y., Wang, Z., He, J., Ke, M., Liu, S., Wang, Q., & Zhang, L. (2023). Mitochondrial Regulation of Ferroptosis in Cancer Therapy. International Journal of Molecular Sciences, 24(12), 10037. https://doi.org/10.3390/ijms241210037