
Chondrocyte Thrombomodulin Protects against Osteoarthritis

 ,
,  ,
,  , , ,
, , ,  and
and 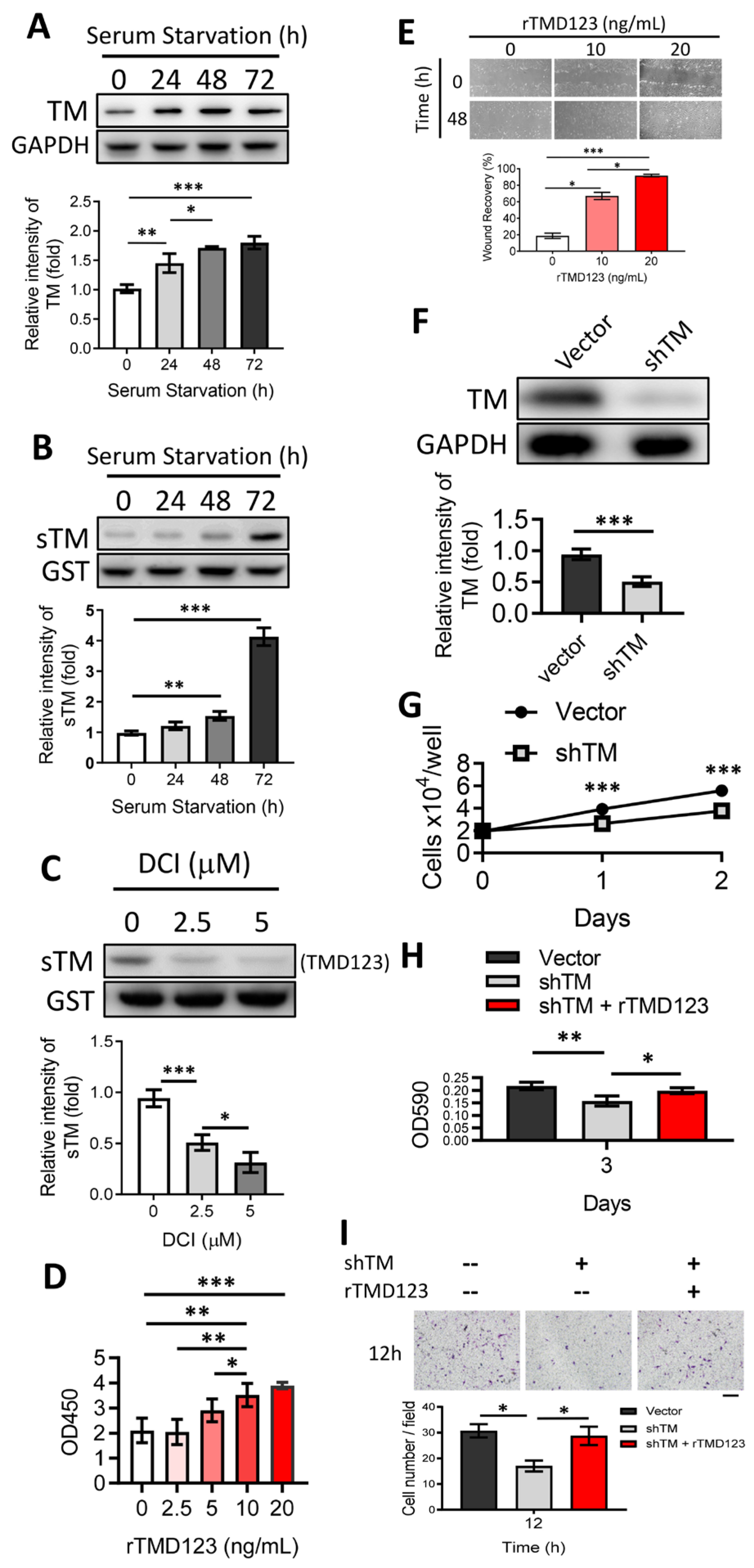{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Chondrocyte Exposure to the Rhomboid Protease RHBDL2 Liberates Soluble TM (sTM), Inducing Cell Proliferation and Migration
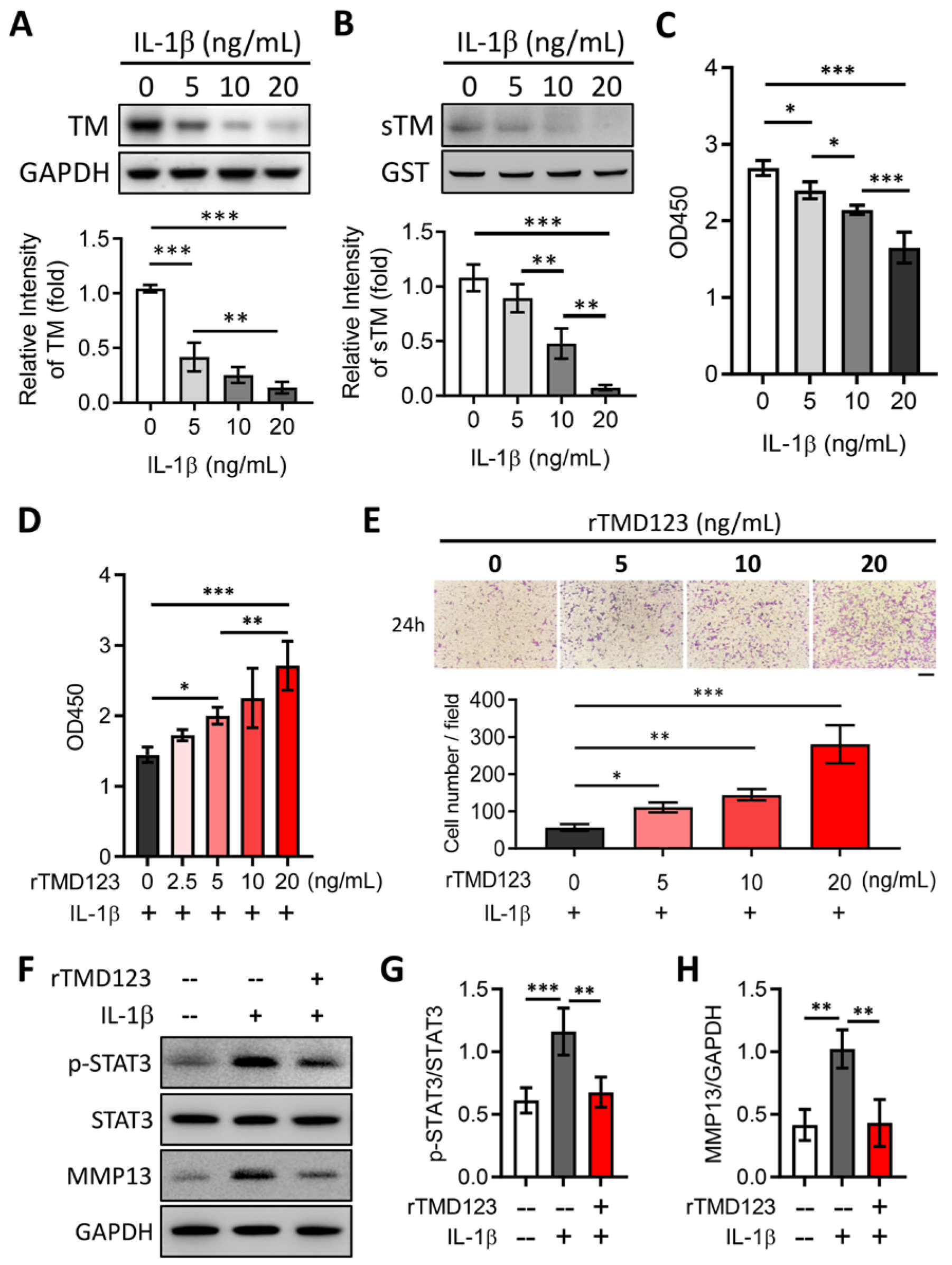
2.2. rTMD123 Inhibits STAT3/MMP 13 Signaling and IL-1β-Mediated Suppression of TM Expression in Chondrocytes
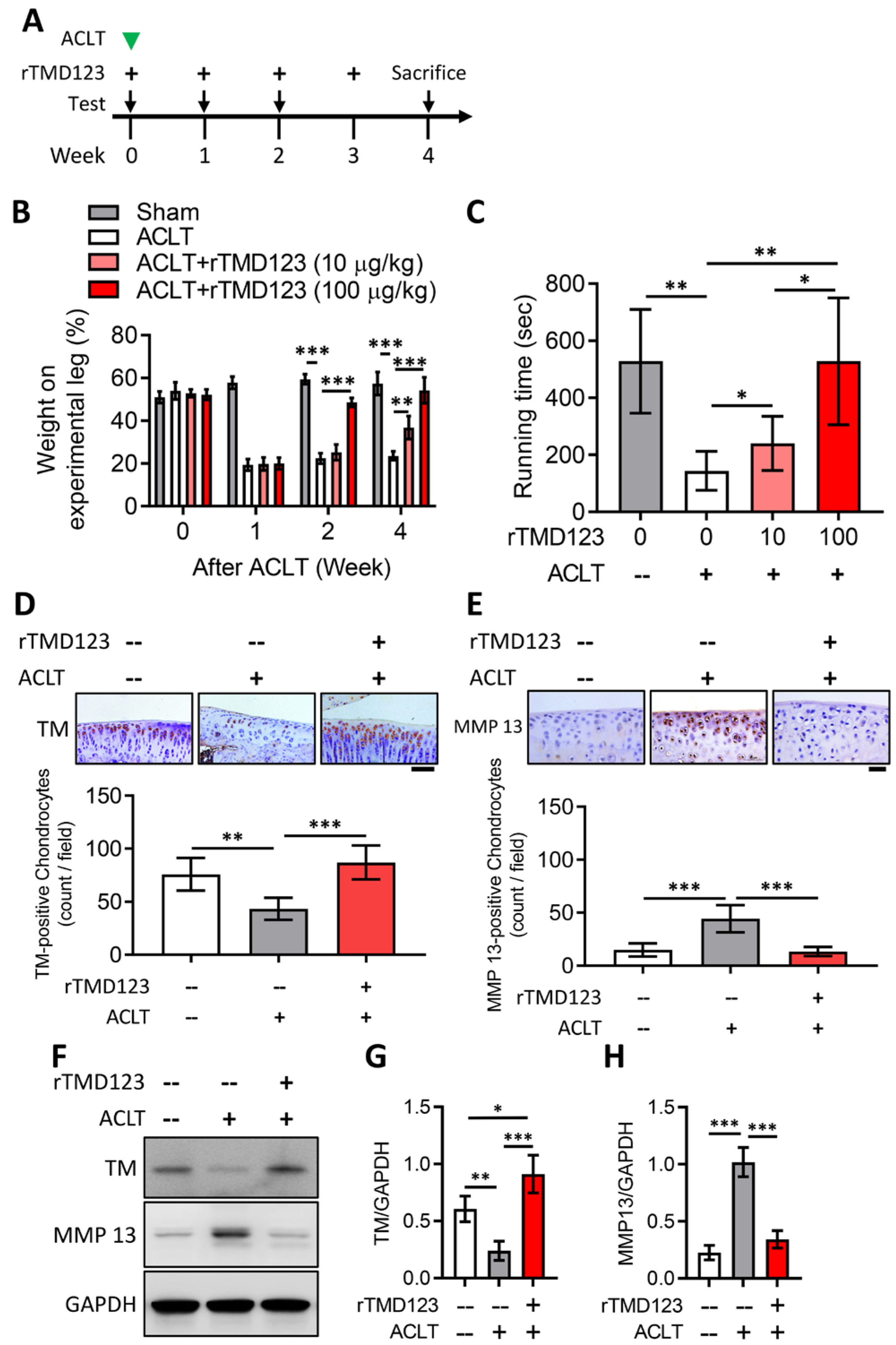
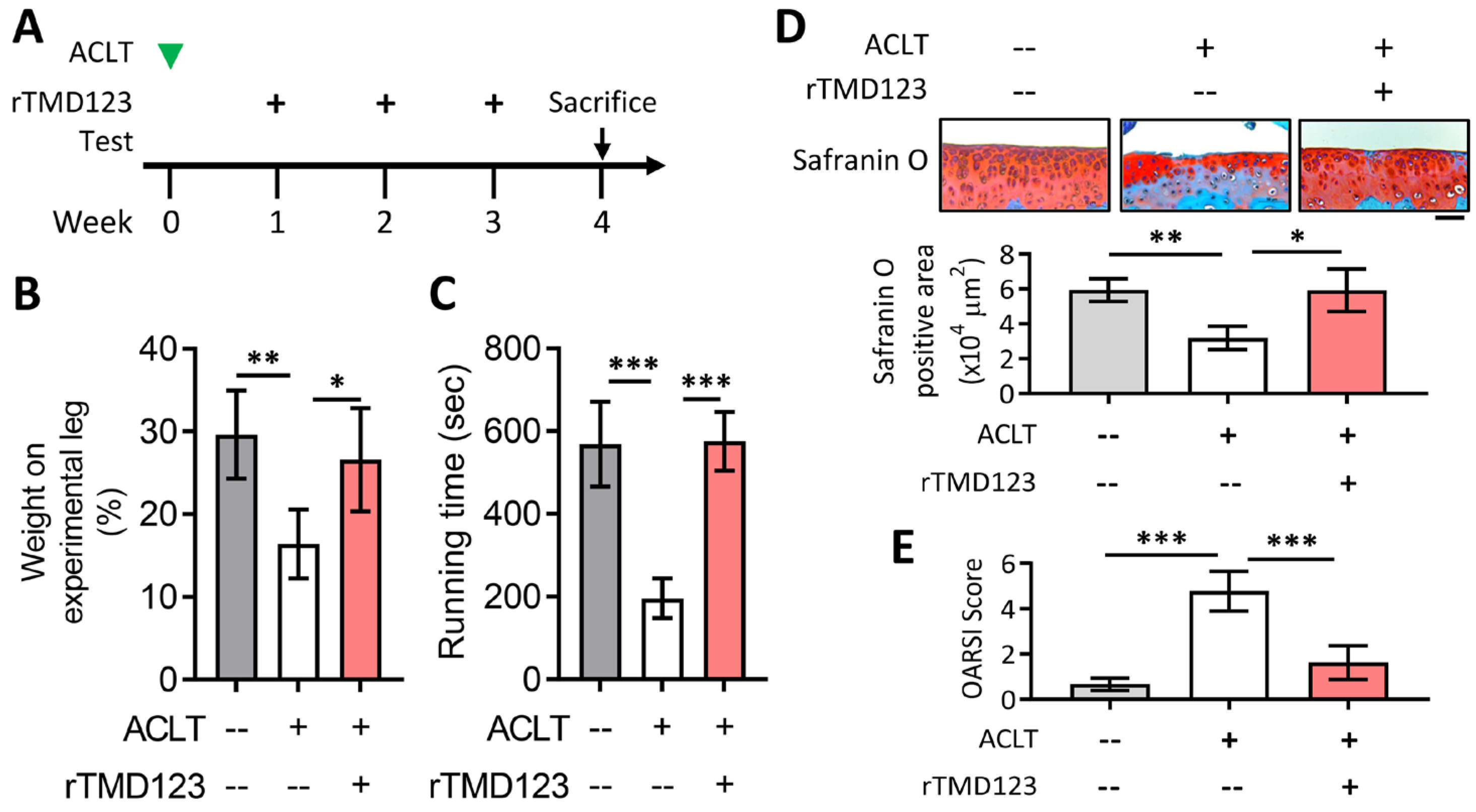
2.3. rTMD123 Protects Mouse Knees from ACLT-Induced OA and Dysfunction, Increases Articular Cartilage TM and Reduces MMP 13
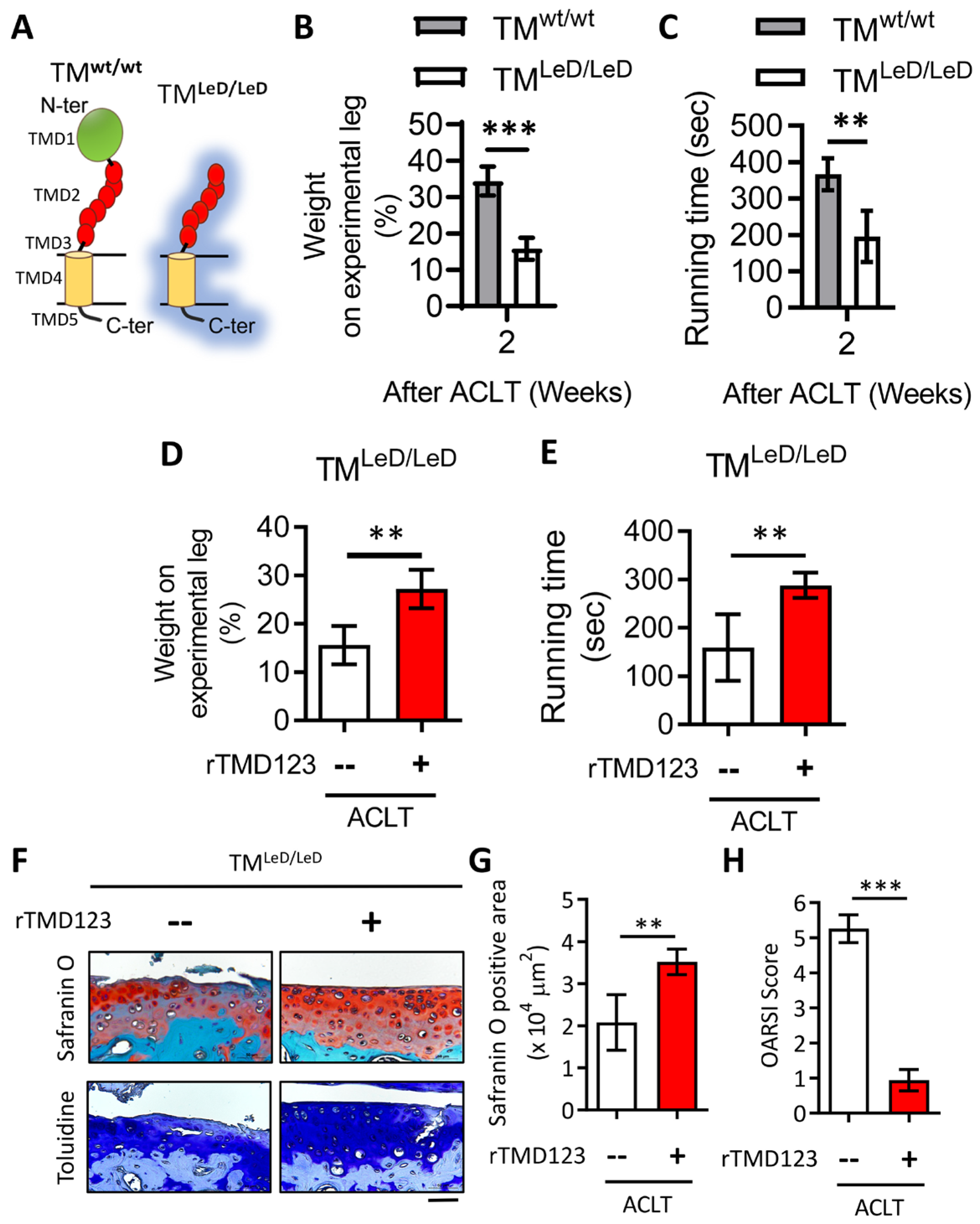
2.4. rTMD123 Rescues Knee Dysfunction and Chondro-Cartilage Joint Damage in TMLeD/LeD Mice with OA
2.5. rTMD123 Protects against Knee Dysfunction and Articular Cartilage Loss after OA-Injury Induction
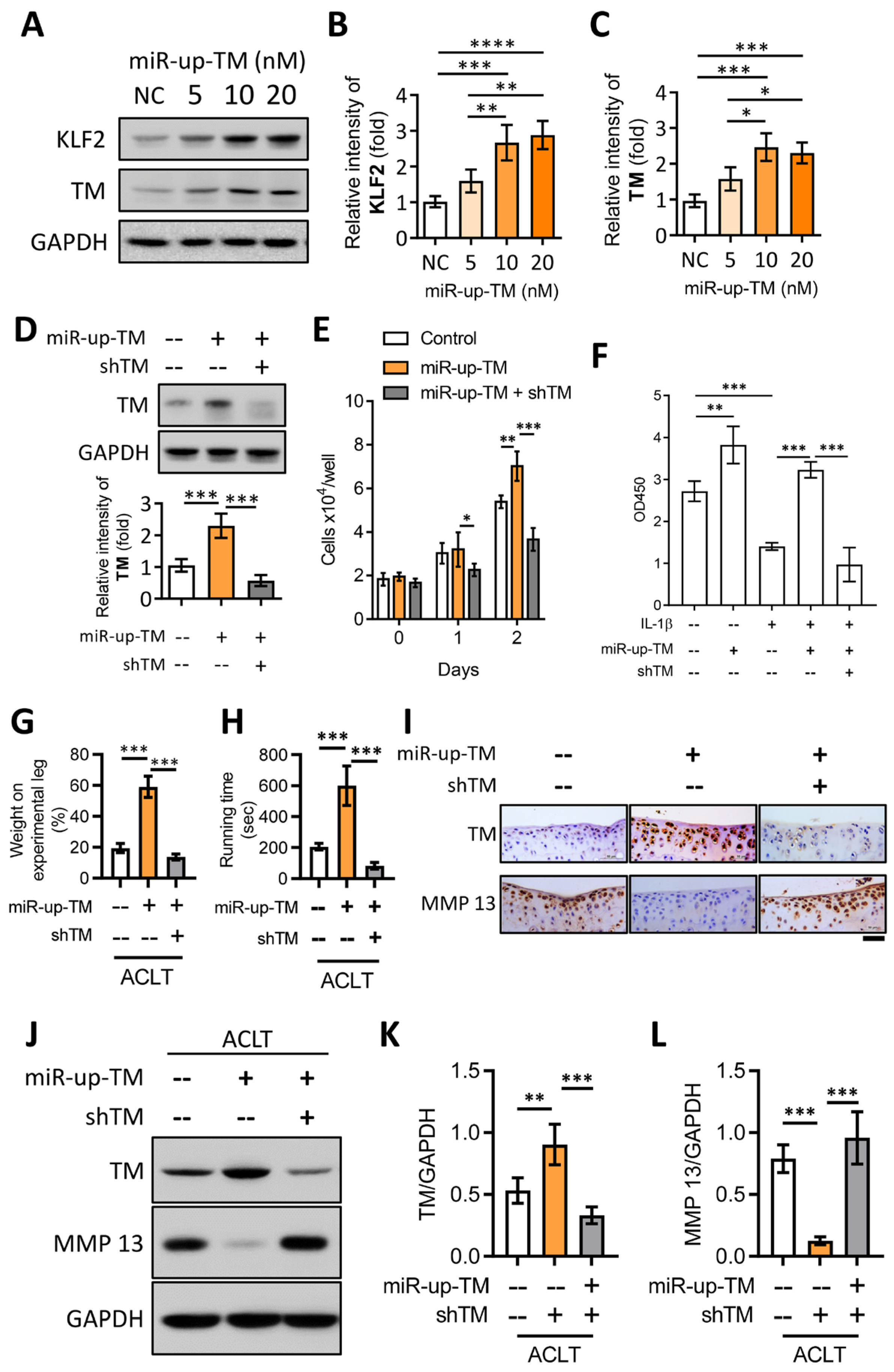
2.6. TM Silencing Inhibits miR-150 Antagomir (miR-up-TM)-Increased KLF2/TM Expression and Abolishes the TM-Mediated Protective Effects on Knee Dysfunction in the OA Model
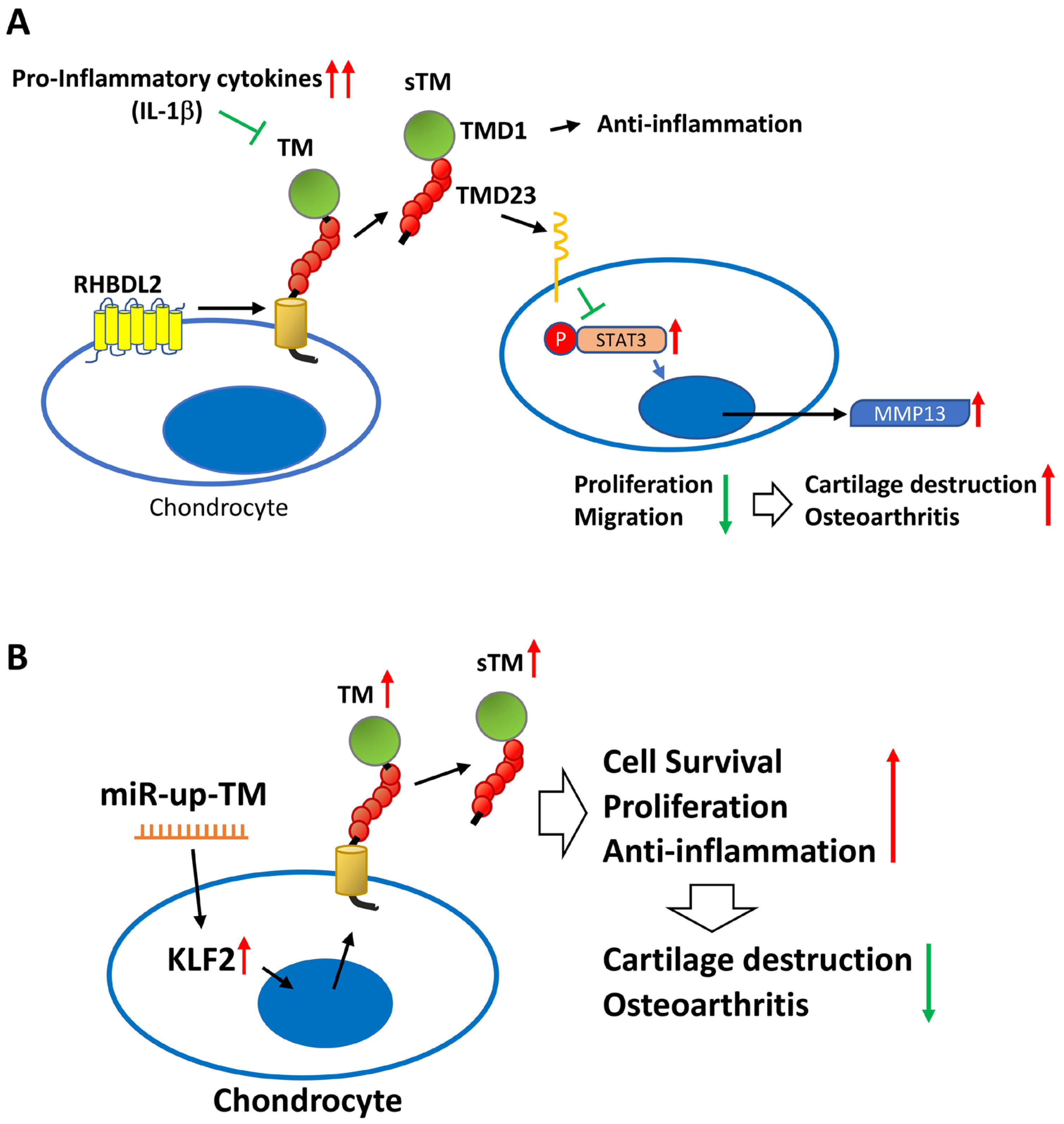
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Expression of Recombinant TM Domains
4.3. Cell Culture
4.4. Western Blotting
4.5. Cell Proliferation/Viability Assay
4.6. In Vitro Wound Healing Assay
4.7. Transwell Cell Migration Assay
4.8. Animals
4.9. Anterior Cruciate Ligament Transection (ACLT)-Induced Knee OA
4.10. Intra-Articular Injection in OA Mice Model
4.11. Weight-Bearing Distribution Test
4.12. Treadmill Test
4.13. Histology and Immunohistochemistry
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- GBD 2017 Disease and Injury Incidence and Prevalence Collaborators; James, S.L.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar] [CrossRef]
- Loeser, R.F.; Goldring, S.R.; Scanzello, C.R.; Goldring, M.B. Osteoarthritis: A disease of the joint as an organ. Arthritis Rheumatol. 2012, 64, 1697–1707. [Google Scholar] [CrossRef]
- Archer, C.W.; Francis-West, P. The chondrocyte. Int. J. Biochem. Cell Biol. 2003, 35, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Maillard, C.; Berruyer, M.; Serre, C.M.; Amiral, J.; Dechavanne, M.; Delmas, P.D. Thrombomodulin is synthesized by osteoblasts, stimulated by 1,25-(OH)2D3 and activates protein C at their cell membrane. Endocrinology 1993, 133, 668–674. [Google Scholar] [CrossRef] [PubMed]
- McCachren, S.S.; Diggs, J.; Weinberg, J.B.; Dittman, W.A. Thrombomodulin expression by human blood monocytes and by human synovial tissue lining macrophages. Blood 1991, 78, 3128–3132. [Google Scholar] [CrossRef] [PubMed]
- Raife, T.J.; Lager, D.J.; Madison, K.C.; Piette, W.W.; Howard, E.J.; Sturm, M.T.; Chen, Y.; Lentz, S.R. Thrombomodulin expression by human keratinocytes. Induction of cofactor activity during epidermal differentiation. J. Clin. Investig. 1994, 93, 1846–1851. [Google Scholar] [CrossRef]
- Stearns-Kurosawa, D.J.; Kurosawa, S.; Mollica, J.S.; Ferrell, G.L.; Esmon, C.T. The endothelial cell protein C receptor augments protein C activation by the thrombin-thrombomodulin complex. Proc. Natl. Acad. Sci. USA 1996, 93, 10212–10216. [Google Scholar] [CrossRef]
- Jackson, M.T.; Smith, M.M.; Smith, S.M.; Jackson, C.J.; Xue, M.; Little, C.B. Activation of cartilage matrix metalloproteinases by activated protein C. Arthritis Rheumatol. 2009, 60, 780–791. [Google Scholar] [CrossRef]
- Weiler, H.; Isermann, B.H. Thrombomodulin. J. Thromb. Haemost. 2003, 1, 1515–1524. [Google Scholar] [CrossRef]
- Esmon, C.T. Thrombomodulin as a model of molecular mechanisms that modulate protease specificity and function at the vessel surface. FASEB J. 1995, 9, 946–955. [Google Scholar] [CrossRef]
- Hamada, H.; Ishii, H.; Sakyo, K.; Horie, S.; Nishiki, K.; Kazama, M. The epidermal growth factor-like domain of recombinant human thrombomodulin exhibits mitogenic activity for Swiss 3T3 cells. Blood 1995, 86, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.C.; Shi, G.Y.; Jiang, S.J.; Shi, C.S.; Wu, C.M.; Yang, H.Y.; Wu, H.L. Thrombomodulin-mediated cell adhesion: Involvement of its lectin-like domain. J. Biol. Chem. 2003, 278, 46750–46759. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.S.; Shi, G.Y.; Chang, Y.S.; Han, H.S.; Kuo, C.H.; Liu, C.; Huang, H.C.; Chang, Y.J.; Chen, P.S.; Wu, H.L. Evidence of human thrombomodulin domain as a novel angiogenic factor. Circulation 2005, 111, 1627–1636. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.S.; Shi, G.Y.; Hsiao, H.M.; Kao, Y.C.; Kuo, K.L.; Ma, C.Y.; Kuo, C.H.; Chang, B.I.; Chang, C.F.; Lin, C.H.; et al. Lectin-like domain of thrombomodulin binds to its specific ligand Lewis Y antigen and neutralizes lipopolysaccharide-induced inflammatory response. Blood 2008, 112, 3661–3670. [Google Scholar] [CrossRef]
- Kuo, C.H.; Chen, P.K.; Chang, B.I.; Sung, M.C.; Shi, C.S.; Lee, J.S.; Chang, C.F.; Shi, G.Y.; Wu, H.L. The recombinant lectin-like domain of thrombomodulin inhibits angiogenesis through interaction with Lewis Y antigen. Blood 2012, 119, 1302–1313. [Google Scholar] [CrossRef]
- Cheng, T.L.; Wu, Y.T.; Lin, H.Y.; Hsu, F.C.; Liu, S.K.; Chang, B.I.; Chen, W.S.; Lai, C.H.; Shi, G.Y.; Wu, H.L. Functions of rhomboid family protease RHBDL2 and thrombomodulin in wound healing. J. Investig. Dermatol. 2011, 131, 2486–2494. [Google Scholar] [CrossRef]
- Chen, C.H.; Lai, C.H.; Hong, Y.K.; Lu, J.M.; Lin, S.Y.; Lee, T.C.; Chang, L.Y.; Ho, M.L.; Conway, E.M.; Wu, H.L.; et al. Thrombomodulin Functional Domains Support Osteoblast Differentiation and Bone Healing in Diabetes in Mice. J. Bone Miner. Res. 2020, 35, 1812–1823. [Google Scholar] [CrossRef]
- Cheng, T.L.; Lai, C.H.; Shieh, S.J.; Jou, Y.B.; Yeh, J.L.; Yang, A.L.; Wang, Y.H.; Wang, C.Z.; Chen, C.H.; Shi, G.Y.; et al. Myeloid thrombomodulin lectin-like domain inhibits osteoclastogenesis and inflammatory bone loss. Sci. Rep. 2016, 6, 28340. [Google Scholar] [CrossRef]
- Lohi, O.; Urban, S.; Freeman, M. Diverse substrate recognition mechanisms for rhomboids; Thrombomodulin is cleaved by Mammalian rhomboids. Curr. Biol. 2004, 14, 236–241. [Google Scholar]
- Conway, E.M.; Rosenberg, R.D. Tumor necrosis factor suppresses transcription of the thrombomodulin gene in endothelial cells. Mol. Cell. Biol. 1988, 8, 5588–5592. [Google Scholar]
- Gracia-Sancho, J.; Russo, L.; Garcia-Caldero, H.; Garcia-Pagan, J.C.; Garcia-Cardena, G.; Bosch, J. Endothelial expression of transcription factor Kruppel-like factor 2 and its vasoprotective target genes in the normal and cirrhotic rat liver. Gut 2011, 60, 517–524. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, Q.; Gao, Z.; Yu, C.; Zhang, L. Down-Regulation of MiR-150 Alleviates Inflammatory Injury Induced by Interleukin 1 via Targeting Kruppel-Like Factor 2 in Human Chondrogenic Cells. Cell. Physiol. Biochem. 2018, 47, 2579–2588. [Google Scholar] [CrossRef]
- Conway, E.M.; Nowakowski, B. Biologically active thrombomodulin is synthesized by adherent synovial fluid cells and is elevated in synovial fluid of patients with rheumatoid arthritis. Blood 1993, 81, 726–733. [Google Scholar] [CrossRef]
- Hopper, N.; Henson, F.; Brooks, R.; Ali, E.; Rushton, N.; Wardale, J. Peripheral blood derived mononuclear cells enhance osteoarthritic human chondrocyte migration. Arthritis Res. Ther. 2015, 17, 199. [Google Scholar] [CrossRef]
- Hopper, N.; Wardale, J.; Brooks, R.; Power, J.; Rushton, N.; Henson, F. Peripheral Blood Mononuclear Cells Enhance Cartilage Repair in in vivo Osteochondral Defect Model. PLoS ONE 2015, 10, e0133937. [Google Scholar] [CrossRef]
- Wojdasiewicz, P.; Poniatowski, L.A.; Szukiewicz, D. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of osteoarthritis. Mediat. Inflamm. 2014, 2014, 561459. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Chen, T.; Qiu, J.; Gao, W.; Qiu, X.; Zhu, Y.; Wang, X.; Chen, Y.; Zhou, H.; Deng, Z.; et al. Inhibition of nuclear receptor RORalpha attenuates cartilage damage in osteoarthritis by modulating IL-6/STAT3 pathway. Cell Death Dis. 2021, 12, 886. [Google Scholar] [CrossRef] [PubMed]
- Van de Wouwer, M.; Plaisance, S.; De Vriese, A.; Waelkens, E.; Collen, D.; Persson, J.; Daha, M.R.; Conway, E.M. The lectin-like domain of thrombomodulin interferes with complement activation and protects against arthritis. J. Thromb. Haemost. 2006, 4, 1813–1824. [Google Scholar] [CrossRef]
- Martin, F.A.; Murphy, R.P.; Cummins, P.M. Thrombomodulin and the vascular endothelium: Insights into functional, regulatory, and therapeutic aspects. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1585–H1597. [Google Scholar] [CrossRef]
- Conway, E.M. Thrombomodulin and its role in inflammation. Semin. Immunopathol. 2012, 34, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Tonge, D.P.; Pearson, M.J.; Jones, S.W. The hallmarks of osteoarthritis and the potential to develop personalised disease-modifying pharmacological therapeutics. Osteoarthr. Cartil. 2014, 22, 609–621. [Google Scholar] [CrossRef]
- Aigner, T.; Soder, S.; Gebhard, P.M.; McAlinden, A.; Haag, J. Mechanisms of disease: Role of chondrocytes in the pathogenesis of osteoarthritis—Structure, chaos and senescence. Nat. Clin. Pract. Rheumatol. 2007, 3, 391–399. [Google Scholar] [CrossRef]
- Cohen, S.B.; Proudman, S.; Kivitz, A.J.; Burch, F.X.; Donohue, J.P.; Burstein, D.; Sun, Y.N.; Banfield, C.; Vincent, M.S.; Ni, L.; et al. A randomized, double-blind study of AMG 108 (a fully human monoclonal antibody to IL-1R1) in patients with osteoarthritis of the knee. Arthritis Res. Ther. 2011, 13, R125. [Google Scholar] [CrossRef]
- Wei, Y.; Luo, L.; Gui, T.; Yu, F.; Yan, L.; Yao, L.; Zhong, L.; Yu, W.; Han, B.; Patel, J.M.; et al. Targeting cartilage EGFR pathway for osteoarthritis treatment. Sci. Transl. Med. 2021, 13, eabb3946. [Google Scholar] [CrossRef]
- Conway, E.M.; Van de Wouwer, M.; Pollefeyt, S.; Jurk, K.; Van Aken, H.; De Vriese, A.; Weitz, J.I.; Weiler, H.; Hellings, P.W.; Schaeffer, P.; et al. The lectin-like domain of thrombomodulin confers protection from neutrophil-mediated tissue damage by suppressing adhesion molecule expression via nuclear factor kappaB and mitogen-activated protein kinase pathways. J. Exp. Med. 2002, 196, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Thachil, J.; Asakura, H.; Levy, J.H.; Iba, T. Thrombomodulin in disseminated intravascular coagulation and other critical conditions-a multi-faceted anticoagulant protein with therapeutic potential. Crit. Care 2019, 23, 280. [Google Scholar] [CrossRef]
- Turpaev, K.T. Transcription Factor KLF2 and Its Role in the Regulation of Inflammatory Processes. Biochemistry 2020, 85, 54–67. [Google Scholar] [CrossRef]
- Kao, Y.C.; Wu, L.W.; Shi, C.S.; Chu, C.H.; Huang, C.W.; Kuo, C.P.; Sheu, H.M.; Shi, G.Y.; Wu, H.L. Downregulation of thrombomodulin, a novel target of Snail, induces tumorigenesis through epithelial-mesenchymal transition. Mol. Cell. Biol. 2010, 30, 4767–4785. [Google Scholar] [CrossRef] [PubMed]
- Kamekura, S.; Hoshi, K.; Shimoaka, T.; Chung, U.; Chikuda, H.; Yamada, T.; Uchida, M.; Ogata, N.; Seichi, A.; Nakamura, K.; et al. Osteoarthritis development in novel experimental mouse models induced by knee joint instability. Osteoarthr. Cartil. 2005, 13, 632–641. [Google Scholar] [CrossRef]
- Wen, Z.H.; Tang, C.C.; Chang, Y.C.; Huang, S.Y.; Chen, C.H.; Wu, S.C.; Hsieh, S.P.; Hsieh, C.S.; Wang, K.Y.; Lin, S.Y.; et al. Intra-articular injection of the selective cyclooxygenase-2 inhibitor meloxicam (Mobic) reduces experimental osteoarthritis and nociception in rats. Osteoarthr. Cartil. 2013, 21, 1976–1986. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Ho, M.L.; Chang, L.H.; Kang, L.; Lin, Y.S.; Lin, S.Y.; Wu, S.C.; Chang, J.K. Parathyroid hormone-(1-34) ameliorated knee osteoarthritis in rats via autophagy. J. Appl. Physiol. 2018, 124, 1177–1185. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, L.; Yang, A.-L.; Lai, C.-H.; Chen, T.-J.; Lin, S.-Y.; Wang, Y.-H.; Wang, C.-Z.; Conway, E.M.; Wu, H.-L.; Ho, M.-L.; et al. Chondrocyte Thrombomodulin Protects against Osteoarthritis. Int. J. Mol. Sci. 2023, 24, 9522. https://doi.org/10.3390/ijms24119522
Kang L, Yang A-L, Lai C-H, Chen T-J, Lin S-Y, Wang Y-H, Wang C-Z, Conway EM, Wu H-L, Ho M-L, et al. Chondrocyte Thrombomodulin Protects against Osteoarthritis. International Journal of Molecular Sciences. 2023; 24(11):9522. https://doi.org/10.3390/ijms24119522
Chicago/Turabian StyleKang, Lin, Ai-Lun Yang, Chao-Han Lai, Tsan-Ju Chen, Sung-Yen Lin, Yan-Hsiung Wang, Chau-Zen Wang, Edward M. Conway, Hua-Lin Wu, Mei-Ling Ho, and et al. 2023. "Chondrocyte Thrombomodulin Protects against Osteoarthritis" International Journal of Molecular Sciences 24, no. 11: 9522. https://doi.org/10.3390/ijms24119522
APA StyleKang, L., Yang, A.-L., Lai, C.-H., Chen, T.-J., Lin, S.-Y., Wang, Y.-H., Wang, C.-Z., Conway, E. M., Wu, H.-L., Ho, M.-L., Chang, J.-K., Chen, C.-H., & Cheng, T.-L. (2023). Chondrocyte Thrombomodulin Protects against Osteoarthritis. International Journal of Molecular Sciences, 24(11), 9522. https://doi.org/10.3390/ijms24119522