
Non-Haemodynamic Mechanisms Underlying Hypertension-Associated Damage in Target Kidney Components
 , ,
, ,  , and
, and
Abstract
1. Introduction
2. Mechanisms Underlying Kidney Injury in Arterial Hypertension
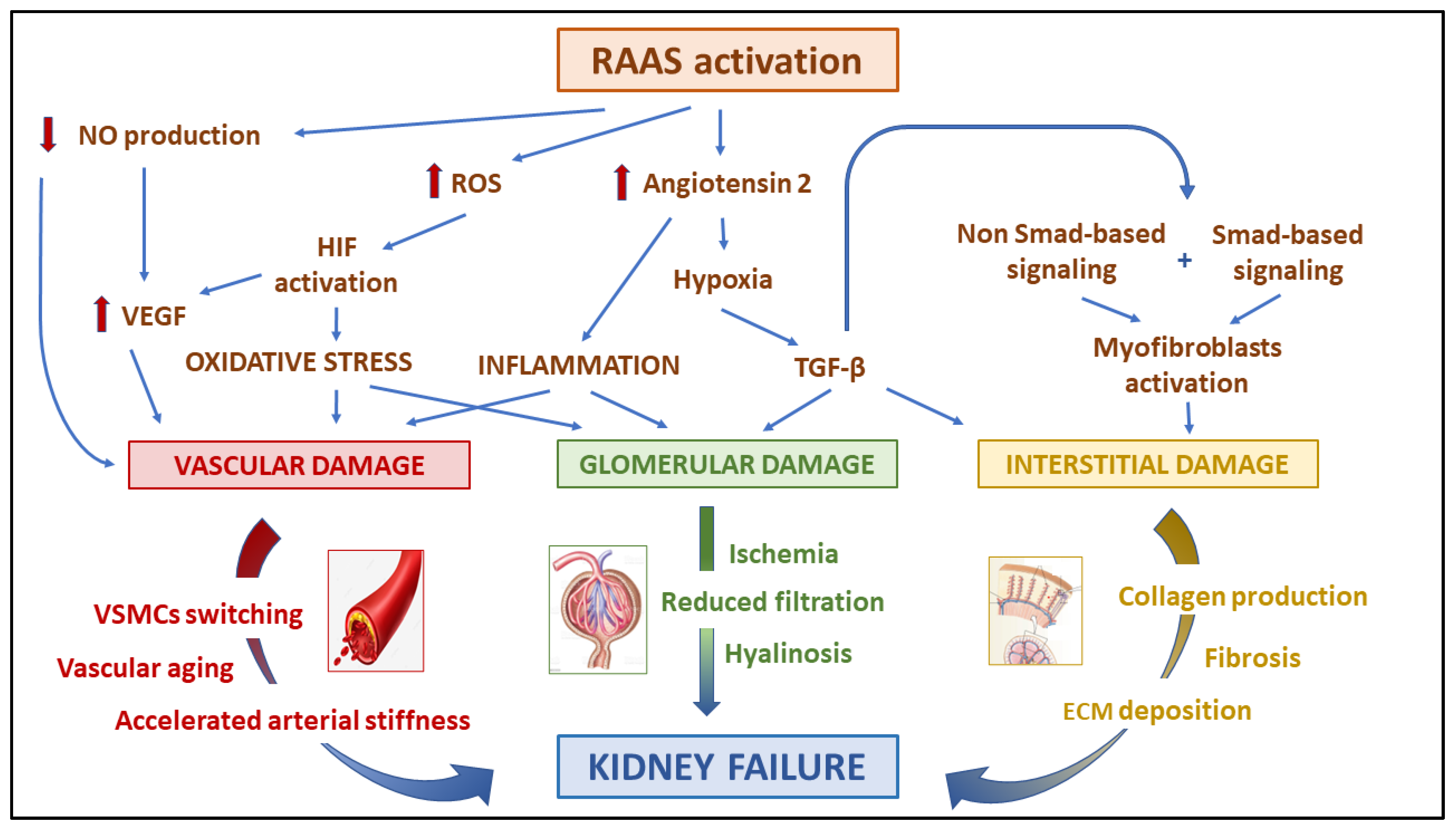
2.1. Renin–Angiotensin–Aldosterone System
2.2. Endothelin-1 and Its Signaling Pathway
2.3. Innate Immunity Response and Oxidative Stress
2.4. Hypoxia
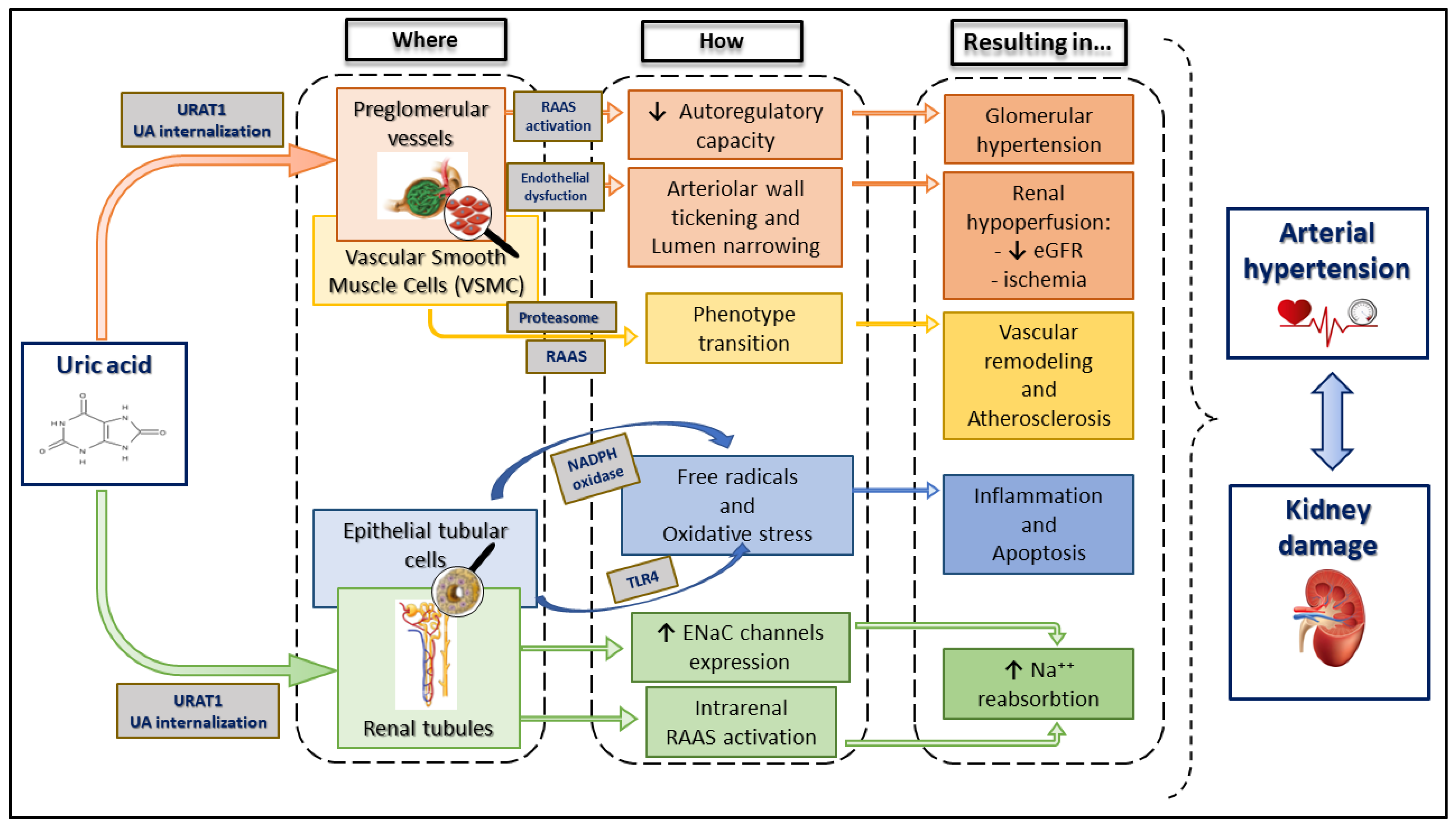
2.5. Uric Acid
2.6. Ageing
2.7. miRNA
3. Histopathological Kidney Changes during Arterial Hypertension
3.1. Smooth Muscle Cell Phenotypic Switching
3.2. Epithelial-to-Mesenchymal Transition and Endothelial-to-Mesenchymal Transition
3.3. Renal Congestion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mancia, G.; Fagard, R.; Narkiewicz, K.; Redón, J.; Zanchetti, A.; Böhm, M.; Christiaens, T.; Cifkova, R.; De Backer, G.; Dominiczak, A.; et al. 2013 ESH/ESC Guidelines for the management of arterial hypertension: The Task Force for the management of arterial hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). J. Hypertens. 2013, 31, 1281–1357. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.C.; Becker, G.J. Summary of KDIGO guideline. What do we really know about management of blood pressure in patients with chronic kidney disease? Kidney Int. 2013, 83, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Meyrier, A. Nephrosclerosis: Update on a centenarian. Nephrol. Dial. Transpl. 2015, 30, 1833–1841. [Google Scholar] [CrossRef] [PubMed]
- Bidani, A.K.; Griffin, K.A. Pathophysiology of hypertensive renal damage: Implications for therapy. Hypertension 2004, 44, 595–601. [Google Scholar] [CrossRef]
- Viazzi, F.; Bonino, B.; Mirijello, A.; Fioretto, P.; Giorda, C.; Ceriello, A.; Guida, P.; Russo, G.T.; De Cosmo, S.; Pontremoli, R.; et al. Long-term blood pressure variability and development of chronic kidney disease in type 2 diabetes. J. Hypertens. 2019, 37, 805–813. [Google Scholar] [CrossRef]
- Viazzi, F.; Russo, E.; Mirijello, A.; Fioretto, P.; Giorda, C.; Ceriello, A.; Copetti, M.; Russo, G.T.; Di Bartolo, P.; Manicardi, V.; et al. Long-term blood pressure variability, incidence of hypertension and changes in renal function in type 2 diabetes. J. Hypertens. 2020, 38, 2279–2286. [Google Scholar] [CrossRef]
- Theofilis, P.; Sagris, M.; Oikonomou, E.; Antonopoulos, A.S.; Siasos, G.; Tsioufis, C.; Tousoulis, D. Inflammatory Mechanisms Contributing to Endothelial Dysfunction. Biomedicines 2021, 9, 781. [Google Scholar] [CrossRef]
- Guzik, T.J.; Touyz, R.M. Oxidative Stress, Inflammation, and Vascular Aging in Hypertension. Hypertension 2017, 70, 660–667. [Google Scholar] [CrossRef]
- Leoncini, G.; Viazzi, F.; Conti, N.; Baratto, E.; Tomolillo, C.; Bezante, G.P.; Deferrari, G.; Pontremoli, R. Renal and cardiac abnormalities in primary hypertension. J. Hypertens. 2009, 27, 1064–1073. [Google Scholar] [CrossRef]
- Leoncini, G.; Viazzi, F.; De Cosmo, S.; Russo, G.; Fioretto, P.; Pontremoli, R. Blood pressure reduction and RAAS inhibition in diabetic kidney disease: Therapeutic potentials and limitations. J. Nephrol. 2020, 33, 949–963. [Google Scholar] [CrossRef]
- Leoncini, G.; Viazzi, F.; Pontremoli, R. RAAS inhibition and renal protection. Curr. Pharm. Des. 2012, 18, 971–980. [Google Scholar] [CrossRef]
- Koszegi, S.; Molnar, A.; Lenart, L.; Hodrea, J.; Balogh, D.B.; Lakat, T.; Szkibinszkij, E.; Hosszu, A.; Sparding, N.; Genovese, F.; et al. RAAS inhibitors directly reduce diabetes-induced renal fibrosis via growth factor inhibition. J. Physiol. 2019, 597, 193–209. [Google Scholar] [CrossRef]
- Maggioni, A.P.; Iervolino, A.; Andreotti, F. Is it time to introduce anti-inflammatory drugs into secondary cardiovascular prevention: Evidence from clinical trials? Vessel Plus 2021, 5, 14. [Google Scholar] [CrossRef]
- Márquez, E.; Riera, M.; Pascual, J.; Soler, M.J. Renin-angiotensin system within the diabetic podocyte. Am. J. Physiol. Renal Physiol. 2015, 308, F1–F10. [Google Scholar] [CrossRef]
- Unger, T.; Borghi, C.; Charchar, F.; Khan, N.A.; Poulter, N.R.; Prabhakaran, D.; Ramirez, A.; Schlaich, M.; Stergiou, G.S.; Tomaszewski, M.; et al. 2020 International Society of Hypertension Global Hypertension Practice Guidelines. Hypertension 2020, 75, 1334–1357. [Google Scholar] [CrossRef]
- Leoncini, G.; Russo, E.; Bussalino, E.; Barnini, C.; Viazzi, F.; Pontremoli, R. SGLT2is and Renal Protection: From Biological Mechanisms to Real-World Clinical Benefits. Int. J. Mol. Sci. 2021, 22, 4441. [Google Scholar] [CrossRef]
- Palmer, B.F.; Clegg, D.J. Kidney-Protective Effects of SGLT2 Inhibitors. Clin. J. Am. Soc. Nephrol. 2023, 18, 279–289. [Google Scholar] [CrossRef]
- Tang, J.; Ye, L.; Yang, Q.; Zhang, X.; Wang, L. Effects of Sodium-Glucose Cotransporter 2 Inhibitors on Water and Sodium Metabolism. Front. Pharmacol. 2022, 13, 800490. [Google Scholar] [CrossRef]
- Winiarska, A.; Knysak, M.; Nabrdalik, K.; Gumprecht, J.; Stompór, T. Inflammation and Oxidative Stress in Diabetic Kidney Disease: The Targets for SGLT2 Inhibitors and GLP-1 Receptor Agonists. Int. J. Mol. Sci. 2021, 22, 10822. [Google Scholar] [CrossRef]
- Macconi, D.; Remuzzi, G.; Benigni, A. Key fibrogenic mediators: Old players. Renin-angiotensin system. Kidney Int. Suppl. 2014, 4, 58–64. [Google Scholar] [CrossRef]
- Mezzano, S.A.; Ruiz-Ortega, M.; Egido, J. Angiotensin II and renal fibrosis. Hypertension 2001, 38, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Te Riet, L.; van Esch, J.H.; Roks, A.J.; van den Meiracker, A.H.; Danser, A.H. Hypertension: Renin-angiotensin-aldosterone system alterations. Circ. Res. 2015, 116, 960–975. [Google Scholar] [CrossRef] [PubMed]
- Andersen, S.; van Nieuwenhoven, F.A.; Tarnow, L.; Rossing, P.; Rossing, K.; Wieten, L.; Goldschmeding, R.; Parving, H.H. Reduction of urinary connective tissue growth factor by Losartan in type 1 patients with diabetic nephropathy. Kidney Int. 2005, 67, 2325–2329. [Google Scholar] [CrossRef] [PubMed]
- Ruperez, M.; Lorenzo, O.; Blanco-Colio, L.M.; Esteban, V.; Egido, J.; Ruiz-Ortega, M. Connective tissue growth factor is a mediator of angiotensin II-induced fibrosis. Circulation 2003, 108, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Yuan, W.; Varga, J. Transforming growth factor-β repression of matrix metalloproteinase-1 in dermal fibroblasts involves Smad3. J. Biol. Chem. 2001, 276, 38502–38510. [Google Scholar] [CrossRef]
- Das, R.; Xu, S.; Quan, X.; Nguyen, T.T.; Kong, I.D.; Chung, C.H.; Lee, E.Y.; Cha, S.K.; Park, K.S. Upregulation of mitochondrial Nox4 mediates TGF-β-induced apoptosis in cultured mouse podocytes. Am. J. Physiol. Renal Physiol. 2014, 306, F155–F167. [Google Scholar] [CrossRef]
- Mack, M.; Yanagita, M. Origin of myofibroblasts and cellular events triggering fibrosis. Kidney Int. 2015, 87, 297–307. [Google Scholar] [CrossRef]
- Jain, M.; Chauhan, A.K. Role of Integrins in Modulating Smooth Muscle Cell Plasticity and Vascular Remodeling: From Expression to Therapeutic Implications. Cells 2022, 11, 646. [Google Scholar] [CrossRef]
- Liao, W.; Liang, P.; Liu, B.; Xu, Z.; Zhang, L.; Feng, M.; Tang, Y.; Xu, A. MicroRNA-140-5p Mediates Renal Fibrosis through TGF-β1/Smad Signaling Pathway by Directly Targeting TGFBR1. Front. Physiol. 2020, 11, 1093. [Google Scholar] [CrossRef]
- Droebner, K.; Pavkovic, M.; Grundmann, M.; Hartmann, E.; Goea, L.; Nordlohne, J.; Klar, J.; Eitner, F.; Kolkhof, P. Direct Blood Pressure-Independent Anti-Fibrotic Effects by the Selective Nonsteroidal Mineralocorticoid Receptor Antagonist Finerenone in Progressive Models of Kidney Fibrosis. Am. J. Nephrol. 2021, 52, 588–601. [Google Scholar] [CrossRef]
- Weldon, S.M.; Brown, N.F. Inhibitors of aldosterone synthase. Vitam. Horm. 2019, 109, 211–239. [Google Scholar]
- Pham, T.D.; Verlander, J.W.; Wang, Y.; Romero, C.A.; Yue, Q.; Chen, C.; Thumova, M.; Eaton, D.C.; Lazo-Fernandez, Y.; Wall, S.M. Aldosterone Regulates Pendrin and Epithelial Sodium Channel Activity through Intercalated Cell Mineralocorticoid Receptor-Dependent and -Independent Mechanisms over a Wide Range in Serum Potassium. J. Am. Soc. Nephrol. 2020, 31, 483–499. [Google Scholar] [CrossRef]
- Miyata, K.N.; Lo, C.S.; Zhao, S.; Liao, M.C.; Pang, Y.; Chang, S.Y.; Peng, J.; Kretzler, M.; Filep, J.G.; Ingelfinger, J.R.; et al. Angiotensin II up-regulates sodium-glucose co-transporter 2 expression and SGLT2 inhibitor attenuates Ang II-induced hypertensive renal injury in mice. Clin. Sci. 2021, 135, 943–961. [Google Scholar] [CrossRef]
- Ruiz-Ortega, M.; Lamas, S.; Ortiz, A. Antifibrotic Agents for the Management of CKD: A Review. Am. J. Kidney Dis. 2022, 80, 251–263. [Google Scholar] [CrossRef]
- Pirklbauer, M.; Schupart, R.; Fuchs, L.; Staudinger, P.; Corazza, U.; Sallaberger, S.; Leierer, J.; Mayer, G.; Schramek, H. Unraveling reno-protective effects of SGLT2 inhibition in human proximal tubular cells. Am. J. Physiol. Renal Physiol. 2019, 316, F449–F462. [Google Scholar] [CrossRef]
- EMahfooz, K.; Najeed, S.; Tun, H.N.; Khamosh, M.; Grewal, D.; Hussain, A.; Ong, K.; Dharmarajan, L.; Vasavada, A. New Dual Endothelin Receptor Antagonist Aprocitentan in Hypertension: A Systematic Review and Meta-Analysis. Curr. Probl. Cardiol. 2023, 48, 101686. [Google Scholar] [CrossRef]
- Martínez-Díaz, I.; Martos, N.; Llorens-Cebrià, C.; Álvarez, F.J.; Bedard, P.W.; Vergara, A.; Jacobs-Cachá, C.; Soler, M.J. Endothelin Receptor Antagonists in Kidney Disease. Int. J. Mol. Sci. 2023, 24, 3427. [Google Scholar] [CrossRef]
- Jung, R.; Wild, J.; Ringen, J.; Karbach, S.; Wenzel, P. Innate Immune Mechanisms of Arterial Hypertension and Autoimmune Disease. Am. J. Hypertens. 2021, 34, 143–153. [Google Scholar] [CrossRef]
- Wen, Y.; Crowley, S.D. Renal effects of cytokines in hypertension. Curr. Opin. Nephrol. Hypertens. 2018, 27, 70–76. [Google Scholar] [CrossRef]
- Lu, X.; Crowley, S.D. Inflammation in Salt-Sensitive Hypertension and Renal Damage. Curr. Hypertens. Rep. 2018, 20, 103. [Google Scholar] [CrossRef] [PubMed]
- Amador, C.A.; Barrientos, V.; Peña, J.; Herrada, A.A.; González, M.; Valdés, S.; Carrasco, L.; Alzamora, R.; Figueroa, F.; Kalergis, A.M.; et al. Spironolactone decreases DOCA-salt-induced organ damage by blocking the activation of T helper 17 and the downregulation of regulatory T lymphocytes. Hypertension 2014, 63, 797–803. [Google Scholar] [CrossRef] [PubMed]
- McMaster, W.G.; Kirabo, A.; Madhur, M.S.; Harrison, D.G. Inflammation, Immunity, and Hypertensive End-Organ Damage. Circ. Res. 2015, 116, 1022–1033. [Google Scholar] [CrossRef]
- Davis, G.K.; Fehrenbach, D.J.; Madhur, M.S. Interleukin 17A: Key Player in the Pathogenesis of Hypertension and a Potential Therapeutic Target. Curr. Hypertens. Rep. 2021, 23, 13. [Google Scholar] [CrossRef] [PubMed]
- Lavoz, C.; Matus, Y.S.; Orejudo, M.; Carpio, J.D.; Droguett, A.; Egido, J.; Mezzano, S.; Ruiz-Ortega, M. Interleukin-17A blockade reduces albuminuria and kidney injury in an accelerated model of diabetic nephropathy. Kidney Int. 2019, 95, 1418–1432. [Google Scholar] [CrossRef]
- Mulay, S.R.; Anders, H.J. Crystal nephropathies: Mechanisms of crystal-induced kidney injury. Nat. Rev. Nephrol. 2017, 13, 226–240. [Google Scholar] [CrossRef]
- Mulay, S.R. Multifactorial functions of the inflammasome component NLRP3 in pathogenesis of chronic kidney diseases. Kidney Int. 2019, 96, 58–66. [Google Scholar] [CrossRef]
- Wen, L.; Yang, H.; Ma, L.; Fu, P. The roles of NLRP3 inflammasome-mediated signaling pathways in hyperuricemic nephropathy. Mol. Cell. Biochem. 2021, 476, 1377–1386. [Google Scholar] [CrossRef]
- Ke, Q.; Shi, C.; Lv, Y.; Wang, L.; Luo, J.; Jiang, L.; Yang, J.; Zhou, Y. SGLT2 inhibitor counteracts NLRP3 inflammasome via tubular metabolite itaconate in fibrosis kidney. FASEB J. 2022, 36, e22078. [Google Scholar] [CrossRef]
- Evans, R.G.; Gardiner, B.S.; Smith, D.W.; O’Connor, P.M. Intrarenal oxygenation: Unique challenges and the biophysical basis of homeostasis. Am. J. Physiol. Renal Physiol. 2008, 295, F1259–F1270. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Reeves, W.B.; Awad, A.S. Pathophysiology of diabetic kidney disease: Impact of SGLT2 inhibitors. Nat. Rev. Nephrol. 2021, 17, 319–334. [Google Scholar] [CrossRef]
- Shu, S.; Wang, Y.; Zheng, M.; Liu, Z.; Cai, J.; Tang, C.; Dong, Z. Hypoxia and Hypoxia-Inducible Factors in Kidney Injury and Repair. Cells 2019, 8, 207. [Google Scholar] [CrossRef]
- Honda, T.; Hirakawa, Y.; Nangaku, M. The role of oxidative stress and hypoxia in renal disease. Kidney Res. Clin. Pract. 2019, 38, 414–426. [Google Scholar] [CrossRef]
- Yu, J.; Wang, S.; Shi, W.; Zhou, W.; Niu, Y.; Huang, S.; Zhang, Y.; Zhang, A.; Jia, Z. Roxadustat prevents Ang II hypertension by targeting angiotensin receptors and eNOS. JCI Insight 2021, 6, e133690. [Google Scholar] [CrossRef]
- Packer, M. Mechanisms Leading to Differential Hypoxia-Inducible Factor Signaling in the Diabetic Kidney: Modulation by SGLT2 Inhibitors and Hypoxia Mimetics. Am. J. Kidney Dis. 2021, 77, 280–286. [Google Scholar] [CrossRef]
- Piani, F.; Cicero, A.F.G.; Borghi, C. Uric Acid and Hypertension: Prognostic Role and Guide for Treatment. J. Clin. Med. 2021, 10, 448. [Google Scholar] [CrossRef]
- Russo, E.; Verzola, D.; Cappadona, F.; Leoncini, G.; Garibotto, G.; Pontremoli, R.; Viazzi, F. The role of uric acid in renal damage—A history of inflammatory pathways and vascular remodeling. Vessel Plus 2021, 5, 15. [Google Scholar] [CrossRef]
- Russo, E.; Viazzi, F.; Pontremoli, R.; Barbagallo, C.M.; Bombelli, M.; Casiglia, E.; Cicero, A.F.G.; Cirillo, M.; Cirillo, P.; Desideri, G.; et al. Serum Uric Acid and Kidney Disease Measures Independently Predict Cardiovascular and Total Mortality: The Uric Acid Right for Heart Health (URRAH) Project. Front. Cardiovasc. Med. 2021, 8, 713652. [Google Scholar] [CrossRef]
- Del Pinto, R.; Viazzi, F.; Pontremoli, R.; Ferri, C.; Carubbi, F.; Russo, E. The URRAH study. Panminerva Med. 2021, 63, 416–423. [Google Scholar] [CrossRef]
- Russo, E.; Viazzi, F.; Pontremoli, R.; Barbagallo, C.M.; Bombelli, M.; Casiglia, E.; Cicero, A.F.G.; Cirillo, M.; Cirillo, P.; Desideri, G.; et al. Association of uric acid with kidney function and albuminuria: The Uric Acid Right for heArt Health (URRAH) Project. J. Nephrol. 2022, 35, 211–221. [Google Scholar] [CrossRef]
- Bao, D.; Lv, N.; Duan, X.; Zhang, X.; Wang, J.; Wang, S.; Wang, Y.; Zhao, M.H. Prevalence and clinical association of hyperechoic crystal deposits on ultrasonography in patients with chronic kidney disease: A cross-sectional study from a single center. J. Nephrol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Bardin, T.; Nguyen, Q.D.; Tran, K.M.; Le, N.H.; Do, M.D.; Richette, P.; Letavernier, E.; Correas, J.M.; Resche-Rigon, M. A cross-sectional study of 502 patients found a diffuse hyperechoic kidney medulla pattern in patients with severe gout. Kidney Int. 2021, 99, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Russo, E.; Verzola, D.; Leoncini, G.; Cappadona, F.; Esposito, P.; Pontremoli, R.; Viazzi, F. Treating Hyperuricemia: The Last Word Hasn’t Been Said Yet. J. Clin. Med. 2021, 10, 819. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.J.; Sanchez Lozada, L.G.; Lanaspa, M.A.; Piani, F.; Borghi, C. Uric Acid and Chronic Kidney Disease: Still More to Do. Kidney Int. Rep. 2022, 8, 229–239. [Google Scholar] [CrossRef]
- Piani, F.; Agnoletti, D.; Borghi, C. Advances in pharmacotherapies for hyperuricemia. Expert Opin. Pharmacother. 2023, 24, 737–745. [Google Scholar] [CrossRef]
- Zhang, M.; Cui, R.; Zhou, Y.; Ma, Y.; Jin, Y.; Gou, X.; Yang, J.; Wu, X. Uric acid accumulation in the kidney triggers mast cell degranulation and aggravates renal oxidative stress. Toxicology 2023, 483, 153387. [Google Scholar] [CrossRef]
- Milanesi, S.; Verzola, D.; Cappadona, F.; Bonino, B.; Murugavel, A.; Pontremoli, R.; Garibotto, G.; Viazzi, F. Uric acid and angiotensin II additively promote inflammation and oxidative stress in human proximal tubule cells by activation of toll-like receptor 4. J. Cell. Physiol. 2019, 234, 10868–10876. [Google Scholar] [CrossRef]
- Verzola, D.; Ratto, E.; Villaggio, B.; Parodi, E.L.; Pontremoli, R.; Garibotto, G.; Viazzi, F. Uric acid promotes apoptosis in human proximal tubule cells by oxidative stress and the activation of NADPH oxidase NOX 4. PLoS ONE 2014, 9, e115210. [Google Scholar] [CrossRef]
- Mulè, G.; Castiglia, A.; Morreale, M.; Geraci, G.; Cusumano, C.; Guarino, L.; Altieri, D.; Panzica, M.; Vaccaro, F.; Cottone, S. Serum uric acid is not independently associated with plasma renin activity and plasma aldosterone in hypertensive adults. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 350–359. [Google Scholar] [CrossRef]
- Dumor, K.; Shoemaker-Moyle, M.; Nistala, R.; Whaley-Connell, A. Arterial Stiffness in Hypertension: An Update. Curr. Hypertens. Rep. 2018, 20, 72. [Google Scholar] [CrossRef]
- Russo, E.; Bertolotto, M.; Zanetti, V.; Picciotto, D.; Esposito, P.; Carbone, F.; Montecucco, F.; Pontremoli, R.; Garibotto, G.; Viazzi, F.; et al. Role of Uric Acid in Vascular Remodeling: Cytoskeleton Changes and Migration in VSMCs. Int. J. Mol. Sci. 2023, 24, 2960. [Google Scholar] [CrossRef]
- Sapankaew, T.; Thadanipon, K.; Ruenroengbun, N.; Chaiyakittisopon, K.; Ingsathit, A.; Numthavaj, P.; Chaiyakunapruk, N.; McKay, G.; Attia, J.; Thakkinstian, A. Efficacy and safety of urate-lowering agents in asymptomatic hyperuricemia: Systematic review and network meta-analysis of randomized controlled trials. BMC Nephrol. 2022, 23, 223. [Google Scholar] [CrossRef]
- Suijk, D.L.S.; van Baar, M.J.B.; van Bommel, E.J.M.; Iqbal, Z.; Krebber, M.M.; Vallon, V.; Touw, D.; Hoorn, E.J.; Nieuwdorp, M.; Kramer, M.M.H.; et al. SGLT2 Inhibition and Uric Acid Excretion in Patients with Type 2 Diabetes and Normal Kidney Function. Clin. J. Am. Soc. Nephrol. 2022, 17, 663–671. [Google Scholar] [CrossRef]
- Novikov, A.; Fu, Y.; Huang, W.; Freeman, B.; Patel, R.; van Ginkel, C.; Koepsell, H.; Busslinger, M.; Onishi, A.; Nespoux, J.; et al. SGLT2 inhibition and renal urate excretion: Role of luminal glucose, GLUT9, and URAT1. Am. J. Physiol. Renal Physiol. 2019, 316, F173–F185. [Google Scholar] [CrossRef]
- Lu, Y.H.; Chang, Y.P.; Li, T.; Han, F.; Li, C.J.; Li, X.Y.; Xue, M.; Cheng, Y.; Meng, Z.Y.; Han, Z.; et al. Empagliflozin Attenuates Hyperuricemia by Upregulation of ABCG2 via AMPK/AKT/CREB Signaling Pathway in Type 2 Diabetic Mice. Int. J. Biol. Sci. 2020, 16, 529–542. [Google Scholar] [CrossRef]
- Verzola, D.; Saio, M.; Picciotto, D.; Viazzi, F.; Russo, E.; Cipriani, L.; Carta, A.; Costigliolo, F.; Gaggero, G.; Salvidio, G.; et al. Cellular Senescence Is Associated with Faster Progression of Focal Segmental Glomerulosclerosis. Am. J. Nephrol. 2020, 51, 950–958. [Google Scholar] [CrossRef]
- Matjusaitis, M.; Chin, G.; Sarnoski, E.A.; Stolzing, A. Biomarkers to identify and isolate senescent cells. Ageing Res. Rev. 2016, 29, 1–12. [Google Scholar] [CrossRef]
- Saito, Y.; Nakamura, T.; Ohyama, Y.; Suzuki, T.; Iida, A.; Shiraki-Iida, T.; Kuro-o, M.; Nabeshima, Y.; Kurabayashi, M.; Nagai, R. In vivo klotho gene delivery protects against endothelial dysfunction in multiple risk factor syndrome. Biochem. Biophys. Res. Commun. 2000, 276, 767–772. [Google Scholar] [CrossRef]
- Shimada, T.; Takeshita, Y.; Murohara, T.; Sasaki, K.I.; Egami, K.; Shintani, S.; Katsuda, Y.; Ikeda, H.; Nabeshima, Y.I.; Imaizumi, T. Angiogenesis and vasculogenesis are impaired in the precocious-aging klotho mouse. Circulation 2004, 110, 1148–1155. [Google Scholar] [CrossRef]
- Park, M.Y.; Herrmann, S.M.; Saad, A.; Eirin, A.; Tang, H.; Lerman, A.; Textor, S.C.; Lilach, O. Lerman Biomarkers of Kidney Injury and Klotho in Patients with Atherosclerotic Renovascular Disease. CJASN 2015, 10, 443–451. [Google Scholar] [CrossRef]
- Xu, Y.; Sun, Z. Molecular Basis of Klotho: From Gene to Function in Aging. Endocr. Rev. 2015, 36, 174–193. [Google Scholar] [CrossRef] [PubMed]
- Westhoff, J.H.; Hilgers, K.F.; Steinbach, M.P.; Hartner, A.; Klanke, B.; Amann, K.; Melk, A. Hypertension induces somatic cellular senescence in rats and humans by induction of cell cycle inhibitor p16INK4a. Hypertension 2008, 52, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Gomes, P.; Simão, S.; Silva, E.; Pinto, V.; Amaral, J.S.; Afonso, J.; Serrão, M.P.; Pinho, M.J.; Soares-da-Silva, P. Aging increases oxidative stress and renal expression of oxidant and antioxidant enzymes that are associated with an increased trend in systolic blood pressure. Oxid. Med. Cell. Longev. 2009, 2, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Musso, C.G.; Jauregui, J.R. Renin-angiotensin-aldosteronesystem and the aging kidney. Expert Rev. Endocrinol. Metab. 2014, 6, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Benigni, A.; Corna, D.; Zoja, C.; Sonzogni, A.; Latini, R.; Salio, M.; Conti, S.; Rottoli, D.; Longaretti, L.; Cassis, P.; et al. Disruption of the Ang II type 1 receptor promotes longevity in mice. J. Clin. Investig. 2009, 119, 524–530. [Google Scholar] [CrossRef]
- Packer, M. Critical Reanalysis of the Mechanisms Underlying the Cardiorenal Benefits of SGLT2 Inhibitors and Reaffirmation of the Nutrient Deprivation Signaling/Autophagy Hypothesis. Circulation 2022, 146, 1383–1405. [Google Scholar] [CrossRef]
- Cheng, Y.; Wang, D.; Wang, F.; Liu, J.; Huang, B.; Baker, M.A.; Yin, J.; Wu, R.; Liu, X.; Regner, K.R.; et al. Endogenous miR-204 Protects the Kidney against Chronic Injury in Hypertension and Diabetes. J. Am. Soc. Nephrol. 2020, 31, 1539–1554. [Google Scholar] [CrossRef]
- Peters, L.J.F.; Floege, J.; Biessen, E.A.L.; Jankowski, J.; van der Vorst, E.P.C. MicroRNAs in Chronic Kidney Disease: Four Candidates for Clinical Application. Int. J. Mol. Sci. 2020, 21, 6547. [Google Scholar] [CrossRef]
- Briet, M.; Boutouyrie, P.; Laurent, S.; London, G.M. Arterial stiffness and pulse pressure in CKD and ESRD. Kidney Int. 2012, 82, 388–400. [Google Scholar] [CrossRef]
- Ahmed, S.; Warren, D.T. Vascular smooth muscle cell contractile function and mechanotransduction. Vessel Plus 2018, 2, 36. [Google Scholar] [CrossRef]
- Jadli, A.S.; Ballasy, N.N.; Gomes, K.P.; Mackay, C.D.A.; Meechem, M.; Wijesuriya, T.M.; Belke, D.; Thompson, J.; Fedak, P.W.M.; Patel, V.B. Attenuation of Smooth Muscle Cell Phenotypic Switching by Angiotensin 1–7 Protects against Thoracic Aortic Aneurysm. Int. J. Mol. Sci. 2022, 23, 15566. [Google Scholar] [CrossRef]
- Li, Y.; Song, B.; Ruan, C.; Xue, W.; Zhao, J. AdipoRon Attenuates Hypertension-Induced Epithelial-Mesenchymal Transition and Renal Fibrosis via Promoting Epithelial Autophagy. J. Cardiovasc. Transl. Res. 2021, 14, 538–545. [Google Scholar] [CrossRef]
- Li, Y.; Lui, K.O.; Zhou, B. Reassessing endothelial-to-mesenchymal transition in cardiovascular diseases. Nat. Rev. Cardiol. 2018, 15, 445–456. [Google Scholar] [CrossRef]
- He, J.; Xu, Y.; Koya, D.; Kanasaki, K. Role of the endothelial-to-mesenchymal transition in renal fibrosis of chronic kidney disease. Clin. Exp. Nephrol. 2013, 17, 488–497. [Google Scholar] [CrossRef]
- Chapman, A.B.; Devuyst, O.; Eckardt, K.U.; Gansevoort, R.T.; Harris, T.; Horie, S.; Kasiske, B.L.; Odland, D.; Pei, Y.; Perrone, R.D.; et al. Autosomal-dominant polycystic kidney disease (ADPKD): Executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2015, 88, 17–27. [Google Scholar] [CrossRef]
- Shimada, S.; Hirose, T.; Takahashi, C.; Sato, E.; Kinugasa, S.; Ohsaki, Y.; Kisu, K.; Sato, H.; Ito, S.; Mori, T. Pathophysiological and molecular mechanisms involved in renal congestion in a novel rat model. Sci. Rep. 2018, 8, 16808. [Google Scholar] [CrossRef]
- Rigato, M.; Carraro, G.; Cirella, I.; Dian, S.; Di Vico, V.; Stefanelli, L.F.; Ravarotto, V.; Bertoldi, G.; Nalesso, F.; Calò, L.A. Effects of Tolvaptan on Oxidative Stress in ADPKD: A Molecular Biological Approach. J. Clin. Med. 2022, 11, 402. [Google Scholar] [CrossRef]



| Mechanisms by Which ah Affects the Kidney | Emerging Therapies Targeting Specific Kidney Pathways |
|---|---|
|
|
|
|
|
|
|
|
|
|
|
|

{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Russo, E.; Bussalino, E.; Macciò, L.; Verzola, D.; Saio, M.; Esposito, P.; Leoncini, G.; Pontremoli, R.; Viazzi, F. Non-Haemodynamic Mechanisms Underlying Hypertension-Associated Damage in Target Kidney Components. Int. J. Mol. Sci. 2023, 24, 9422. https://doi.org/10.3390/ijms24119422
Russo E, Bussalino E, Macciò L, Verzola D, Saio M, Esposito P, Leoncini G, Pontremoli R, Viazzi F. Non-Haemodynamic Mechanisms Underlying Hypertension-Associated Damage in Target Kidney Components. International Journal of Molecular Sciences. 2023; 24(11):9422. https://doi.org/10.3390/ijms24119422
Chicago/Turabian StyleRusso, Elisa, Elisabetta Bussalino, Lucia Macciò, Daniela Verzola, Michela Saio, Pasquale Esposito, Giovanna Leoncini, Roberto Pontremoli, and Francesca Viazzi. 2023. "Non-Haemodynamic Mechanisms Underlying Hypertension-Associated Damage in Target Kidney Components" International Journal of Molecular Sciences 24, no. 11: 9422. https://doi.org/10.3390/ijms24119422
APA StyleRusso, E., Bussalino, E., Macciò, L., Verzola, D., Saio, M., Esposito, P., Leoncini, G., Pontremoli, R., & Viazzi, F. (2023). Non-Haemodynamic Mechanisms Underlying Hypertension-Associated Damage in Target Kidney Components. International Journal of Molecular Sciences, 24(11), 9422. https://doi.org/10.3390/ijms24119422