
Exploring the Therapeutic Potential of Targeting GH and IGF-1 in the Management of Obesity: Insights from the Interplay between These Hormones and Metabolism


Abstract
1. Introduction
2. Method
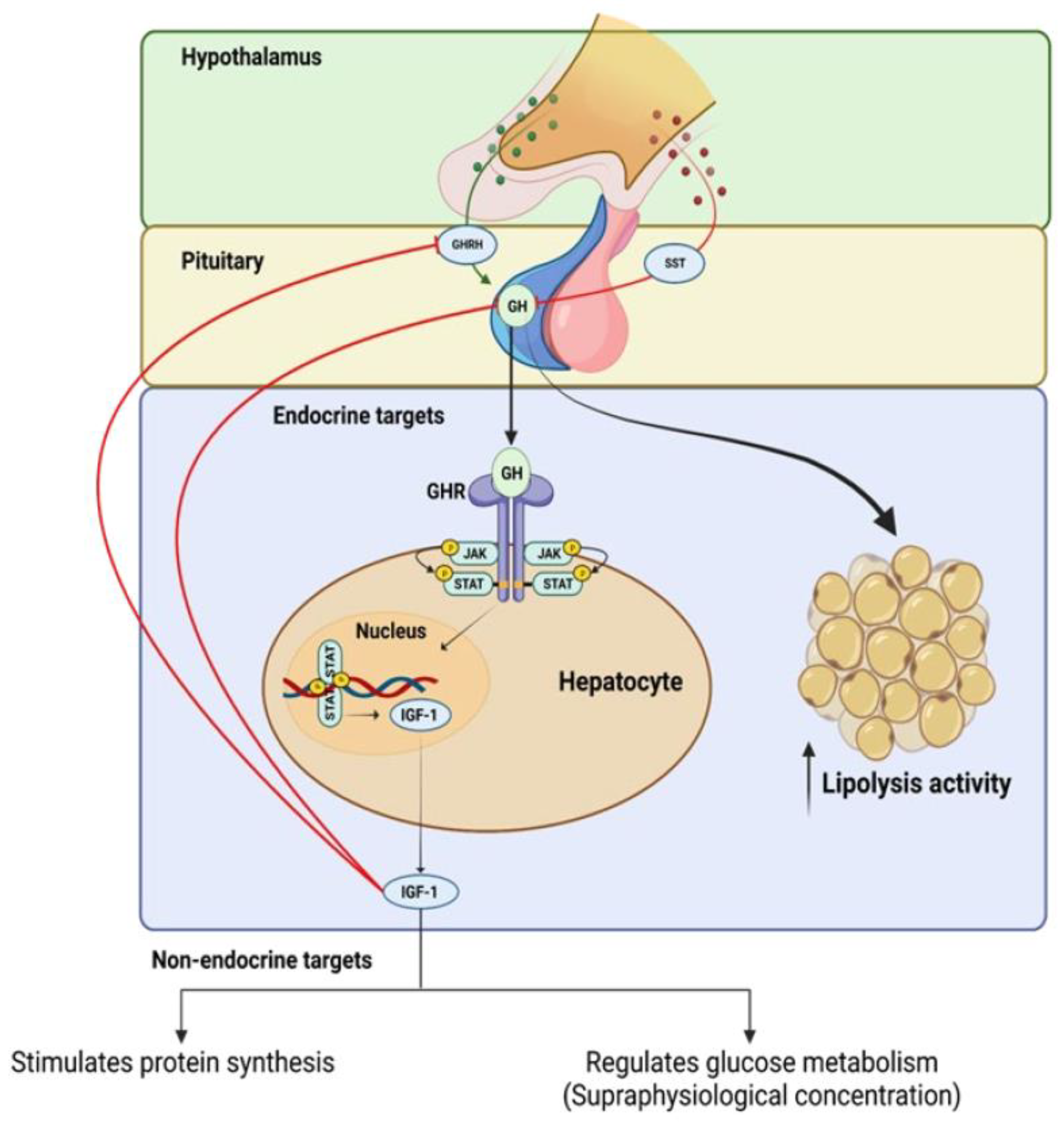
3. Organ Involvement in the Regulation of GH and IGF-1 Axis in Obesity
3.1. The Hypothalamus and Pituitary Gland (Hypothalamic–Pituitary Axis)
3.2. The Liver and Adipose Tissue
4. The Advancements in the Understanding GH Secretion: Mechanisms, Modulators, and Consequences
5. The Negative and Positive Feedback Regulating GH Production
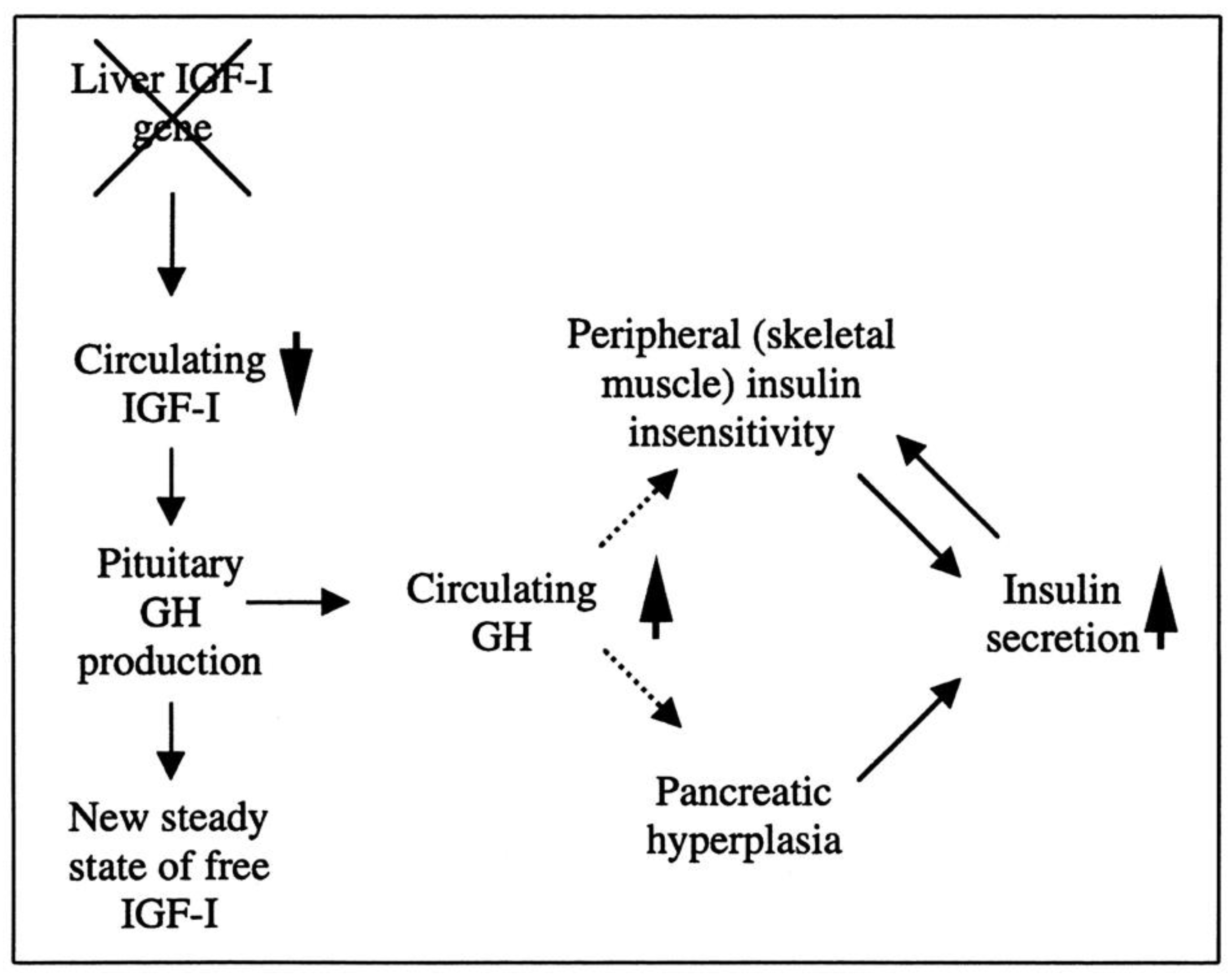
6. Overview of the Current Understanding of the Relationship between the GH–IGF-1 Axis and Obesity
7. Mouse Models Were Used to Study the Role of GH and IGF-1 in Obesity
7.1. The Snell Dwarf Mouse
7.2. The Ames Dwarf Mouse
7.3. The Metallothionein-I Human Growth Hormone Transgenic Mouse Model (MT1-hGH)
7.4. The Adult-Onset, Isolated, Growth Hormone Deficiency (AOiGHD) Mouse Model
7.5. The GH−/− Mouse Model
7.6. The GHR−/− (Laron) Mouse Model
7.7. The Adipocyte-Specific GHR Knockout (AdGHRKO) Mouse Model
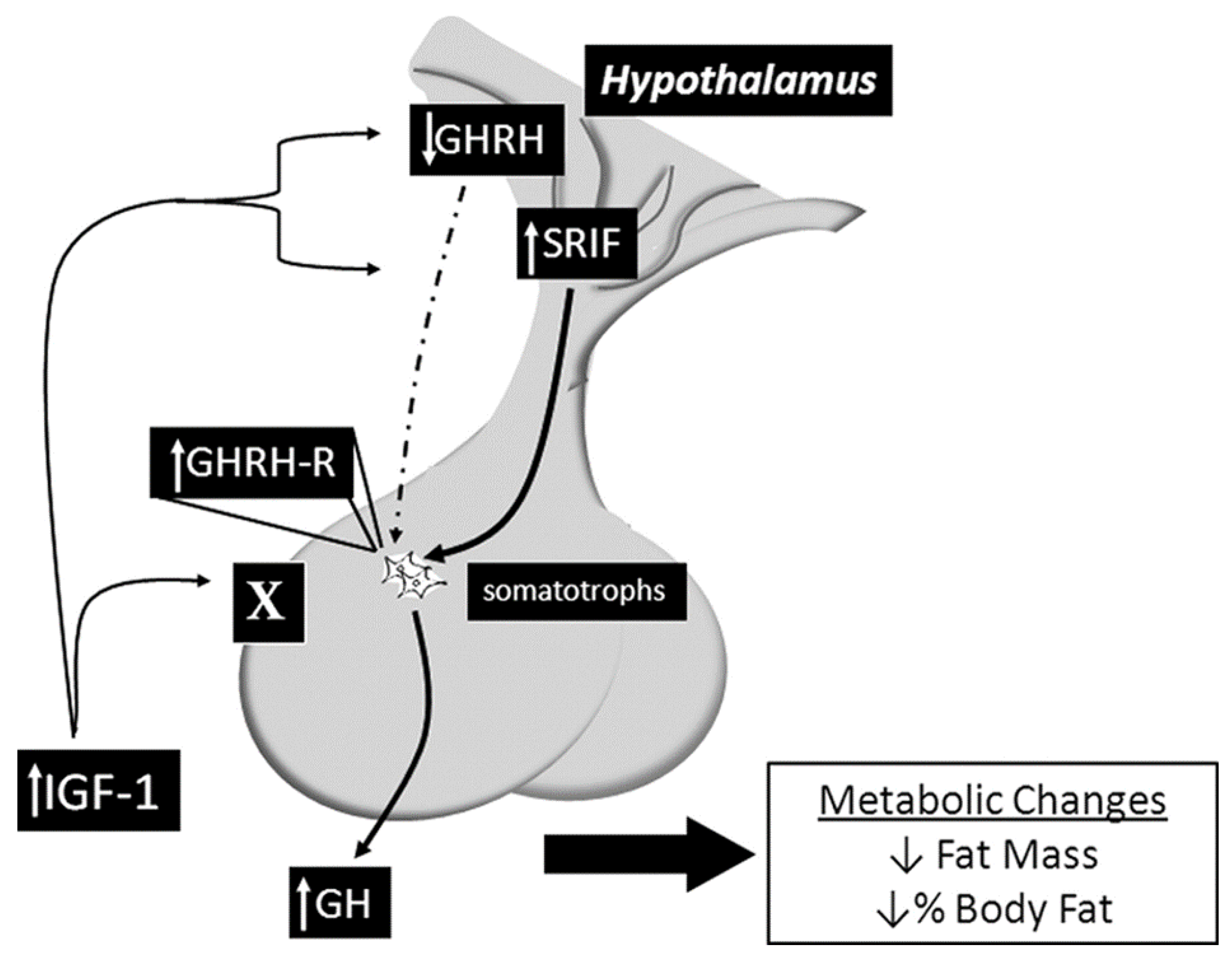
7.8. The Somatotroph IGF-1R Knockout Mouse Model (SIGFRKO)
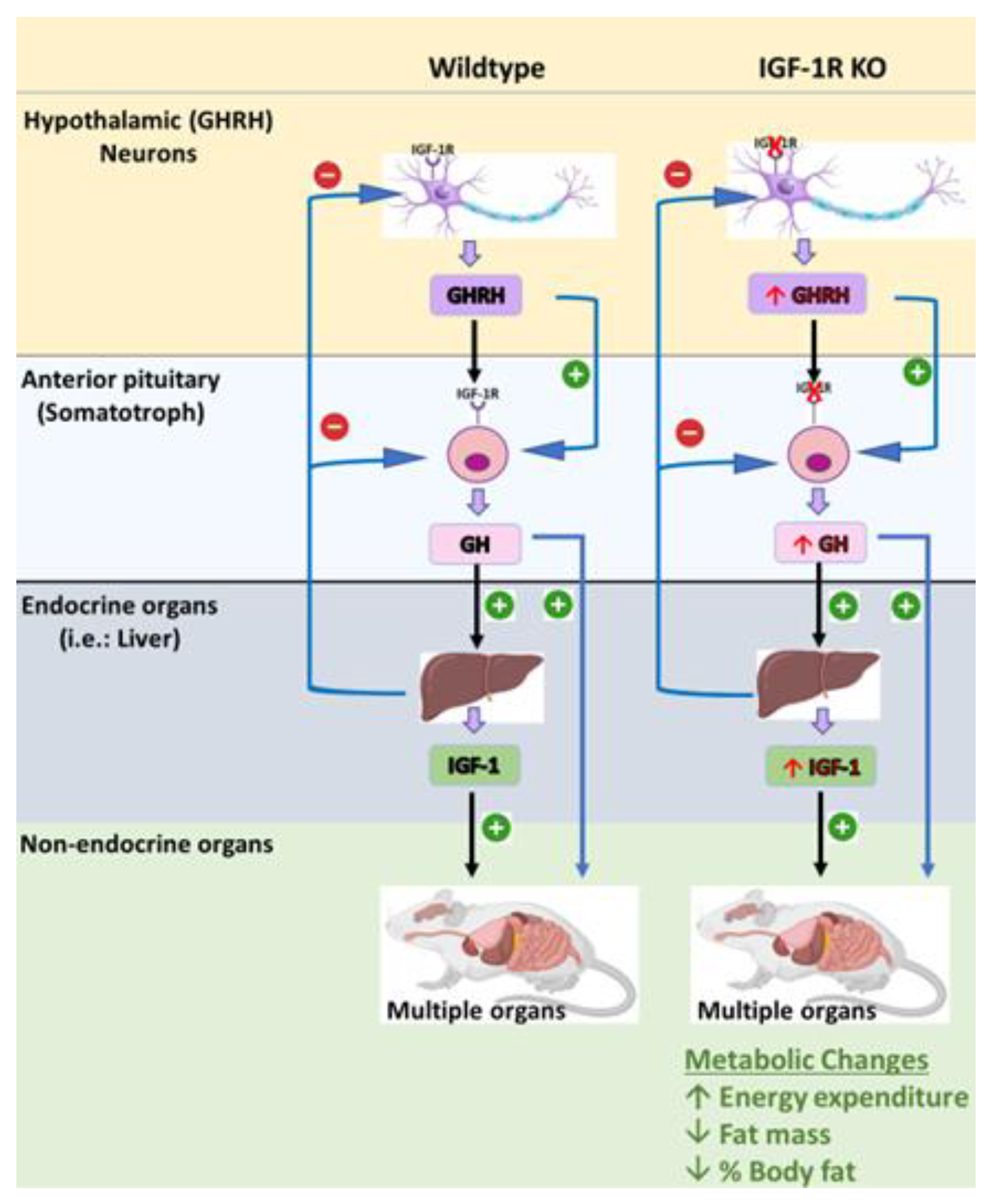
7.9. The Somatotroph GHRH Neurons IGF-1R Knockout (S-GIGFRKO) Mouse Model
8. The Effect of Obesity on GH and IGF-1 Production
9. Conclusions and Further Study
Funding
Conflicts of Interest
References
- Berryman, D.E.; Glad, C.A.M.; List, E.O.; Johannsson, G. The GH/IGF-1 axis in obesity: Pathophysiology and therapeutic considerations. Nat. Rev. Endocrinol. 2013, 9, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Romieu, I.; Dossus, L.; Barquera, S.; Blottière, H.M.; Franks, P.W.; Gunter, M.; Hwalla, N.; Hursting, S.D.; Leitzmann, M.; Margetts, B.; et al. Obesity. Energy balance and obesity: What are the main drivers? Cancer Causes Control CCC 2017, 28, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.O.; Wyatt, H.R.; Peters, J.C. Energy balance and obesity. Circulation 2012, 126, 126–132. [Google Scholar] [CrossRef]
- Nillni, E.A. Textbook of Energy Balance, Neuropeptide Hormones, and Neuroendocrine Function, 1st ed.; Springer International Publishing: Cham, Switzerland, 2018. [Google Scholar]
- Al-Samerria, S.; Radovick, S. The Role of Insulin-like Growth Factor-1 (IGF-1) in the Control of Neuroendocrine Regulation of Growth. Cells 2021, 10, 2664. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Bolado, G. Development of neuroendocrine neurons in the mammalian hypothalamus. Cell Tissue Res. 2019, 375, 23–39. [Google Scholar] [CrossRef]
- Huang, Z.; Huang, L.; Waters, M.J.; Chen, C. Insulin and Growth Hormone Balance: Implications for Obesity. Trends Endocrinol. Metab. 2020, 31, 642–654. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Flanagan, J.U.; Langley, R.J.; Hay, M.P.; Perry, J.K. Targeting growth hormone function: Strategies and therapeutic applications. Signal Transduct. Target. Ther. 2019, 4, 3. [Google Scholar] [CrossRef]
- Kraemer, W.J.; Ratamess, N.A.; Hymer, W.C.; Nindl, B.C.; Fragala, M.S. Growth Hormone(s), Testosterone, Insulin-Like Growth Factors, and Cortisol: Roles and Integration for Cellular Development and Growth with Exercise. Front. Endocrinol. 2020, 11, 33. [Google Scholar] [CrossRef]
- Berryman, D.E.; List, E.O. Growth Hormone’s Effect on Adipose Tissue: Quality versus Quantity. Int. J. Mol. Sci. 2017, 18, 1621. [Google Scholar] [CrossRef]
- Dehkhoda, F.; Lee, C.M.; Medina, J.; Brooks, A.J. The growth hormone receptor: Mechanism of receptor activation, cell signaling, and physiological aspects. Front. Endocrinol. 2018, 9, 35. [Google Scholar] [CrossRef]
- De Feo, P.; Perriello, G.; Torlone, E.; Ventura, M.M.; Santeusanio, F.; Brunetti, P.; Gerich, J.E.; Bolli, G. Demonstration of a role for growth hormone in glucose counterregulation. Am. J. Physiol.-Endocrinol. Metab. 1989, 256, E835–E843. [Google Scholar] [CrossRef] [PubMed]
- Rizza, R.A.; Mandarino, L.J.; Gerich, J.E. Effects of growth hormone on insulin action in man: Mechanisms of insulin resistance, impaired suppression of glucose production, and impaired stimulation of glucose utilization. Diabetes 1982, 31, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Krusenstjerna-Hafstrøm, T.; Clasen, B.; Møller, N.; Jessen, N.; Pedersen, S.; Christiansen, J.; Jørgensen, J. Growth hormone (GH)-induced insulin resistance is rapidly reversible: An experimental study in GH-deficient adults. J. Clin. Endocrinol. Metab. 2011, 96, 2548–2557. [Google Scholar] [CrossRef] [PubMed]
- Møller, N.; Jørgensen, J.O.L. Effects of growth hormone on glucose, lipid, and protein metabolism in human subjects. Endocr. Rev. 2009, 30, 152–177. [Google Scholar] [CrossRef]
- Yakar, S.; Setser, J.; Zhao, H.; Stannard, B.; Haluzik, M.; Glatt, V.; Bouxsein, M.L.; Kopchick, J.J.; LeRoith, D. Inhibition of growth hormone action improves insulin sensitivity in liver IGF-1–deficient mice. J. Clin. Investig. 2004, 113, 96–105. [Google Scholar] [CrossRef]
- List, E.O.; Berryman, D.E.; Slyby, J.; Duran-Ortiz, S.; Funk, K.; Bisset, E.S.; Howlett, S.E.; Kopchick, J.J. Disruption of growth hormone receptor in adipocytes improves insulin sensitivity and lifespan in mice. Endocrinology 2022, 163, bqac129. [Google Scholar] [CrossRef]
- Romero, C.J.; Ng, Y.; Luque, R.M.; Kineman, R.D.; Koch, L.; Bruning, J.C.; Radovick, S. Targeted Deletion of Somatotroph Insulin-Like Growth Factor-I Signaling in a Cell-Specific Knockout Mouse Model. Mol. Endocrinol. 2010, 24, 1077–1089. [Google Scholar] [CrossRef]
- Vitale, G.; Pellegrino, G.; Vollery, M.; Hofland, L.J. ROLE of IGF-1 System in the Modulation of Longevity: Controversies and New Insights from a Centenarians’ Perspective. Front. Endocrinol. 2019, 10, 27. [Google Scholar] [CrossRef]
- Hakuno, F.; Takahashi, S.I. IGF1 receptor signaling pathways. J. Mol. Endocrinol. 2018, 61, T69–T86. [Google Scholar] [CrossRef]
- Yakar, S.; Adamo, M.L. Insulin-like growth factor 1 physiology: Lessons from mouse models. Endocrinol. Metab. Clin. N. Am. 2012, 41, 231–247. [Google Scholar] [CrossRef]
- Arcopinto, M.; Salzano, A.; Giallauria, F.; Bossone, E.; Isgaard, J.; Marra, A.M.; Cittadini, A. Growth Hormone Deficiency Is Associated with Worse Cardiac Function, Physical Performance, and Outcome in Chronic Heart Failure: Insights from the T.O.S.CA. GHD Study. PLoS ONE 2017, 12, e0170058. [Google Scholar] [CrossRef]
- Kim, S.H.; Park, M.J. Effects of growth hormone on glucose metabolism and insulin resistance in human. Ann. Pediatr. Endocrinol. Metab. 2017, 22, 145–152. [Google Scholar] [CrossRef]
- Ranke, M.B.; Wit, J.M. Growth hormone—Past, present and future. Nat. Rev. Endocrinol. 2018, 14, 285–300. [Google Scholar] [CrossRef] [PubMed]
- Devesa, J.; Almengló, C.; Devesa, P. Multiple effects of growth hormone in the body: Is it really the hormone for growth? Clin. Med. Insights Endocrinol. Diabetes 2016, 9, 47–71. [Google Scholar] [CrossRef] [PubMed]
- Lechan, R.M.; Toni, R. Functional Anatomy of the Hypothalamus and Pituitary. 2016. Available online: https://www.ncbi.nlm.nih.gov/books/NBK279126/ (accessed on 26 April 2023).
- Fink, G.; Pfaff, D.W.; Levine, J. Handbook of Neuroendocrinology; Elsevier Science & Technology: San Diego, CA, USA, 2011. [Google Scholar]
- Junnila, R.K.; List, E.O.; Berryman, D.E.; Murrey, J.W.; Kopchick, J.J. The GH/IGF-1 axis in ageing and longevity. Nat. Rev. Endocrinol. 2013, 9, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Hochberg, I.; Hochberg, Z. Hypothalamic obesity. Endocr. Dev. 2010, 17, 185–196. [Google Scholar] [CrossRef]
- Sohn, J.W. Network of hypothalamic neurons that control appetite. BMB Rep. 2015, 48, 229–233. [Google Scholar] [CrossRef]
- Cowley, M.A.; Smith, R.G.; Diano, S.; Tschöp, M.; Pronchuk, N.; Grove, K.L.; Strasburger, C.J.; Bidlingmaier, M.; Esterman, M.; Heiman, M.L. The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron 2003, 37, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Harno, E.; Ramamoorthy, T.G.; Coll, A.P.; White, A. POMC: The Physiological Power of Hormone Processing. Physiol. Rev. 2018, 98, 2381–2430. [Google Scholar] [CrossRef]
- Baldini, G.; Phelan, K.D. The melanocortin pathway and control of appetite-progress and therapeutic implications. J. Endocrinol. 2019, 241, R1–R33. [Google Scholar] [CrossRef]
- Huvenne, H.; Dubern, B.; Clément, K.; Poitou, C. Rare Genetic Forms of Obesity: Clinical Approach and Current Treatments in 2016. Obes. Facts 2016, 9, 158–173. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Choi, J.H. Pathophysiology and clinical characteristics of hypothalamic obesity in children and adolescents. Ann. Pediatr. Endocrinol. Metab. 2013, 18, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Tran, L.T.; Park, S.; Kim, S.K.; Lee, J.S.; Kim, K.W.; Kwon, O. Hypothalamic control of energy expenditure and thermogenesis. Exp. Mol. Med. 2022, 54, 358–369. [Google Scholar] [CrossRef]
- El Sayed, S.F.M.; Schwartz, J. Physiology, Pituitary Gland; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Sadiq, N.M.; Tadi, P. Physiology, Pituitary Hormones; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Rucker, P.; Ikuta, T. Pituitary Gland Functional Connectivity and BMI. Front. Neurosci. 2019, 13, 120. [Google Scholar] [CrossRef] [PubMed]
- Romero, C.J.; Wolfe, A.; Law, Y.Y.; Costelloe, C.Z.; Miller, R.; Wondisford, F.; Radovick, S. Altered somatotroph feedback regulation improves metabolic efficiency and limits adipose deposition in male mice. Metabolism 2016, 65, 557–568. [Google Scholar] [CrossRef]
- Mauras, N.; Haymond, M.W. Are the metabolic effects of GH and IGF-I separable? Growth Horm. IGF Res. 2005, 15, 19–27. [Google Scholar] [CrossRef]
- Mullur, R.; Liu, Y.Y.; Brent, G.A. Thyroid hormone regulation of metabolism. Physiol. Rev. 2014, 94, 355–382. [Google Scholar] [CrossRef]
- Motomura, K.; Brent, G.A. Mechanisms of thyroid hormone action. Implications for the clinical manifestation of thyrotoxicosis. Endocrinol. Metab. Clin. N. Am. 1998, 27, 1–23. [Google Scholar] [CrossRef]
- Azzu, V.; Vacca, M.; Virtue, S.; Allison, M.; Vidal-Puig, A. Adipose Tissue-Liver Cross Talk in the Control of Whole-Body Metabolism: Implications in Nonalcoholic Fatty Liver Disease. Gastroenterology 2020, 158, 1899–1912. [Google Scholar] [CrossRef]
- Abdel-Misih, S.R.; Bloomston, M. Liver anatomy. Surg. Clin. N. Am. 2010, 90, 643–653. [Google Scholar] [CrossRef]
- Sibulesky, L. Normal liver anatomy. Clin. Liver Dis. 2013, 2 (Suppl. S1), S1–S3. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Li, Y.; Guo, F.; Jiang, Y.; Ying, W.; Li, D.; Yang, D.; Xia, X.; Liu, W.; Zhao, Y. A Cell-type-resolved Liver Proteome. Mol. Cell Proteom. 2016, 15, 3190–3202. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, R.W. Adipose tissue and the physiologic underpinnings of metabolic disease. Surg. Obes. Relat. Dis. 2018, 14, 1755–1763. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.C.; Stenesen, D.; Zeve, D.; Graff, J.M. The developmental origins of adipose tissue. Development 2013, 140, 3939–3949. [Google Scholar] [CrossRef]
- Qureshi, K.; Abrams, G.A. Metabolic liver disease of obesity and role of adipose tissue in the pathogenesis of nonalcoholic fatty liver disease. World J. Gastroenterol. 2007, 13, 3540–3553. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yun, K.; Mu, R. A review on the biology and properties of adipose tissue macrophages involved in adipose tissue physiological and pathophysiological processes. Lipids Health Dis. 2020, 19, 164. [Google Scholar] [CrossRef]
- Cypess, A.M. Reassessing Human Adipose Tissue. N. Engl. J. Med. 2022, 386, 768–779. [Google Scholar] [CrossRef] [PubMed]
- Zwick, R.K.; Guerrero-Juarez, C.F.; Horsley, V.; Plikus, M.V. Anatomical, Physiological, and Functional Diversity of Adipose Tissue. Cell Metab. 2018, 27, 68–83. [Google Scholar] [CrossRef]
- Saito, M.; Matsushita, M.; Yoneshiro, T.; Okamatsu-Ogura, Y. Brown Adipose Tissue, Diet-Induced Thermogenesis, and Thermogenic Food Ingredients: From Mice to Men. Front. Endocrinol. 2020, 11, 222. [Google Scholar] [CrossRef]
- Cheng, L.; Wang, J.; Dai, H.; Duan, Y.; An, Y.; Shi, L.; Lv, Y.; Li, H.; Wang, C.; Ma, Q. Brown and beige adipose tissue: A novel therapeutic strategy for obesity and type 2 diabetes mellitus. Adipocyte 2021, 10, 48–65. [Google Scholar] [CrossRef]
- Wu, J.; Boström, P.; Sparks, L.M.; Ye, L.; Choi, J.H.; Giang, A.H.; Khandekar, M.; Virtanen, K.A.; Nuutila, P.; Schaart, G.; et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 2012, 150, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Scheja, L.; Heeren, J. The endocrine function of adipose tissues in health and cardiometabolic disease. Nat. Rev. Endocrinol. 2019, 15, 507–524. [Google Scholar] [CrossRef] [PubMed]
- Newell-Fugate, A.E. The role of sex steroids in white adipose tissue adipocyte function. Reproduction 2017, 153, R133–R149. [Google Scholar] [CrossRef]
- Luo, L.; Liu, M. Adipose tissue in control of metabolism. J. Endocrinol. 2016, 231, R77–R99. [Google Scholar] [CrossRef]
- Popko, K.; Gorska, E.; Stelmaszczyk-Emmel, A.; Plywaczewski, R.; Stoklosa, A.; Gorecka, D.; Pyrzak, B.; Demkow, U. Proinflammatory cytokines Il-6 and TNF-α and the development of inflammation in obese subjects. Eur. J. Med. Res. 2010, 15 (Suppl. S2), 120–122. [Google Scholar] [CrossRef]
- Kern, L.; Mittenbühler, M.J.; Vesting, A.J.; Ostermann, A.L.; Wunderlich, C.M.; Wunderlich, F.T. Obesity-Induced TNFα and IL-6 Signaling: The Missing Link between Obesity and Inflammation-Driven Liver and Colorectal Cancers. Cancers 2018, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, H.; Liang, X.; Roberts, M.S. Chapter 30—Hepatic Metabolism in Liver Health and Disease. In Liver Pathophysiology; Muriel, P., Ed.; Academic Press: Boston, MA, USA, 2017; pp. 391–400. [Google Scholar]
- Scherer, T.; Lindtner, C.; O’Hare, J.; Hackl, M.; Zielinski, E.; Freudenthaler, A.; Baumgartner-Parzer, S.; Tödter, K.; Heeren, J.; Krššák, M.; et al. Insulin Regulates Hepatic Triglyceride Secretion and Lipid Content via Signaling in the Brain. Diabetes 2016, 65, 1511–1520. [Google Scholar] [CrossRef]
- Glick, S.M.; Roth, J.; Yalow, R.S.; Berson, S.A. Immunoassay of Human Growth Hormone in Plasma. Nature 1963, 199, 784–787. [Google Scholar] [CrossRef]
- Hjelholt, A.; Høgild, M.; Bak, A.M.; Arlien-Søborg, M.C.; Bæk, A.; Jessen, N.; Richelsen, B.; Pedersen, S.B.; Møller, N.; Lunde Jørgensen, J.O. Growth Hormone and Obesity. Endocrinol. Metab. Clin. N. Am. 2020, 49, 239–250. [Google Scholar] [CrossRef]
- Qian, Y.; Berryman, D.E.; Basu, R.; List, E.O.; Okada, S.; Young, J.A.; Jensen, E.A.; Bell, S.R.C.; Kulkarni, P.; Duran-Ortiz, S. Mice with gene alterations in the GH and IGF family. Pituitary 2022, 25, 1–51. [Google Scholar] [CrossRef]
- Nass, R.; Gaylinn, B.D.; Thorner, M.O. The role of ghrelin in GH secretion and GH disorders. Mol. Cell Endocrinol. 2011, 340, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Devesa, J. The Complex World of Regulation of Pituitary Growth Hormone Secretion: The Role of Ghrelin, Klotho, and Nesfatins in It. Front. Endocrinol. 2021, 12, 636403. [Google Scholar] [CrossRef]
- Kojima, M.; Hosoda, H.; Date, Y.; Nakazato, M.; Matsuo, H.; Kangawa, K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 1999, 402, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Olarescu, N.C.; Gunawardane, K.; Hansen, T.K.; Moller, N.; Jorgensen, J.O.L. Normal Physiology of Growth Hormone in Adults. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., Dungan, K., Grossman, A., Hershman, J.M., Kaltsas, G., Koch, C., Kopp, P., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Copinschi, G.; Wegienka, L.C.; Hane, S.; Forsham, P.H. Effect of arginine on serum levels of insulin and growth hormone in obese subjects. Metabolism 1967, 16, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Beck, P.; Koumans, J.H.; Winterling, C.A.; Stein, M.F.; Daughaday, W.H.; Kipnis, D.M. Studies of Insulin and Growth Hormone Secretion in Human Obesity. J. Lab. Clin. Med. 1964, 64, 654–667. [Google Scholar] [PubMed]
- Vahl, N.; Jørgensen, J.O.; Skjaerbaek, C.; Veldhuis, J.D.; Orskov, H.; Christiansen, J.S. Abdominal adiposity rather than age and sex predicts mass and regularity of GH secretion in healthy adults. Am. J. Physiol 1997, 272 Pt 1, E1108–E1116. [Google Scholar] [CrossRef]
- Imaki, T.; Shibasaki, T.; Shizume, K.; Masuda, A.; Hotta, M.; Kiyosawa, Y.; Jibiki, K.; Demura, H.; Tsushima, T.; Ling, N. The effect of free fatty acids on growth hormone (GH)-releasing hormone-mediated GH secretion in man. J. Clin. Endocrinol. Metab. 1985, 60, 290–293. [Google Scholar] [CrossRef]
- Casanueva, F.F.; Villanueva, L.; Dieguez, C.; Diaz, Y.; Cabranes, J.A.; Szoke, B.; Scanlon, M.F.; Schally, A.V.; Fernandez-Cruz, A. Free fatty acids block growth hormone (GH) releasing hormone-stimulated GH secretion in man directly at the pituitary. J. Clin. Endocrinol. Metab. 1987, 65, 634–642. [Google Scholar] [CrossRef]
- Qiu, H.; Yang, J.K.; Chen, C. Influence of insulin on growth hormone secretion, level and growth hormone signalling. Sheng Li Xue Bao 2017, 69, 541–556. [Google Scholar]
- Bartke, A. Growth Hormone and Aging: Updated Review. World J. Mens. Health 2019, 37, 19–30. [Google Scholar] [CrossRef]
- Ashpole, N.M.; Sanders, J.E.; Hodges, E.L.; Yan, H.; Sonntag, W.E. Growth hormone, insulin-like growth factor-1 and the aging brain. Exp. Gerontol. 2015, 68, 76–81. [Google Scholar] [CrossRef]
- Czogała, W.; Strojny, W.; Tomasik, P.; Multanowski, M.B.; Wójcik, M.; Miklusiak, K.; Krzysztofik, E.; Wróbel, A.; Miklusiak, K.; Skoczeń, S. The Insight into Insulin-Like Growth Factors and Insulin-Like Growth-Factor-Binding Proteins and Metabolic Profile in Pediatric Obesity. Nutrients 2021, 13, 2432. [Google Scholar] [CrossRef] [PubMed]
- Kubo, H.; Sawada, S.; Satoh, M.; Asai, Y.; Kodama, S.; Sato, T.; Tomiyama, S.; Seike, J.; Takahashi, K.; Kaneko, K. Insulin-like growth factor-1 levels are associated with high comorbidity of metabolic disorders in obese subjects; a Japanese single-center, retrospective-study. Sci. Rep. 2022, 12, 20130. [Google Scholar] [CrossRef]
- Gubbi, S.; Quipildor, G.F.; Barzilai, N.; Huffman, D.M.; Milman, S. 40 YEARS of IGF1: IGF1: The Jekyll and Hyde of the aging brain. J. Mol. Endocrinol. 2018, 61, T171–T185. [Google Scholar] [CrossRef]
- Baik, M.; Yu, J.H.; Hennighausen, L. Growth hormone-STAT5 regulation of growth, hepatocellular carcinoma, and liver metabolism. Ann. N. Y. Acad. Sci. 2011, 1229, 29–37. [Google Scholar] [CrossRef]
- Argetsinger, L.S.; Campbell, G.S.; Yang, X.; Witthuhn, B.A.; Silvennoinen, O.; Ihle, J.N.; Carter-Su, C. Identification of JAK2 as a growth hormone receptor-associated tyrosine kinase. Cell 1993, 74, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Gusmao, D.O.; de Sousa, M.E.; Tavares, M.R.; Donato, J. Increased GH Secretion and Body Growth in Mice Carrying Ablation of IGF-1 Receptor in GH-releasing Hormone Cells. Endocrinology 2022, 163, bqac151. [Google Scholar] [CrossRef] [PubMed]
- Niiori-Onishi, A.; Iwasaki, Y.; Mutsuga, N.; Oiso, Y.; Inoue, K.; Saito, H. Molecular Mechanisms of the Negative Effect of Insulin-Like Growth Factor-I on Growth Hormone Gene Expression in MtT/S Somatotroph Cells. Endocrinology 1999, 140, 344–349. [Google Scholar] [CrossRef][Green Version]
- Romero, C.J.; Pine-Twaddell, E.; Sima, D.I.; Miller, R.S.; He, L.; Wondisford, F.; Radovick, S. Insulin-like growth factor 1 mediates negative feedback to somatotroph GH expression via POU1F1/CREB binding protein interactions. Mol. Cell. Biol. 2012, 32, 4258–4269. [Google Scholar] [CrossRef]
- Goldenberg, N.; Horowitz, J.F.; Gorgey, A.; Sakharova, A.; Barkan, A.L. Role of pulsatile growth hormone (GH) secretion in the regulation of lipolysis in fasting humans. Clin. Diabetes Endocrinol. 2022, 8, 1. [Google Scholar] [CrossRef]
- Friedrich, N.; Thuesen, B.; Jørgensen, T.; Juul, A.; Spielhagen, C.; Wallaschofksi, H.; Linneberg, A. The association between IGF-I and insulin resistance: A general population study in Danish adults. Diabetes Care 2012, 35, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Clemmons, D.R. Role of insulin-like growth factor iin maintaining normal glucose homeostasis. Horm. Res. 2004, 62 (Suppl. S1), 77–82. [Google Scholar] [CrossRef]
- Yakar, S.; Liu, J.L.; Fernandez, A.M.; Wu, Y.; Schally, A.V.; Frystyk, J.; Chernausek, S.D.; Mejia, W.; Le Roith, D. Liver-specific igf-1 gene deletion leads to muscle insulin insensitivity. Diabetes 2001, 50, 1110–1118. [Google Scholar] [CrossRef]
- Bailey-Downs, L.C.; Sosnowska, D.; Toth, P.; Mitschelen, M.; Gautam, T.; Henthorn, J.C.; Ballabh, P.; Koller, A.; Farley, J.A.; Sonntag, W.E.; et al. Growth hormone and IGF-1 deficiency exacerbate high-fat diet-induced endothelial impairment in obese Lewis dwarf rats: Implications for vascular aging. J. Gerontol. A Biol. Sci. Med. Sci. 2012, 67, 553–564. [Google Scholar] [CrossRef]
- Velloso, C.P. Regulation of muscle mass by growth hormone and IGF-I. Br. J. Pharmacol. 2008, 154, 557–568. [Google Scholar] [CrossRef]
- Lewitt, M.S. The Role of the Growth Hormone/Insulin-Like Growth Factor System in Visceral Adiposity. Biochem. Insights 2017, 10, 1178626417703995. [Google Scholar] [CrossRef] [PubMed]
- Ambele, M.A.; Dhanraj, P.; Giles, R.; Pepper, M.S. Adipogenesis: A Complex Interplay of Multiple Molecular Determinants and Pathways. Int. J. Mol. Sci. 2020, 21, 4283. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Li, C.; Lei, Y.; Xu, S.; Zhao, D.; Lu, X.Y. Role of the adipose PPARγ-adiponectin axis in susceptibility to stress and depression/anxiety-related behaviors. Mol. Psychiatry 2017, 22, 1056–1068. [Google Scholar] [CrossRef]
- Bianchi, V.E.; Locatelli, V.; Rizzi, L. Neurotrophic and Neuroregenerative Effects of GH/IGF1. Int. J. Mol. Sci. 2017, 18, 2441. [Google Scholar] [CrossRef]
- Lu, Q.; Tian, X.; Wu, H.; Huang, J.; Li, M.; Mei, Z.; Zhou, L.; Xie, H.; Zheng, S. Metabolic Changes of Hepatocytes in NAFLD. Front. Physiol. 2021, 12, 710420. [Google Scholar] [CrossRef]
- Reed, M.L.; Merriam, G.R.; Kargi, A.Y. Adult growth hormone deficiency—Benefits, side effects, and risks of growth hormone replacement. Front. Endocrinol. 2013, 4, 64. [Google Scholar] [CrossRef] [PubMed]
- Wondmkun, Y.T. Obesity, Insulin Resistance, and Type 2 Diabetes: Associations and Therapeutic Implications. Diabetes Metab. Syndr. Obes. 2020, 13, 3611–3616. [Google Scholar] [CrossRef] [PubMed]
- Lewitt, M.S.; Dent, M.S.; Hall, K. The Insulin-Like Growth Factor System in Obesity, Insulin Resistance and Type 2 Diabetes Mellitus. J. Clin. Med. 2014, 3, 1561–1574. [Google Scholar] [CrossRef]
- Ibrahim Abdalla, M.M. Ghrelin—Physiological Functions and Regulation. Eur. Endocrinol. 2015, 11, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, G.; Samson, S.L.; Sun, Y. Ghrelin: Much more than a hunger hormone. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Giustina, A.; Veldhuis, J.D. Pathophysiology of the neuroregulation of growth hormone secretion in experimental animals and the human. Endocr. Rev. 1998, 19, 717–797. [Google Scholar] [CrossRef] [PubMed]
- Luque, R.M.; Lin, Q.; Córdoba-Chacón, J.; Subbaiah, P.V.; Buch, T.; Waisman, A.; Vankelecom, H.; Kineman, R.D. Metabolic impact of adult-onset, isolated, growth hormone deficiency (AOiGHD) due to destruction of pituitary somatotropes. PLoS ONE 2011, 6, e15767. [Google Scholar] [CrossRef]
- Rimbach, R.; Yamada, Y.; Sagayama, H.; Ainslie, P.N.; Anderson, L.F.; Anderson, L.J.; Arab, L.; Baddou, I.; Bedu-Addo, K.; Blaak, E.E.; et al. Total energy expenditure is repeatable in adults but not associated with short-term changes in body composition. Nat. Commun 2022, 13, 99. [Google Scholar] [CrossRef]
- Hall, K.D.; Farooqi, I.S.; Friedman, J.M.; Klein, S.; Loos, R.J.F.; Mangelsdorf, D.J.; O’Rahilly, S.; Ravussin, E.; Redman, L.M.; Ryan, D.H.; et al. The energy balance model of obesity: Beyond calories in, calories out. Am. J. Clin. Nutr. 2022, 115, 1243–1254. [Google Scholar] [CrossRef]
- Snell, G.D. Dwarf, A New Mendelian Recessive Character of the House Mouse. Proc. Natl. Acad. Sci. USA 1929, 15, 733–734. [Google Scholar] [CrossRef]
- Kano, K. Dwarfism, Mouse. In Brenner’s Encyclopedia of Genetics; Maloy, S., Hughes, K., Eds.; Academic Press: San Diego, CA, USA, 2013; pp. 435–437. [Google Scholar]
- Schaible, R.; Gowen, J. A new dwarf mouse. Genetics 1961, 46, 896. [Google Scholar]
- Palmiter, R.D.; Norstedt, G.; Gelinas, R.E.; Hammer, R.E.; Brinster, R.L. Metallothionein-Human GH Fusion Genes Stimulate Growth of Mice. Science 1983, 222, 809–814. [Google Scholar] [CrossRef]
- List, E.O.; Berryman, D.E.; Buchman, M.; Jensen, E.A.; Funk, K.; Duran-Ortiz, S.; Qian, Y.; Young, J.A.; Slyby, J.; McKenna, S.; et al. GH Knockout Mice Have Increased Subcutaneous Adipose Tissue with Decreased Fibrosis and Enhanced Insulin Sensitivity. Endocrinology 2019, 160, 1743–1756. [Google Scholar] [CrossRef] [PubMed]
- Pilcher, H. Money for old mice. Nature 2003, in press. [Google Scholar] [CrossRef]
- Zhou, Y.; Xu, B.C.; Maheshwari, H.G.; He, L.; Reed, M.; Lozykowski, M.; Okada, S.; Cataldo, L.; Coschigamo, K.; Wagner, T.E.; et al. A mammalian model for Laron syndrome produced by targeted disruption of the mouse growth hormone receptor/binding protein gene (the Laron mouse). Proc. Natl. Acad. Sci. USA 1997, 94, 13215–13220. [Google Scholar] [CrossRef]
- Al-Samerria, S.; Nandankar, N.; Negrón, A.; Radovick, S. PMON53 Resistance to weight gain and obesity in mice lacking the IGF-1 receptor in GHRH neurons. J. Endocr. Soc. 2022, 6 (Suppl. S1), A555. [Google Scholar] [CrossRef]
- Scacchi, M.; Pincelli, A.I.; Cavagnini, F. Growth hormone in obesity. Int. J. Obes. Relat. Metab. Disord. 1999, 23, 260–271. [Google Scholar] [CrossRef]
- Frystyk, J.; Brick, D.J.; Gerweck, A.V.; Utz, A.L.; Miller, K.K. Bioactive insulin-like growth factor-I in obesity. J. Clin. Endocrinol. Metab. 2009, 94, 3093–3097. [Google Scholar] [CrossRef]
- Mekala, K.C.; Tritos, N.A. Effects of Recombinant Human Growth Hormone Therapy in Obesity in Adults: A Metaanalysis. J. Clin. Endocrinol. Metab. 2009, 94, 130–137. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GH–IGF-1 Axis and Obesity | Summary of Current Understanding | References |
|---|---|---|
| GH deficiency and obesity | GH deficiency can lead to an increase in body fat mass, while GH replacement therapy can decrease it. However, the relationship between GH deficiency and obesity is complex and is influenced by various factors. | [97,98] |
| GH excess and obesity | GH excess is associated with decreased body fat mass but also with insulin resistance and glucose intolerance. | [99] |
| IGF-1 and obesity | IGF-1 levels are positively correlated with body fat mass, and low IGF-1 levels have been linked to obesity-related complications such as insulin resistance and T2DM. However, the relationship between IGF-1 and obesity is not fully understood and may be influenced by other factors such as age and gender. | [80,100] |
| GH–IGF-1 axis and adipocyte differentiation | The GH–IGF-1 axis plays a crucial role in regulating adipocyte proliferation and differentiation. GH promotes the differentiation of preadipocytes into mature adipocytes, while IGF-1 promotes the proliferation of preadipocytes. Dysregulation of this process may contribute to the development of obesity. | [94] |
| GH–IGF-1 axis and appetite regulation | GH and IGF-1 have been shown to affect appetite regulation through various mechanisms, such as stimulating the production of leptin and ghrelin. However, the exact role of the GH–IGF-1 axis in appetite regulation and its contribution to obesity is still unclear. | [101,102] |
| Mouse Model | Characteristics | Applications |
|---|---|---|
| Snell dwarf mouse | Naturally occurring mouse model with dwarfism caused by mutations in the Pit-1 gene, which regulates the production of GH, TSH, and PRL. Lack of Pit-1 results in decreased GH and IGF-1 production. | A valuable tool for understanding the GH–IGF-1 axis; extensively utilized to investigate the role of GH and IGF-1 in various physiological processes, such as bone growth, metabolism, and longevity. |
| Ames dwarf mouse | Naturally occurring mouse model with dwarfism caused by mutations in the Pit-1 gene, resulting in proportionate dwarfism with adult mice that are half the size of their control littermates. Almost complete absence of pituitary somatotrophs, lactotrophs, and thyrotropes. | Useful for understanding the GH–IGF-1 axis; studied the role of GH and IGF-1 in bone growth, metabolism, and longevity, as well as the effect of GH on brain development, behavior, and cognitive function; shown to have an elevation in fat mass, making it a useful model for studying the effects of GH on obesity. |
| MT1-hGH transgenic mouse | The transgenic mouse model was generated by fusing the promoter or regulatory region of the mouse gene for metallothionein-I (MT1) with the structural gene coding for human growth hormone (hGH). Exhibits increased size compared with control mice due to elevated levels of GH and a subsequent elevation in IGF-1. | Extensively utilized to study the role of the GH–IGF-1 axis in growth, development, and metabolism; has been used to investigate the effects of alterations in GH and IGF-1 in various tissues and organs involved in energy metabolism such as the liver, adipose tissue, and muscle. |
| (AOiGHD) mouse model | The model was developed by crossbreeding rat GH promoter-driven Cre recombinase mice with inducible diphtheria toxin receptor mice (iDTR) to selectively destroy the somatotroph population of the anterior pituitary gland, leading to a reduction in circulating GH and IGF-1 levels. | This model has been used in various studies to investigate the role of GH in metabolic regulation and to understand the mechanisms underlying metabolic disorders such as obesity, insulin resistance, and diabetes |
| GH−/− mouse model | A genetically engineered mouse model that lacks GH production in the pituitary gland due to a genetic deletion of the GH gene. These mice exhibit a phenotype of reduced body size, muscle mass, and bone density, as well as increased fat mass. | This model is useful for studying the effects of GH deficiency in various physiological processes and for developing therapies for GH deficiency in humans. |
| The GHR−/− (Laron) mouse model | A mouse model with a targeted disruption of the gene encoding the GH receptor (GHR), resulting in a lack of functional GHR. | Useful in understanding the role of the GHR in various physiological processes such as growth, metabolism, and immune function; provides insights into the mechanisms of GHR signaling and its downstream effects on energy metabolism. |
| (AdGHRKO) mouse model | This model was specifically designed to ablate GHR expression from adipose tissue, which caused increased adiposity, enhanced insulin sensitivity, increased fat mass, reduced circulating levels of insulin, c-peptide, adiponectin, and resistin, improved frailty scores with increased grip strength at advanced ages in both sexes, and increased lifespan in male AdGHRKO mice. | Study the specific effects of GH on energy metabolism and the development of obesity; identify potential therapeutic targets for the treatment of obesity and related metabolic disorders. |
| Somatotroph IGF-1R knockout mouse model (SIGFRKO) | Uses the Cre/lox system to specifically delete the IGF-1R from the somatotroph cells. Increased GH expression and secretion, as well as increased serum IGF-1 levels as compensatory changes in the expression of GHRH and SST; normal linear growth in adulthood, elevation in metabolic activity associated with an elevation in energy expenditure reducing the total fat mass due to increased lipolytic activity | A valuable tool for understanding the mechanisms of IGF-1 regulation in the hypothalamic–pituitary axis and the compensatory mechanisms that mediate growth and metabolic function in mammals. |
| Somatotroph GHRH neurons IGF-1R knockout (S-GIGFRKO) mouse mode | Selective deletion of the IGF-1R in GHRH neurons and somatotrophs resulted in a modest increase in serum GH levels and GH gene mRNA expression, as well as a modest increase in serum IGF-1 levels; elevation of GHRH and SST in the hypothalamus normal growth, but adult mice had a reduction in weight gain compared with control littermates; reduction in total fat mass but no changes in lean mass. Elevation in metabolic activity associated with increased energy expenditure | Provides new insights into the role of IGF-1R signaling in GH production and growth regulation and the potential use of this mouse model for further research on GH-related disorders. Can be used to understand the role of the IGF-1R–GHRH pathway in the regulation of body weight and energy balance. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Samerria, S.; Radovick, S. Exploring the Therapeutic Potential of Targeting GH and IGF-1 in the Management of Obesity: Insights from the Interplay between These Hormones and Metabolism. Int. J. Mol. Sci. 2023, 24, 9556. https://doi.org/10.3390/ijms24119556
Al-Samerria S, Radovick S. Exploring the Therapeutic Potential of Targeting GH and IGF-1 in the Management of Obesity: Insights from the Interplay between These Hormones and Metabolism. International Journal of Molecular Sciences. 2023; 24(11):9556. https://doi.org/10.3390/ijms24119556
Chicago/Turabian StyleAl-Samerria, Sarmed, and Sally Radovick. 2023. "Exploring the Therapeutic Potential of Targeting GH and IGF-1 in the Management of Obesity: Insights from the Interplay between These Hormones and Metabolism" International Journal of Molecular Sciences 24, no. 11: 9556. https://doi.org/10.3390/ijms24119556
APA StyleAl-Samerria, S., & Radovick, S. (2023). Exploring the Therapeutic Potential of Targeting GH and IGF-1 in the Management of Obesity: Insights from the Interplay between These Hormones and Metabolism. International Journal of Molecular Sciences, 24(11), 9556. https://doi.org/10.3390/ijms24119556