
Revealing the Role of Alternariol in the Local Steroidogenesis in Human Prostate Normal and Cancer Cells

 ,
,  ,
,  , ,
, ,  and
and
Abstract
1. Introduction
2. Results
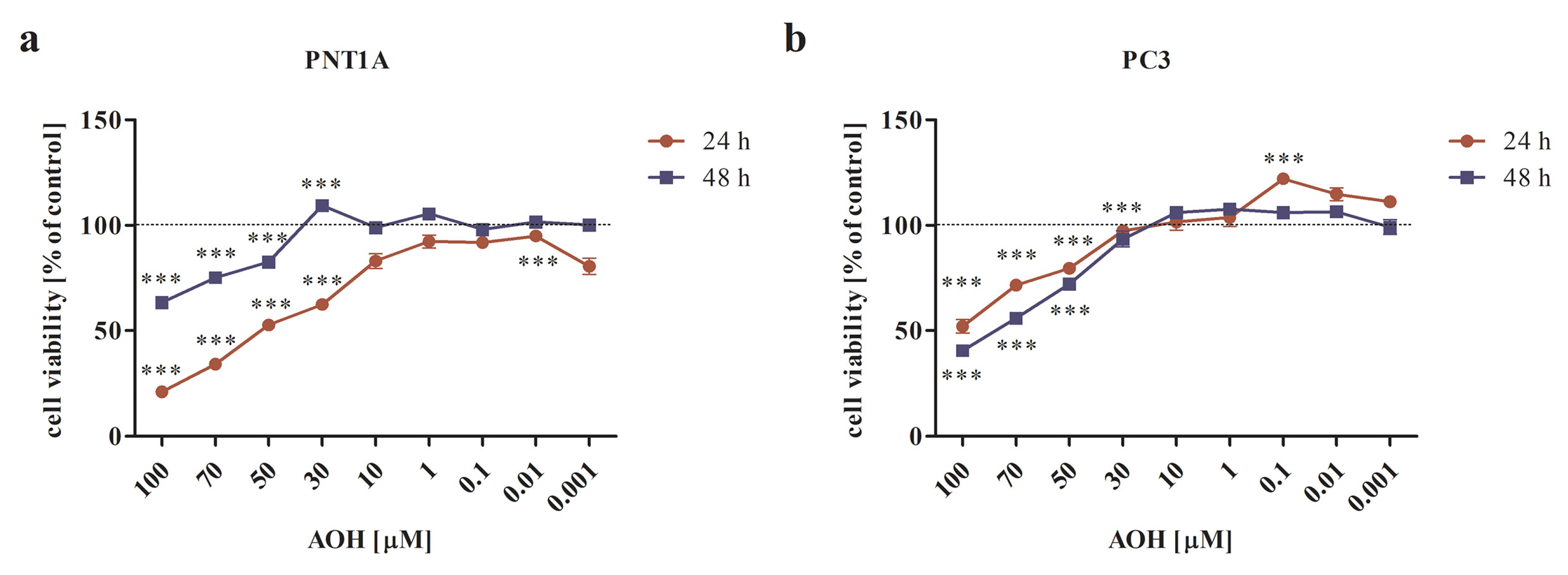
2.1. AOH Modulates the Viability of Prostate Normal and Cancer Cell Lines in a Dose- and Time-Dependent Manner
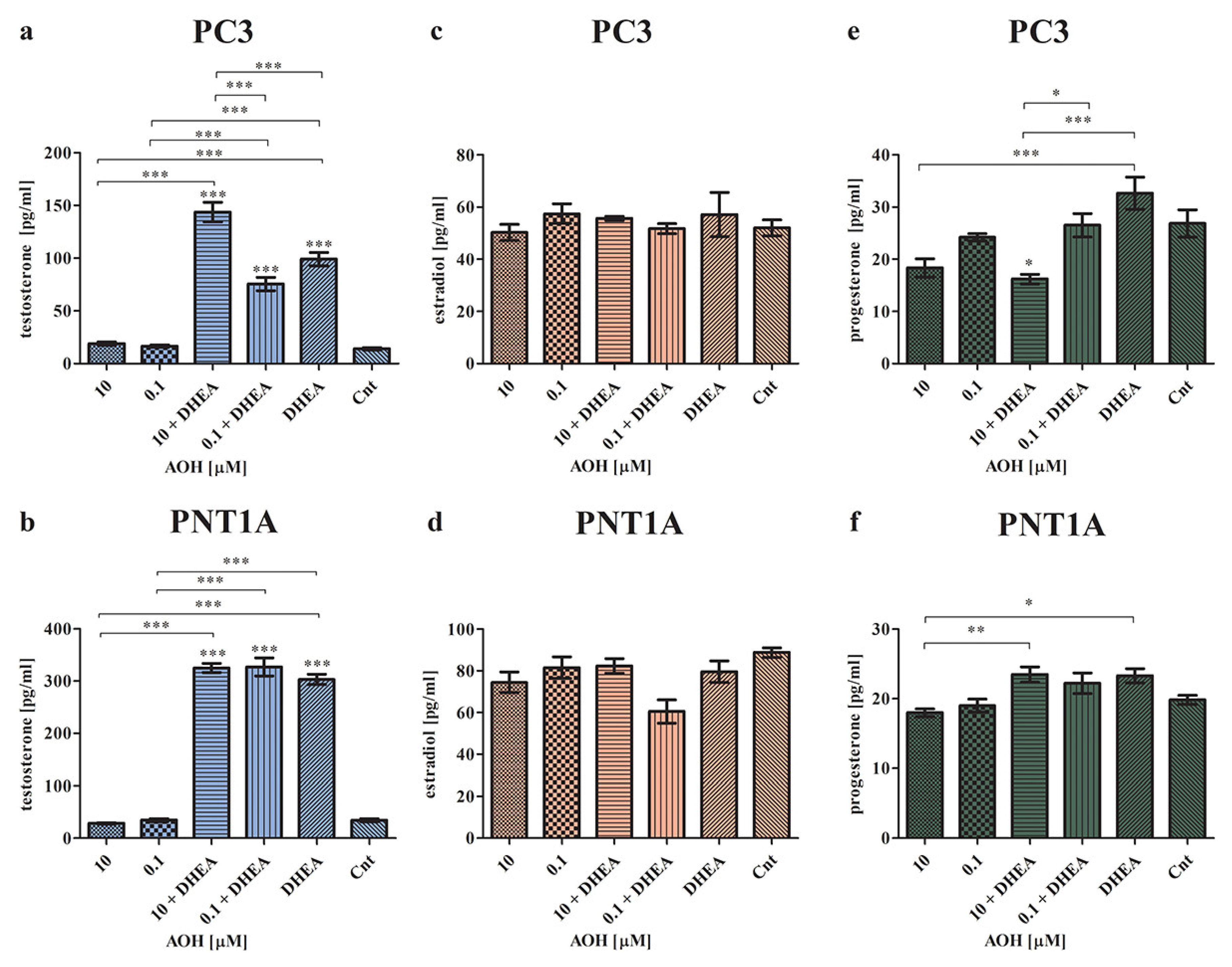
2.2. AOH Modulates the Local Steroidogenesis in Prostate Normal and Cancer Cells in the Presence of DHEA
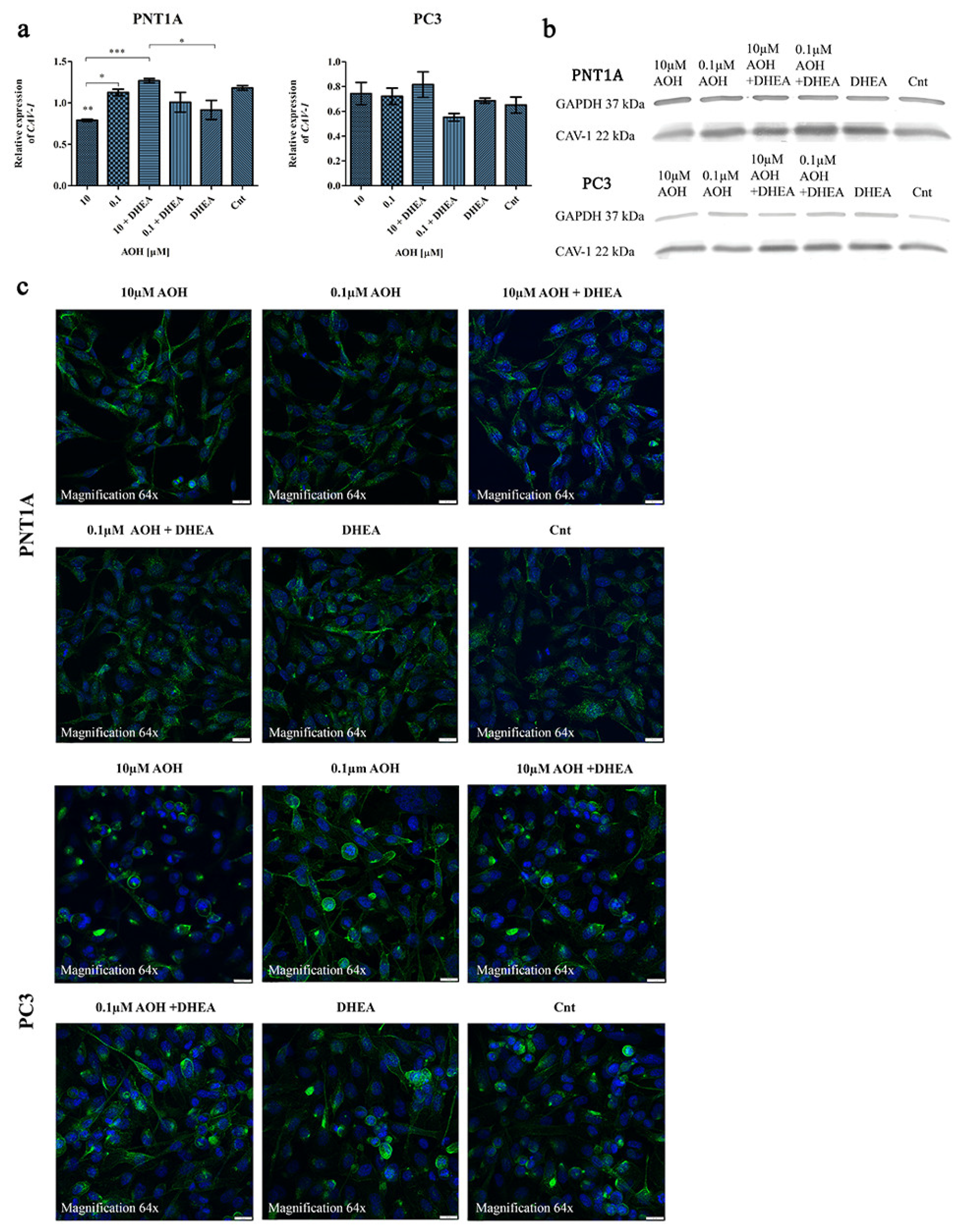
2.3. AOH Affects the Expression of Caveolin-1 (CAV-1) in Prostate Cells
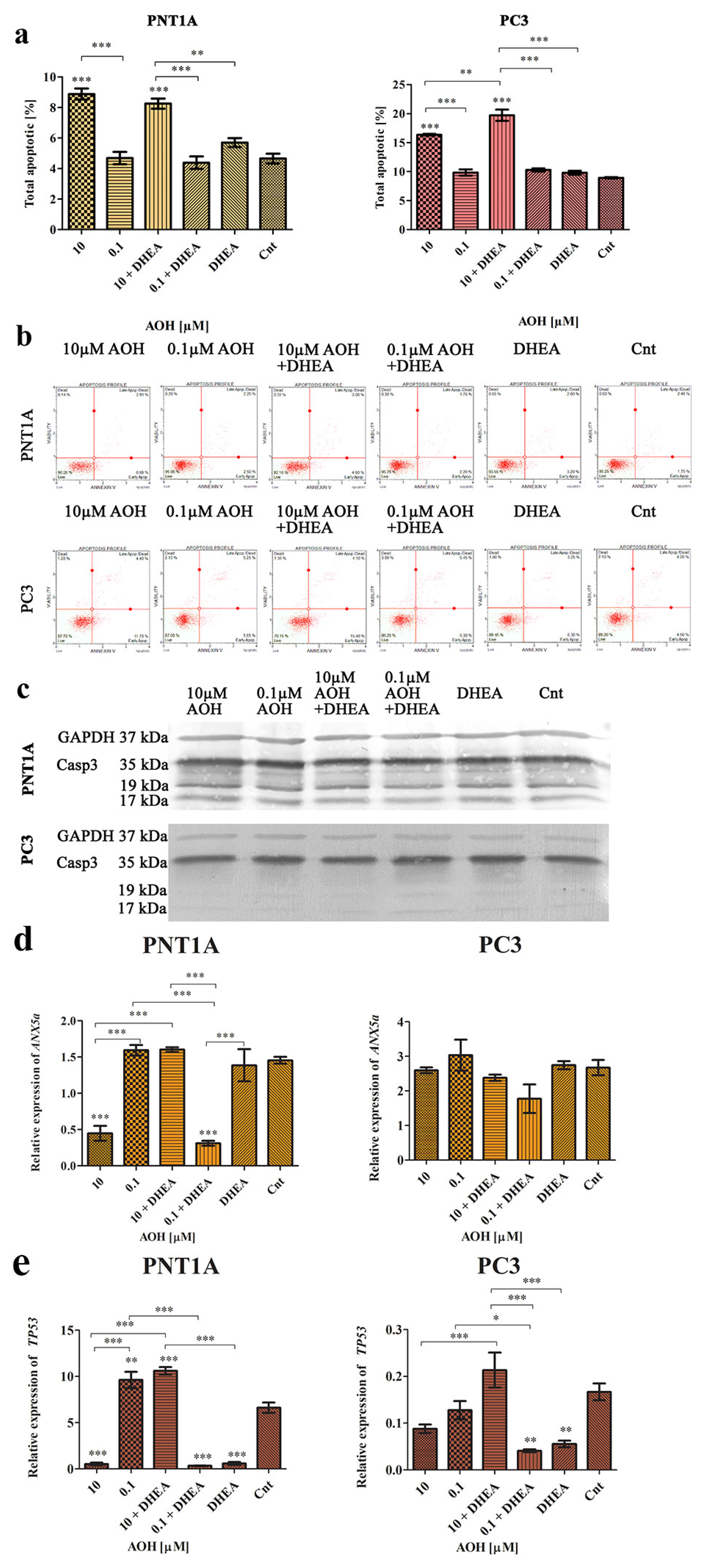
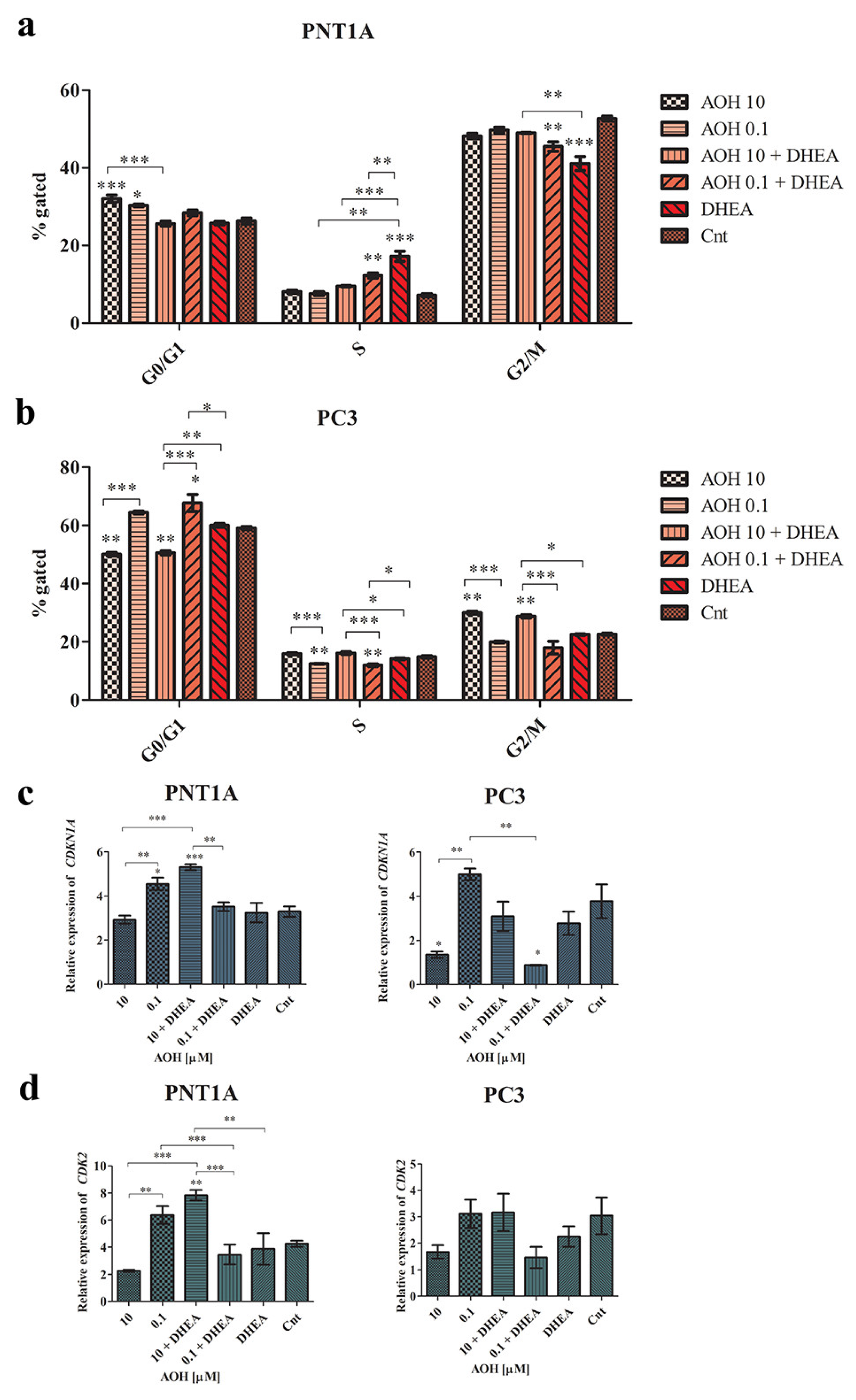
2.4. AOH Modulates Cell Cycle and Apoptosis in Prostate Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Experimental Treatments
4.2. Cell Viability Assay
4.3. Annexin V Staining Assay
4.4. Cell Cycle
4.5. Steroid Assays
4.6. RNA Extraction and Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
4.7. Western Blot
4.8. Immunohistochemistry Staining and Confocal Microscopy
4.9. Statistical Analysis
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Holanda, D.M.; Kim, S.W. Mycotoxin Occurrence, Toxicity, and Detoxifying Agents in Pig Production with an Emphasis on Deoxynivalenol. Toxins 2021, 13, 171. [Google Scholar] [CrossRef]
- Eskola, M.; Kos, G.; Elliott, C.T.; Hajšlová, J.; Mayar, S.; Krska, R. Worldwide contamination of food-crops with mycotoxins: Validity of the widely cited ‘FAO estimate’ of 25%. Crit. Rev. Food Sci. Nutr. 2020, 60, 2773–2789. [Google Scholar] [CrossRef] [PubMed]
- Solhaug, A.; Eriksen, G.S.; Holme, J.A. Mechanisms of Action and Toxicity of the Mycotoxin Alternariol: A Review. Basic Clin. Pharmacol. Toxicol. 2016, 119, 533–539. [Google Scholar] [CrossRef]
- Crudo, F.; Aichinger, G.; Dellafiora, L.; Kiss, E.; Mihajlovic, J.; Del Favero, G.; Berry, D.; Dall’Asta, C.; Marko, D. Persistence of the antagonistic effects of a natural mixture of Alternaria mycotoxins on the estrogen-like activity of human feces after anaerobic incubation. Toxicol. Lett. 2022, 358, 88–99. [Google Scholar] [CrossRef]
- Den Hollander, D.; Holvoet, C.; Demeyere, K.; De Zutter, N.; Audenaert, K.; Meyer, E.; Croubels, S. Cytotoxic Effects of Alternariol, Alternariol Monomethyl-Ether, and Tenuazonic Acid and Their Relevant Combined Mixtures on Human Enterocytes and Hepatocytes. Front. Microbiol. 2022, 13, 9243. [Google Scholar] [CrossRef] [PubMed]
- Scientific Opinion on the risks for animal and public health related to the presence of Alternaria toxins in feed and food. EFSA J. 2011, 9, 2407. [CrossRef]
- Fraeyman, S.; Croubels, S.; Devreese, M.; Antonissen, G. Emerging Fusarium and Alternaria Mycotoxins: Occurrence, Toxicity and Toxicokinetics. Toxins 2017, 9, 228. [Google Scholar] [CrossRef] [PubMed]
- Frizzell, C.; Ndossi, D.; Verhaegen, S.; Dahl, E.; Eriksen, G.; Sørlie, M.; Ropstad, E.; Muller, M.; Elliott, C.T.; Connolly, L. Endocrine disrupting effects of zearalenone, alpha- and beta-zearalenol at the level of nuclear receptor binding and steroidogenesis. Toxicol. Lett. 2011, 206, 210–217. [Google Scholar] [CrossRef]
- Kalayou, S.; Hamre, A.G.; Ndossi, D.; Connolly, L.; Sørlie, M.; Ropstad, E.; Verhaegen, S. Using SILAC proteomics to investigate the effect of the mycotoxin, alternariol, in the human H295R steroidogenesis model. Cell Biol. Toxicol. 2014, 30, 361–376. [Google Scholar] [CrossRef]
- Del Favero, G.; Mayer, R.M.; Dellafiora, L.; Janker, L.; Niederstaetter, L.; Dall’Asta, C.; Gerner, C.; Marko, D. Structural Similarity with Cholesterol Reveals Crucial Insights into Mechanisms Sustaining the Immunomodulatory Activity of the Mycotoxin Alternariol. Cells 2020, 9, 847. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P. Epidemiology of Prostate Cancer. World J. Oncol. 2019, 10, 63–89. [Google Scholar] [CrossRef] [PubMed]
- Shafi, A.A.; Yen, A.E.; Weigel, N.L. Androgen receptors in hormone-dependent and castration-resistant prostate cancer. Pharmacol. Ther. 2013, 140, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Chung, Y.-M.; Sergeeva, O.; Kepe, V.; Berk, M.; Li, J.; Ko, H.-K.; Li, Z.; Petro, M.; DiFilippo, F.P.; et al. Loss of dihydrotestosterone-inactivation activity promotes prostate cancer castration resistance detectable by functional imaging. J. Biol. Chem. 2018, 293, 17829–17837. [Google Scholar] [CrossRef] [PubMed]
- Frycz, B.A.; Jagodziński, P.P. Expressions of genes encoding steroidogenic enzymes and their role in prostate carcinogenesis. J. Med. Sci. 2014, 83, 73–80. [Google Scholar] [CrossRef]
- Fokidis, H.B.; Adomat, H.H.; Kharmate, G.; Hosseini-Beheshti, E.; Guns, E.S.; Soma, K.K. Regulation of local steroidogenesis in the brain and in prostate cancer: Lessons learned from interdisciplinary collaboration. Front. Neuroendocrinol. 2015, 36, 108–129. [Google Scholar] [CrossRef]
- Škara, L.; Huđek Turković, A.; Pezelj, I.; Vrtarić, A.; Sinčić, N.; Krušlin, B.; Ulamec, M. Prostate Cancer—Focus on Cholesterol. Cancers 2021, 13, 4696. [Google Scholar] [CrossRef]
- Deb, S.; Pham, S.; Ming, D.-S.; Chin, M.; Adomat, H.; Hurtado-Coll, A.; Gleave, M.; Guns, E. Characterization of Precursor-Dependent Steroidogenesis in Human Prostate Cancer Models. Cancers 2018, 10, 343. [Google Scholar] [CrossRef]
- Miyoshi, Y.; Uemura, H.; Umemoto, S.; Sakamaki, K.; Taguri, M.; Suzuki, K.; Shibata, Y.; Masumori, N.; Ichikawa, T.; Mizokami, A.; et al. Low serum dehydroepiandrosterone examined by liquid chromatography-tandem mass spectrometry correlates with poor prognosis in hormone-naïve prostate cancer. Prostate 2016, 76, 376–382. [Google Scholar] [CrossRef]
- Urbanek, K.A.; Kowalska, K.; Habrowska-Górczyńska, D.E.; Domińska, K.; Sakowicz, A.; Piastowska-Ciesielska, A.W. In Vitro Analysis of Deoxynivalenol Influence on Steroidogenesis in Prostate. Toxins 2021, 13, 685. [Google Scholar] [CrossRef]
- Kowalska, K.; Kozieł, M.J.; Urbanek, K.A.; Habrowska-Górczyńska, D.E.; Domińska, K.; Piastowska-Ciesielska, A.W. Estrogen receptor β participates in alternariol-induced oxidative stress in normal prostate epithelial cells. Toxins 2021, 13, 766. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Xu, H.; Li, Z.; Li, F. Interactions of caveolin-1 scaffolding and intramembrane regions containing a CRAC motif with cholesterol in lipid bilayers. Biochim. Biophys. Acta—Biomembr. 2014, 1838, 2588–2599. [Google Scholar] [CrossRef] [PubMed]
- Chan, N.N.; Yamazaki, M.; Maruyama, S.; Abé, T.; Haga, K.; Kawaharada, M.; Izumi, K.; Kobayashi, T.; Tanuma, J. Cholesterol Is a Regulator of CAV1 Localization and Cell Migration in Oral Squamous Cell Carcinoma. Int. J. Mol. Sci. 2023, 24, 6035. [Google Scholar] [CrossRef] [PubMed]
- Thompson, T.C.; Tahir, S.A.; Li, L.; Watanabe, M.; Naruishi, K.; Yang, G.; Kadmon, D.; Logothetis, C.J.; Troncoso, P.; Ren, C.; et al. The role of caveolin-1 in prostate cancer: Clinical implications. Prostate Cancer Prostatic Dis. 2010, 13, 6–11. [Google Scholar] [CrossRef]
- Ayala, G.; Morello, M.; Frolov, A.; You, S.; Li, R.; Rosati, F.; Bartolucci, G.; Danza, G.; Adam, R.M.; Thompson, T.C.; et al. Loss of caveolin-1 in prostate cancer stroma correlates with reduced relapse-free survival and is functionally relevant to tumour progression. J. Pathol. 2013, 231, 77–87. [Google Scholar] [CrossRef]
- Maxfield, F.R.; Wüstner, D. Intracellular cholesterol transport. J. Clin. Investig. 2002, 110, 891–898. [Google Scholar] [CrossRef]
- Kowalska, K.; Nowakowska, M.; Dominska, K.; Piastowska-Ciesielska, A.W. Coexpression of CAV-1, AT1-R and FOXM1 in prostate and breast cancer and normal cell lines and their influence on metastatic properties. Acta Biochim. 2016, 63, 493–499. [Google Scholar] [CrossRef]
- Ali, A.; Kulik, G. Signaling Pathways That Control Apoptosis in Prostate Cancer. Cancers 2021, 13, 937. [Google Scholar] [CrossRef]
- Small, E.J.; Ryan, C.J. The Case for Secondary Hormonal Therapies in the Chemotherapy Age. J. Urol. 2006, 176, S66–S71. [Google Scholar] [CrossRef]
- Montgomery, R.B.; Mostaghel, E.A.; Vessella, R.; Hess, D.L.; Kalhorn, T.F.; Higano, C.S.; True, L.D.; Nelson, P.S. Maintenance of intratumoral androgens in metastatic prostate cancer: A mechanism for castration-resistant tumor growth. Cancer Res. 2008, 68, 4447–4454. [Google Scholar] [CrossRef]
- Fehr, M.; Pahlke, G.; Fritz, J.; Christensen, M.O.; Boege, F.; Altemöller, M.; Podlech, J.; Marko, D. Alternariol acts as a topoisomerase poison, preferentially affecting the IIα isoform. Mol. Nutr. Food Res. 2009, 53, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, L.; Wagner, J.; Metzler, M. Estrogenic and clastogenic potential of the mycotoxin alternariol in cultured mammalian cells. Food Chem. Toxicol. 2006, 44, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Stypuła-Trębas, S.; Minta, M.; Radko, L.; Jedziniak, P.; Posyniak, A. Nonsteroidal mycotoxin alternariol is a full androgen agonist in the yeast reporter androgen bioassay. Environ. Toxicol. Pharmacol. 2017, 55, 208–211. [Google Scholar] [CrossRef]
- Dellafiora, L.; Warth, B.; Schmidt, V.; Del Favero, G.; Mikula, H.; Fröhlich, J.; Marko, D. An integrated in silico/in vitro approach to assess the xenoestrogenic potential of Alternaria mycotoxins and metabolites. Food Chem. 2018, 248, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Arcella, D.; Eskola, M.; Gómez Ruiz, J.A. Dietary exposure assessment to Alternaria toxins in the European population. EFSA J. 2016, 14, e04654. [Google Scholar] [CrossRef]
- Puntscher, H.; Aichinger, G.; Grabher, S.; Attakpah, E.; Krüger, F.; Tillmann, K.; Motschnig, T.; Hohenbichler, J.; Braun, D.; Plasenzotti, R.; et al. Bioavailability, metabolism, and excretion of a complex Alternaria culture extract versus altertoxin II: A comparative study in rats. Arch. Toxicol. 2019, 93, 3153–3167. [Google Scholar] [CrossRef]
- Ndossi, D.G.; Frizzell, C.; Tremoen, N.H.; Fæste, C.K.; Verhaegen, S.; Dahl, E.; Eriksen, G.S.; Sørlie, M.; Connolly, L.; Ropstad, E. An in vitro investigation of endocrine disrupting effects of trichothecenes deoxynivalenol (DON), T-2 and HT-2 toxins. Toxicol. Lett. 2012, 214, 268–278. [Google Scholar] [CrossRef]
- Rebhahn, V.I.C.; Kiss, E.; Marko, D.; Del Favero, G. Foodborne compounds that alter plasma membrane architecture can modify the response of intestinal cells to shear stress in vitro. Toxicol. Appl. Pharmacol. 2022, 446, 116034. [Google Scholar] [CrossRef]
- Huang, C.-H.; Wang, F.-T.; Chan, W.-H. Alternariol exerts embryotoxic and immunotoxic effects on mouse blastocysts through ROS-mediated apoptotic processes. Toxicol. Res. 2021, 10, 719–732. [Google Scholar] [CrossRef]
- Fernández-Blanco, C.; Juan-García, A.; Juan, C.; Font, G.; Ruiz, M.-J. Alternariol induce toxicity via cell death and mitochondrial damage on Caco-2 cells. Food Chem. Toxicol. 2016, 88, 32–39. [Google Scholar] [CrossRef]
- Bensassi, F.; Gallerne, C.; Sharaf El Dein, O.; Hajlaoui, M.R.; Bacha, H.; Lemaire, C. Cell death induced by the Alternaria mycotoxin Alternariol. Toxicol. Vitr. 2012, 26, 915–923. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Treatment | CAV-1 |
|---|---|---|
| PNT1A | 10 μM AOH | 0.62 |
| 0.1 μM AOH | 1.06 | |
| 100 nM DHEA | 1.08 | |
| 10 μM AOH + 100 nM DHEA | 1.61 | |
| 0.1 μM AOH + 100 nM DHEA | 1.48 | |
| Cnt | 1.00 | |
| PC3 | 10 μM AOH | 0.67 |
| 0.1 μM AOH | 0.44 | |
| 100 nM DHEA | 0.95 | |
| 10 μM AOH + 100 nM DHEA | 0.62 | |
| 0.1 μM AOH + 100 nM DHEA | 0.57 | |
| Cnt | 1.00 |
| Cell Line | Treatment | Casp3 | Cleaved Casp3 |
|---|---|---|---|
| PNT1A | 10 μM AOH | 0.94 | 0.70 |
| 0.1 μM AOH | 0.89 | 0.66 | |
| 100 nM DHEA | 1.33 | 1.03 | |
| 10 μM AOH + 100 nM DHEA | 0.92 | 0.71 | |
| 0.1 μM AOH + 100 nM DHEA | 1.31 | 0.80 | |
| Cnt | 1.00 | 1.00 | |
| PC3 | 10 μM AOH | 2.15 | 1.83 |
| 0.1 μM AOH | 1.07 | 1.42 | |
| 100 nM DHEA | 1.51 | 1.92 | |
| 10 μM AOH + 100 nM DHEA | 0.98 | 0.98 | |
| 0.1 μM AOH + 100 nM DHEA | 0.99 | 1.38 | |
| Cnt | 1.00 | 1.00 |
| Gene | Sequence (5′-3′) | Product Size (bp) |
|---|---|---|
| CYP11A1 | For CCAGAACGATTCCTCATCC | 126 |
| Rev CATCACCTCCTGGTTCAG | ||
| CYP17A1 | For GAAGTTATCATCAATCTGTGGG | 119 |
| Rev ACTGACGGTGAGATGAGC | ||
| Rev AAGATGTCTGGTTTGATGAGGAG | ||
| HSD3B2 | For CTTGGTGTCACTCACAGAGAG | 128 |
| Rev GTAGATGAAGACTGGCACACTG | ||
| Rev CACCTCCAATTGTGACATAA | ||
| STAR | For CATGGAGAGGCTCTATGAAGA | 128 |
| Rev CAGCCAGCTCGTGAGTAAT | ||
| ANXA5 | For ACCCTCTCGGCTTTATGATGCT | 116 |
| Rev TGGCTCTCAGTTCTTCAGGTGT | ||
| Rev TGCTGGACAGAAATGTGTACACTCCAGA | ||
| ESR2 | For ACACCTGGGCACCTTTCTCCTTTA | 90 |
| Rev TCTTGCTTCACACCAGGGACTCTT | ||
| AR | For GGGAGGTTACACCAAAGGGC | 102 |
| Rev AGAGACAGGGTAGACGGCAG | ||
| CAV-1 | Rev GAACTTGAAATTGGCACCAGG | 139 |
| For ACCCACTCTTTGAAGCTGTTG | ||
| CDK1N1A | Rev CTGAGACTAAGGCAGAAGATGT | 133 |
| For GACAGATTTCTACCACTCCAA | ||
| CDK2 | Rev GAGCAGAGGCATCCATGAATT | 126 |
| For TGCTTAAGGAGCTTAACCATCC | ||
| TP53 | Rev TTTATGGCGGGAGGTAGA | 102 |
| For TTGGAACTCAAGGATGCC | ||
| For GCAAGACTGTTAGCCCTCAA | ||
| RPLP0 | For ACGGATTACACCTTCCCACTTGCTAAAAGGTC | 69 |
| Rev AGCCACAAAGGCAGATGGATCAGCCAAG | ||
| RPS17 | For AAGCGCGTGTGCGAGGAGATCG | 87 |
| Rev TCGCTTCATCAGAT GCGTGACATAACCTG | ||
| H3F3A | For AGGACTTTAAAAGATCTGCGCTTCCAGAG | 74 |
| Rev ACCAGATAGGCCTCACTTGCCTCCTGC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urbanek, K.A.; Kowalska, K.; Habrowska-Górczyńska, D.E.; Kozieł, M.J.; Domińska, K.; Piastowska-Ciesielska, A.W. Revealing the Role of Alternariol in the Local Steroidogenesis in Human Prostate Normal and Cancer Cells. Int. J. Mol. Sci. 2023, 24, 9513. https://doi.org/10.3390/ijms24119513
Urbanek KA, Kowalska K, Habrowska-Górczyńska DE, Kozieł MJ, Domińska K, Piastowska-Ciesielska AW. Revealing the Role of Alternariol in the Local Steroidogenesis in Human Prostate Normal and Cancer Cells. International Journal of Molecular Sciences. 2023; 24(11):9513. https://doi.org/10.3390/ijms24119513
Chicago/Turabian StyleUrbanek, Kinga Anna, Karolina Kowalska, Dominika Ewa Habrowska-Górczyńska, Marta Justyna Kozieł, Kamila Domińska, and Agnieszka Wanda Piastowska-Ciesielska. 2023. "Revealing the Role of Alternariol in the Local Steroidogenesis in Human Prostate Normal and Cancer Cells" International Journal of Molecular Sciences 24, no. 11: 9513. https://doi.org/10.3390/ijms24119513
APA StyleUrbanek, K. A., Kowalska, K., Habrowska-Górczyńska, D. E., Kozieł, M. J., Domińska, K., & Piastowska-Ciesielska, A. W. (2023). Revealing the Role of Alternariol in the Local Steroidogenesis in Human Prostate Normal and Cancer Cells. International Journal of Molecular Sciences, 24(11), 9513. https://doi.org/10.3390/ijms24119513