

Hyperglycemia and Oxidative Stress: An Integral, Updated and Critical Overview of Their Metabolic Interconnections





Abstract
1. Introduction: Oxidative Stress and Aerobic Metabolism
2. Glucose Metabolism, Hyperglycemia, Oxidative Stress, and Pathological Effects
3. Metabolic Links between Hyperglycemia and Oxidative Stress at Molecular Level
3.1. Cell Respiration and Mitochondrial Generation of Superoxide and Other ROS
3.2. Collateral Pathways of Glucose Metabolism Related to Hyperglycemia
3.2.1. Protein Kinase C Route
3.2.2. Polyol Route
3.2.3. Hexosamine Route
3.3. Spontaneous Glucose Reactions
3.3.1. Protein Glycation
3.3.2. Glucose Auto-Oxidative Lysis; AGE Formation and AGE–RAGE Interactions
4. The AGE–RAGE Pathway and the NFκB Activation
4.1. Main Molecular Mediators Related to Hyperglycemic Pathological Effects
4.2. Enzymatic Pro-Oxidant Systems Directly Involved in ROS Generation
4.2.1. NADPH Oxidase (NOX)
4.2.2. Uncoupled Nitric Oxide Synthase (NOS)
4.2.3. Cyclooxygenase (COX)/Prostaglandin G/H Synthase (PGHS)
4.2.4. Lipoxygenases (LPOs)
4.2.5. Xanthin Oxidase (XO)
4.2.6. Heme Oxygenase (HO1)
4.2.7. Myeloperoxidase (MPO)
4.2.8. Cytochrome P450 (CYP)
5. Main Transcription Factors Involved in the Antioxidant Response: Nrf2 and FoxO
5.1. Antioxidant Enzymes and Antioxidant-Related Proteins
5.1.1. Superoxide Dismutases (SODs)
5.1.2. Catalases and Peroxidases (CAT and PO)
5.1.3. Peroxiredoxins (Prx)
5.1.4. Thioredoxin (Trx) and Related Proteins (Trx Reductase and TxNPI)
5.1.5. Paraoxonases (PON)
5.1.6. Sirtuins (Sirt)
6. Concluding Remarks and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- López-Otín, C.; Pietrocola, F.; Roiz-Valle, D.; Galluzzi, L.; Kroemer, G. Meta-hallmarks of aging and cancer. Cell Metab. 2023, 35, 3. [Google Scholar] [CrossRef]
- López-Otín, C.; Kroemer, G. Hallmarks of Health. Cell 2021, 184, 33–63. [Google Scholar] [CrossRef] [PubMed]
- Bandinelli, A. The Isolated System of Quantifiable Experiences in the 1783 “Mémoire sur la chaleur” of Lavoisier and Laplace. Ambix 2007, 54, 274–284. [Google Scholar] [CrossRef]
- Nelson, D.L.; Cox, M.C. Lehninger. Principles of Biochemistry, 4th ed.; W. H. Freeman & Co.: New York, NY, USA, 2004; ISBN 0-7167-4339-6. [Google Scholar]
- Harman, D. The aging process. Proc. Natl. Acad. Sci. USA 1981, 78, 7124–7128. [Google Scholar] [CrossRef] [PubMed]
- Scudellari, M. The science myths that will not die. Nature 2015, 528, 322–325. [Google Scholar] [CrossRef]
- Chow, C.K.; Ibrahim, W.; Wei, Z.; Chan, A.C. Vitamin E regulates mitochondrial hydrogen peroxide generation. Free Radic. Biol. Med. 1999, 27, 580–587. [Google Scholar] [CrossRef]
- Burgos-Morón, E.; Abad-Jiménez, Z.; Marañón, A.M.; Iannantuoni, F.; Escribano-López, I.; López-Domènech, S.; Salom, C.; Jover, A.; Mora, V.; Roldan, I.; et al. Relationship Between Oxidative Stress, ER Stress, and Inflammation in Type 2 Diabetes: The Battle Continues. J. Clin. Med. 2019, 8, 1385. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef]
- Fujiwara, N.; Kobayashi, K. Macrophages in inflammation. Curr. Drug. Targets Inflamm. Allergy 2005, 4, 281–286. [Google Scholar] [CrossRef]
- Brune, B.; Dehne, N.; Grossmann, N.; Jung, M.; Namgaladze, D.; Schmid, T.; von Knethen, A.; Weigert, A. Redox control of inflammation in macrophages. Antioxid. Redox Signal. 2013, 19, 595–637. [Google Scholar] [CrossRef]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2017, 114, 1752–1761. [Google Scholar] [CrossRef] [PubMed]
- Yaribeygi, H.; Atkin, S.L.; Sahebkar, A. A review of the molecular mechanisms of hyperglycemia-induced free radical generation leading to oxidative stress. J. Cell. Physiol. 2019, 234, 1300–1312. [Google Scholar] [CrossRef] [PubMed]
- Lima, J.E.B.F.; Moreira, N.C.S.; Sakamoto-Hojo, E.T. Mechanisms underlying the pathophysiology of type 2 diabetes: From risk factors to oxidative stress, metabolic dysfunction, and hyperglycemia. Mutat. Res./Gen. Toxicol. Environ. Mutagen. 2022, 874–875, 503437. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Wang, Q.; Rao, Z.; Shi, Y.; Wei, S.; Wang, H.; Lu, X.; Wang, P.; Lu, L.; Zhou, H.; et al. Diabetes induces hepatocyte pyroptosis by promoting oxidative stress-mediated NLRP3 inflammasome activation during liver ischaemia and reperfusion injury. Ann. Transl. Med. 2020, 8, 739. [Google Scholar] [CrossRef]
- Ziolkowska, S.; Binienda, A.; Jabłkowski, M.; Szemraj, J.; Czarny, P. The Interplay between Insulin Resistance, Inflammation, Oxidative Stress, Base Excision Repair and Metabolic Syndrome in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2021, 22, 11128. [Google Scholar] [CrossRef]
- Maslov, L.N.; Naryzhnaya, N.V.; Boshchenko, A.A.; Popov, S.V.; Ivanov, V.V.; Oeltgen, P.R. Is oxidative stress of adipocytes a cause or a consequence of the metabolic syndrome? J. Clin. Transl. Endocrinol. 2019, 15, 1–5. [Google Scholar] [CrossRef]
- El Assar, M.; Angulo, J.; Rodríguez-Mañas, L. Frailty as a phenotypic manifestation of underlying oxidative stress. Free Radic. Biol. Med. 2020, 149, 72–77. [Google Scholar] [CrossRef]
- Furuichi, Y.; Kawabata, Y.; Aoki, M.; Mita, Y.; Fujii, N.L.; Manabe, Y. Excess Glucose Impedes the Proliferation of Skeletal Muscle Satellite Cells Under Adherent Culture Conditions. Front. Cell. Dev. Biol. 2021, 9, 640399. [Google Scholar] [CrossRef]
- Duni, A.; Liakopoulos, V.; Roumeliotis, S.; Peschos, D.; Dounousi, E. Oxidative Stress in the Pathogenesis and Evolution of Chronic Kidney Disease: Untangling Ariadne’s Thread. Int. J. Mol. Sci. 2019, 20, 3711. [Google Scholar] [CrossRef]
- Vodošek Hojs, N.; Bevc, S.; Ekart, R.; Hojs, R. Oxidative Stress Markers in Chronic Kidney Disease with Emphasis on Diabetic Nephropathy. Antioxidants 2020, 9, 925. [Google Scholar] [CrossRef]
- Yuan, T.; Yang, T.; Chen, H.; Fu, D.; Hu, Y.; Wang, J.; Yuan, Q.; Yu, H.; Xu, W.; Xie, X. New Insights into Oxidative Stress and Inflammation during Diabetes Mellitus-Accelerated Atherosclerosis. Redox Biol. 2019, 20, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Charlton, A.; Garzarella, J.; Jandeleit-Dahm, K.A.M.; Jha, J.C. Oxidative Stress and Inflammation in Renal and Cardiovascular Complications of Diabetes. Biology 2021, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Xiong, X.; Wu, X.; Ye, Y.; Jian, Z.; Zhi, Z.; Gu, L. Targeting Oxidative Stress and Inflammation to Prevent Ischemia-Reperfusion Injury. Front. Mol. Neurosci. 2020, 13, 28. [Google Scholar] [CrossRef] [PubMed]
- Kang, Q.; Yang, C. Oxidative stress and diabetic retinopathy: Molecular mechanisms, pathogenetic role and therapeutic implications. Redox Biol. 2020, 37, 101799. [Google Scholar] [CrossRef]
- Ansari, M.Y.; Ahmad, N.; Haqqi, T.M. Oxidative stress and inflammation in osteoarthritis pathogenesis: Role of polyphenols. Biomed. Pharmacother. 2020, 129, 110452. [Google Scholar] [CrossRef]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Chang, K.-H.; Chen, C.-M. The Role of Oxidative Stress in Parkinson’s Disease. Antioxidants 2020, 9, 597. [Google Scholar] [CrossRef]
- Tenorio, M.B.; Raphaela Costa-Ferreira, R.; Moura, F.A.; Bueno, N.B.; Menezes de Oliveira, A.C.; Fonseca-Goulart, M.O. Cross-Talk between Oxidative Stress and Inflammation in Preeclampsia. Oxid. Med. Cel. Longev. 2019, 2019, 8238727. [Google Scholar] [CrossRef]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxid. Med. Cel. Longev. 2018, 2018, 9547613. [Google Scholar] [CrossRef]
- Chen, Y.-F.; Wu, S.-N.; Gao, J.-M.; Liao, Z.-Y.; Tseng, Y.-T.; Fülöp, F.; Chang, F.-R.; Lo, Y.-C. The antioxidant, anti-inflammatory and neuroprotective properties of the synthetic chalcone derivative AN07. Molecules 2020, 25, 2907. [Google Scholar] [CrossRef]
- Sanchez-Margalef, V. La leptina, adipoquinas obesidad y resistencia a insulina. In Actualizaciones en Bioquímica Clínica; Goberna, R., Guerrero, J.M., Eds.; Servicio de Publicaciones de la Universidad de Sevilla: Seville, Spain, 2014; pp. 103–119. [Google Scholar]
- Li, J.; Shen, X. Oxidative stress and adipokine levels were significantly correlated in diabetic patients with hyperglycemic crises. Diabetol. Metab. Syndr. 2019, 11, 13. [Google Scholar] [CrossRef]
- Viña, J.; Borras, C.; Gomez-Cabrera, M.C. A free radical theory of frailty. Free Radic. Biol. Med. 2018, 124, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Rayego-Mateos, S.; Rodrigues-Diez, R.R.; Fernandez-Fernandez, B.; Mora-Ferna, C.; Marchant, V.; Donate-Correa, J.; Navarro-Gonza, J.F.; Ortiz, A.; Ruiz-Ortega, M. Targeting inflammation to treat diabetic kidney disease: The road to 2030. Kidney Intl. 2023, 103, 282–296. [Google Scholar] [CrossRef]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef]
- Olvera-Montaño, C.; Castellanos-González, J.A.; Navarro-Partida, J.; Cardona-Muñoz, E.G.; López-Contreras, A.; Roman-Pintos, L.M.; Robles-Rivera, R.R.; Rodríguez-Carrizalez, A.D. Oxidative Stress as the Main Target in Diabetic Retinopathy Pathophysiology. J. Diabetes Res. 2019, 2019, 8562408. [Google Scholar] [CrossRef]
- Monserrat-Mesquida, M.; Quetglas-Llabrés, M.; Capó, X.; Bouzas, C.; Mateos, D.; Pons, A.; Tur, J.A.; Sureda, A. Metabolic Syndrome Is Associated with Oxidative Stress and Proinflammatory State. Antioxidants 2020, 9, 236. [Google Scholar] [CrossRef] [PubMed]
- Raut, S.K.; Khullar, M. Oxidative stress in metabolic diseases: Current scenario and therapeutic relevance. Mol. Cell. Biochem. 2023, 478, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Dieppe, P.A.; Lohmander, L.S. Pathogenesis and management of pain in osteoarthritis. Lancet 2005, 365, 965–973. [Google Scholar] [CrossRef]
- Greene, M.A.; Loeser, R.F. Aging-related inflammation in osteoarthritis. Osteoarthr. Cartil. 2015, 23, 1966–1971. [Google Scholar] [CrossRef]
- Li, H.; Wang, D.; Yuan, Y.; Min, J. New insights on the MMP-13 regulatory network in the pathogenesis of early osteoarthritis. Arthritis Res. Ther. 2017, 19, 248. [Google Scholar] [CrossRef]
- Ashruf, O.S.; Ansari, M.Y. Natural Compounds: Potential Therapeutics for the Inhibition of Cartilage Matrix Degradation in Osteoarthritis. Life 2023, 13, 102. [Google Scholar] [CrossRef]
- Tossetta, G.; Fantone, S.; Licini, C.; Marzioni, D.; Mattioli-Belmonte, M. The multifaced role of HtrA1 in the development of joint and skeletal disorders. Bone 2022, 157, 116350. [Google Scholar] [CrossRef] [PubMed]
- Durand, M.; Coué, M.; Croyal, M.; Moyon, T.; Tesse, A.; Atger, F.; Ouguerram, K.; Jacobi, D. Changes in Key Mitochondrial Lipids Accompany Mitochondrial Dysfunction and Oxidative Stress in NAFLD. Oxid. Med. Cell. Longev. 2021, 2021, 9986299. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; James, A.M.; Work, L.M.; Saeb-Parsy, K.; Frezza, C.; Krieg, T.; Murphy, M.P. A unifying mechanism for mitochondrial superoxide production during ischemia-reperfusion injury. Cell. Metabol. 2016, 23, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef]
- Fakhruddin, S.; Alanazi, W.; Jackson, K.E. Diabetes-induced reactive oxygen species: Mechanism of their generation and role in renal injury. J. Diabetes Res. 2017, 2017, 8379327. [Google Scholar] [CrossRef]
- Staniek, K.; Nohl, H. Are mitochondria a permanent source of reactive oxygen species? Biochim. Biophys. Acta 2000, 1460, 268–275. [Google Scholar] [CrossRef]
- St-Pierre, J.; Buckingham, J.; Roebuck, S.J.; Brand, M.D. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef]
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef]
- Shah, S.; Iqbal, M.; Karam, J.; Salifu, M.; McFarlane, S.I. Oxidative stress, glucose metabolism, and the prevention of type 2 diabetes: Pathophysiological insights. Antioxid. Redox Sign. 2007, 9, 911–929. [Google Scholar] [CrossRef] [PubMed]
- Bonnard, C.; Durand, A.; Peyrol, S.; Chanseaume, E.; Chauvin, M.A.; Morio, B.; Vidal, H.; Rieusset, J. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J. Clin. Investig. 2008, 118, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Mishra, M. Oxidative stress, mitochondrial damage and diabetic retinopathy. Biochim. Biophys. Acta 2015, 1852, 2474–2483. [Google Scholar] [CrossRef]
- Gerber, P.A.; Rutter, G.A. The Role of Oxidative Stress and Hypoxia in Pancreatic Beta-Cell Dysfunction in Diabetes Mellitus. Antioxid. Redox Signal. 2017, 26, 501–518. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790. [Google Scholar] [CrossRef]
- Feldman, E.L.; Nave, K.A.; Jensen, T.S.; Bennett, D.L.H. New Horizons in Diabetic Neuropathy: Mechanisms, Bioenergetics, and Pain. Neuron 2017, 93, 1296–1313. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Farrokhi, F.R.; Butler, A.E.; Sahebkar, A. Insulin resistance: Review of the underlying molecular mechanisms. J. Cell. Physiol. 2019, 234, 8152–8161. [Google Scholar] [CrossRef]
- Jubaidi, F.F.; Zainalabidin, S.; Taib, I.S.; Abdul Hamid, Z.; Mohamad Anuar, N.N.; Jalil, J.; Mohd Nor, N.A.; Budin, S.B. The Role of PKC-MAPK Signalling Pathways in the Development of Hyperglycemia-Induced Cardiovascular Complications. Int. J. Mol. Sci. 2022, 23, 8582. [Google Scholar] [CrossRef]
- Lien, C.-F.; Chen, S.-J.; Tsai, M.-C.; Lin, C.-S. Potential Role of Protein Kinase C in the Pathophysiology of Diabetes-Associated Atherosclerosis. Front. Pharmacol. 2021, 12, 716332. [Google Scholar] [CrossRef]
- Kay, A.M.; Simpson, C.L.; Steward, J.A., Jr. The role of AGE/RAGE signaling in Diabetes-mediated vascular calcification. J. Diabetes Res. 2016, 2016, 6809703. [Google Scholar] [CrossRef]
- Holmström, K.M.; Finkel, T. Cellular Mechanisms and Physiological Consequences of Redox-dependent Signalling. Nat. Rev. Mol. Cell. Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Batchuluun, B.; Inoguchi, T.; Sonoda, N.; Sasaki, S.; Inoue, T.; Fujimura, Y.; Takayanagi, R. Metformin and liraglutide ameliorate high glucose-induced oxidative stress via inhibition of PKC-NAD(P)H oxidase pathway in human aortic endothelial cells. Atherosclerosis 2014, 232, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Zhao, Y.; Jin, C.; Yu, L.; Ding, F.; Fu, G.; Zhu, J. PKC/NADPH oxidase are involved in the protective effect of pioglitazone in high homocysteine-induced paracrine dysfunction in endothelial progenitor cells. Am. J. Transl. Res. 2017, 9, 1037–1048. [Google Scholar]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Ann. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef]
- Steinberg, S.F. Structural basis of protein kinase C isoform function. Physiol. Rev. 2008, 88, 1341–1378. [Google Scholar] [CrossRef]
- Yabe-Nishimura, C. Aldose reductase in glucose toxicity: A potential target for the prevention of diabetic complications. Pharm. Rev. 1998, 50, 21–33. [Google Scholar]
- Yan, L.J. Redox imbalance stress in diabetes mellitus: Role of the polyol pathway. Anim. Model. Exp. Med. 2018, 1, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Martin, K.A.; Hwa, J. Aldose reductase, oxidative stress, and diabetic mellitus. Front. Pharmacol. 2012, 3, 87. [Google Scholar] [CrossRef]
- El-Kabbani, O.; Ruiz, F.; Darmanin, C.; Chung, R.P. Aldose reductase structures: Implications for mechanism and inhibition. Cell. Mol. Life Sci. 2004, 61, 750–762. [Google Scholar] [CrossRef]
- Bravi, M.C.; Pietrangeli, P.; Laurenti, O.; Basili, S.; Cassone-Faldetta, M.; Ferri, C.; De Mattia, G. Polyol pathway activation and glutathione redox status in non-insulin-dependent diabetic patients. Metabolism 1997, 46, 1194–1198. [Google Scholar] [CrossRef]
- Dong, K.; Ni, H.; Wu, M.; Tang, Z.; Halim, M.; Shi, D. ROS-mediated glucose metabolic reprogram induces insulin resistance in type 2 diabetes. Biochem. Biophys. Res. Commun. 2016, 476, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Hamada, Y.; Araki, N.; Koh, N.; Nakamura, J.; Horiuchi, S.; Hotta, N. Rapid formation of advanced glycation end products by intermediate metabolites of glycolytic pathway and polyol pathway. Biochem. Biophys. Res. Commun. 1996, 228, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Kador, P.F.; Akagi, Y.; Kinoshita, J.H. The effect of aldose reductase and its inhibition on sugar cataract formation. Metabolism 1986, 35 (Suppl. S1), 15–19. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.M.; Gonzalez, R.G. The effect of high glucose and oxidative stress on lens metabolism, aldose reductase and senile cataractogenesis. Metab. Clin. Exp. 1986, 35, 10–14. [Google Scholar] [CrossRef]
- Ramirez, M.A.; Borja, N.L. Epalrestat: An aldose reductase inhibitor for the treatment of diabetic neuropathy. Pharmacotherapy 2008, 28, 646–655. [Google Scholar] [CrossRef]
- McClain, D.A.; Crook, E.D. Hexosamines and insulin resistance. Diabetes 1996, 45, 1003–1009. [Google Scholar] [CrossRef]
- Mizukami, H.; Osonoi, S. Collateral Glucose-Utilizing Pathway in Diabetic Polyneuropathy. Int. J. Mol. Sci. 2021, 22, 94. [Google Scholar] [CrossRef]
- Moore, J.A.; Miller, W.P.; Dennis, M.D. Glucosamine induces REDD1 to suppress insulin action in retinal Müller cells. Cell. Signal. 2016, 28, 384–390. [Google Scholar] [CrossRef]
- Kaneto, H.; Xu, G.; Song, K.-H.; Suzuma, K.; Bonner-Weir, S.; Sharma, A.; Weir, G.C. Activation of the hexosamine pathway leads to deterioration of pancreatic β-cell function through the induction of oxidative stress. J. Biol. Chem. 2001, 276, 31099–31104. [Google Scholar] [CrossRef]
- Morino, K.; Maegawa, H. Role of O-linked N-acetylglucosamine in the homeostasis of metabolic organs, and its potential links with diabetes and its complications. J. Diabetes Investig. 2021, 12, 130–136. [Google Scholar] [CrossRef]
- Horal, M.; Zhang, Z.; Stanton, R.; Virkamäki, A.; Loeken, M.R. Activation of the hexosamine pathway causes oxidative stress and abnormal embryo gene expression: Involvement in diabetic teratogenesis. Birth Defects Res. Part A Clin. Mol. Teratol. 2004, 70, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Kolm-Litty, V.; Sauer, U.; Nerlich, A.; Lehmann, R.; Schleicher, E.D. High glucose-induced transforming growth factor beta 1 production is mediated by the hexosamine pathway in porcine glomerular mesangial cells. J. Clin. Investig. 1998, 101, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Du, X.L.; Edelstein, D.; Rossetti, L.; Fantus, I.G.; Golberg, H.; Ziyadeh, F.; Wu, J.; Brownlee, M. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc. Natl. Acad. Sci. USA 2000, 97, 12222–12226. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Paterson, A.J.; Chin, E.; Kudlow, J.E. Glucose stimulates protein modification by O-linked GlcNAc in pancreatic β cells: Linkage of O-linked GlcNAc to β cell death. Proc. Natl. Acad. Sci. USA 2000, 97, 2820–2825. [Google Scholar] [CrossRef] [PubMed]
- Hunt, J.V.; Dean, R.T.; Wolff, S.P. Hydroxyl radical production and autoxidative glycosylation. Glucose autoxidation as the cause of protein damage in the experimental glycation model of diabetes mellitus and ageing. Biochem. J. 1988, 256, 205–212. [Google Scholar] [CrossRef]
- Rabbania, N.; Thornalley, P.J. Protein glycation–biomarkers of metabolic dysfunction and early-stage decline in health in the era of precision medicine. Redox Biol. 2021, 42, 101920. [Google Scholar] [CrossRef]
- Wheeler, E.; Leong, A.; Liu, C.-T.; Hivert, M.-F.; Strawbridge, R.J.; Podmore, C.; Li, M.; Yao, J.; Sim, X.; Hong, J.; et al. Impact of common genetic determinants of Hemoglobin A1c on type 2 diabetes risk and diagnosis in ancestrally diverse populations: A transethnic genome-wide meta-analysis. PLoS Med. 2017, 14, e1002383. [Google Scholar] [CrossRef]
- Anguizola, J.; Barnaby, O.S.; Hoy, K.S.; Wa, C.; DeBolt, E.; Koke, M.; Hage, D.S. Glycation of human serum albumin. Clin. Chim. Acta 2013, 425, 64–76. [Google Scholar] [CrossRef]
- Qiu, H.Y.; Hou, N.N.; Shi, J.F.; Liu, Y.P.; Kan, C.X.; Han, F.; Sun, X.D. Comprehensive overview of human serum albumin glycation in diabetes mellitus. World J. Diabetes 2021, 12, 1057–1069. [Google Scholar] [CrossRef]
- Morgan, P.E.; Dean, R.T.; Davies, M.J. Inactivation of cellular enzymes by carbonyls and protein-bound glycation/glycoxidation products. Arch. Biochem. Biophys. 2002, 403, 259–269. [Google Scholar] [CrossRef]
- Thonalley, P.J.; Langborg, A.; Minhas, H.S. Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. Biochem. J. 1999, 344, 109–116. [Google Scholar]
- Allaman, I.; Belanger, M.; Magistretti, P.J. Methylglyoxal, the dark side of glycolysis. Front. Neurosci. 2015, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Moraru, A.; Wiederstein, J.; Pfa, D.; Fleming, T.; Miller, A.K.; Nawroth, P.; Teleman, A.A. Elevated Levels of the Reactive Metabolite Methylglyoxal Recapitulate Progression of Type 2 Diabetes. Cell. Metab. 2018, 27, 926–934.e8. [Google Scholar] [CrossRef] [PubMed]
- Wautier, M.P.; Chappey, O.; Corda, S.; Stern, D.M.; Schmidt, A.M.; Wautier, J.-L. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E685–E694. [Google Scholar] [CrossRef] [PubMed]
- Wautier, J.L.; Wautier, M.P.; Schmidt, A.M.; Anderson, G.M.; Hori, O.; Zoukourian, C.; Capron, L.; Chappey, O.; Yan, S.D.; Brett, J. Advanced glycation end products (AGEs) on the surface of diabetic erythrocytes bind to the vessel wall via a specific receptor inducing oxidant stress in the vasculature: A link between surface-associated AGEs and diabetic complications. Proc. Natl. Acad. Sci. USA 1994, 91, 7742–7746. [Google Scholar] [CrossRef] [PubMed]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced glycation end products: Sparking the development of diabetic vascular injury. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Arora, S.; Syal, P.; Sippy, M. A review on role of advanced glycation end products (ages) in rheumatoid arthritis. J. Pharm. Tech. Res. Manag. 2015, 3, 1–10. [Google Scholar] [CrossRef]
- Cepas, V.; Collino, M.; Mayo, J.C.; Sainz, R.M. Redox Signaling and Advanced Glycation End products (AGEs) in Diet-Related Diseases. Antioxidants 2020, 9, 142. [Google Scholar] [CrossRef]
- Nowotny, K.; Jung, T.; Höhn, A.; Weber, D.; Tilman Grune, T. Advanced Glycation End Products and Oxidative Stress in Type 2 Diabetes Mellitus. Biomolecules 2015, 5, 194–222. [Google Scholar] [CrossRef]
- Soman, S.; Raju, R.; Sandhya, V.K.; Advani, J.; Khan, A.A.; Harsha, H.C.; Prasad, T.S.K.; Sudhakaran, P.R.; Pandey, A.; Adishesha, P.K. A multicellular signal transduction network of AGE/RAGE signaling. J. Cell Commun. Signal. 2013, 7, 19–23. [Google Scholar] [CrossRef]
- Shi, M.; Yang, S.; Zhu, X.; Sun, D.; Sun, D.; Jiang, X.; Zhang, C.; Wang, L. The RAGE/STAT5/autophagy axis regulates senescence in mesangial cells. Cell. Signal. 2019, 62, 109334. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Karin, M. Is NFkB the sensor of oxidative stress? FASEB J. 1999, 13, 1137–1143. [Google Scholar] [CrossRef] [PubMed]
- Simard, E.; Sollradl, T.; Maltais, J.-S.; Boucher, J.; D’Orleans-Juste, P.; Grandbois, M. Receptor for advanced glycation end-products signaling interferes with the vascular smooth muscle cell contractile phenotype and function. PLoS ONE 2015, 10, e0128881. [Google Scholar] [CrossRef] [PubMed]
- Lingappan, K. NFkB in oxidative stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef]
- Baker, R.G.; Hayden, M.S.; Ghosh, S. NFκB, Inflammation and Metabolic Disease. Cell. Metab. 2011, 13, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Bierhaus, A.; Schiekofer, S.; Schwaninger, M.; Andrassy, M.; Humpert, P.M.; Chen, J.; Hong, M.; Luther, T.; Henle, T.; Klöting, I.; et al. Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes 2001, 50, 2792–2808. [Google Scholar] [CrossRef] [PubMed]
- Gheibi, S.; Jeddi, S.; Kashfi, K.; Ghasemi, A. Regulation of vascular tone homeostasis by NO and H2S: Implications in hypertension. Biochem. Pharmacol. 2018, 149, 42–59. [Google Scholar] [CrossRef]
- Papayianni, A.; Alexopoulos, E.; Giamalis, P.; Gionanlis, L.; Belechri, A.M.; Koukoudis, P.; Memmos, D. Circulating levels of ICAM-1, VCAM-1, and MCP-1 are increased in haemodialysis patients: Association with inflammation, dyslipidaemia, and vascular events. Nephrol. Dial. Transplant. 2002, 17, 435–441. [Google Scholar] [CrossRef]
- Liao, J.K. Linking endothelial dysfunction with endothelial cell activation. J. Clin. Investig. 2013, 12, 540–541. [Google Scholar] [CrossRef]
- Sena, C.M.; Pereira, A.M.; Seiça, R. Endothelial dysfunction. A major mediator of diabetic vascular disease. Biochim. Biophys. Acta 2013, 1832, 2216–2231. [Google Scholar] [CrossRef]
- Yokoyama, M.; Hirata, K.-I. Endothelial nitric oxide synthase uncoupling: Is it a physiological mechanism of endothelium-dependent relaxation in cerebral artery. Cardiovasc. Res. 2007, 73, 8–9. [Google Scholar] [CrossRef] [PubMed]
- Abudukadier, A.; Fujita, Y.; Obara, A.; Ohashi, A.; Fukushima, M.T.; Sato, Y.; Ogura, M.; Nakamura, Y.; Fujimoto, S.; Hosokawa, M.; et al. Tetrahydrobiopterin Has a Glucose-Lowering Effect by Suppressing Hepatic Gluconeogenesis in an Endothelial Nitric Oxide Synthase–Dependent Manner in Diabetic Mice. Diabetes 2013, 62, 3033–3043. [Google Scholar] [CrossRef] [PubMed]
- Kojda, G.; Harrison, D. Interactions between NO and reactive oxygen species: Pathophysiological importance in atherosclerosis, hypertension, diabetes and heart failure. Cardiovasc. Res. 1999, 43, 562–571. [Google Scholar] [CrossRef]
- Potenza, M.A.; Gagliardi, S.; Nacci, C.; Carratu, M.R.; Montagnani, M. Endothelial dysfunction in diabetes: From mechanisms to therapeutic targets. Curr. Med. Chem. 2009, 16, 94–112. [Google Scholar] [CrossRef]
- Li, J.H.; Huang, X.R.; Zhu, H.J.; Oldfield, M.; Cooper, M.; Truong, L.D.; Johnson, R.J.; Lan, H.Y. Advanced glycation end products activate Smad signaling via TGF-β-dependent and-independent mechanisms: Implications for diabetic renal and vascular disease. FASEB J. 2004, 18, 176–178. [Google Scholar] [CrossRef]
- Liu, R.M.; Gaston Pravia, K.A. Oxidative stress and glutathione in TGF-𝛽-mediated fibrogenesis. Free Radic. Biol. Med. 2010, 48, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.M.; Desai, L.P. Reciprocal regulation of TGF-β and reactive oxygen species: A perverse cycle for fibrosis. Redox Biol. 2015, 6, 565–577. [Google Scholar] [CrossRef]
- Zimmerman, C.M.; Padgett, R.W. Transforming growth factor 𝛽 signaling mediators and modulators. Gene 2000, 249, 17–30. [Google Scholar] [CrossRef]
- Peng, Y.; Kim, J.-M.; Park, H.-S.; Yang, A.; Islam, C.; Lakatta, E.G.; Lin, L. AGE-RAGE signal generates a specific NF-𝜅B RelA ‘barcode’ that directs collagen I expression. Sci. Rep. 2016, 6, 18822. [Google Scholar] [CrossRef]
- Zhang, H.; Qi, S.; Song, Y.; Ling, C. Artemisinin attenuates early renal damage on diabetic nephropathy rats through suppressing TGF-β1 regulator and activating the Nrf2 signaling pathway. Life Sci. 2020, 256, 117966. [Google Scholar] [CrossRef]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef]
- Mishra, M.; Kowluru, R.A. Epigenetic modification of mitochondrial DNA in the development of diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2015, 56, 5133–5142. [Google Scholar] [CrossRef] [PubMed]
- Panday, A.; Sahoo, M.K.; Orsorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity- associated pathologies. Cell. Mol. Immunol. 2015, 12, 523. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Takahito, K.; Ohtsuka, Y.; Fujiwara, Y.; Shimada, M.; Kawakami, Y. Impairment of glutathione metabolism in erythrocytes from patients with diabetes mellitus. Metabolism 1989, 38, 753–758. [Google Scholar] [CrossRef]
- Adamson, G.M.; Harman, A.W. A role for the glutathione peroxidase/reductase enzyme system in the protection from paracetamol toxicity in isolated mouse hepatocytes. Biochem. Pharmacol. 1989, 38, 3323–3330. [Google Scholar] [CrossRef]
- Kattamis, C.A.; Kyriazakou, M.; Chaidas, S. Favism: Clinical and biochemical data. J. Med. Genet. 1969, 6, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Beretta, A.; Manuelli, M.; Cena, H. Favism: Clinical Features at Different Ages. Nutrients 2023, 15, 343. [Google Scholar] [CrossRef]
- Helfinger, V.; Gall FF von Henke, N.; Kunze, M.M.; Schmid, T.; Rezende, F.; Heidler, J.; Wittig, I.; Radeke, H.H.; Marschall, V.; Anderson, K.; et al. Genetic deletion of Nox4 enhances cancerogen-induced formation of solid tumors. Proc. Natl. Acad. Sci. USA 2021, 118, e2020152118. [Google Scholar] [CrossRef]
- Schröder, K.; Zhang, M.; Benkhoff, S.; Mieth, A.; Pliquett, R.; Kosowski, J.; Kruse, C.; Luedike, P.; Michaelis, U.R.; Weissmann, N.; et al. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ. Res. 2012, 110, 1217–1225. [Google Scholar] [CrossRef]
- Tiganis, T. Reactive oxygen species and insulin resistance: The good, the bad and the ugly. Trends Pharm. Sci. 2011, 32, 82–89. [Google Scholar] [CrossRef]
- Juszczak, F.; Caron, N.; Mathew, A.V.; Declèves, A.-E. Critical Role for AMPK in Metabolic Disease-Induced Chronic Kidney Disease. Int. J. Mol. Sci. 2020, 21, 7994. [Google Scholar] [CrossRef]
- Touyz, R.M.; Montezano, A.C. Vascular Nox4 a multifarious NADPH oxidase. Circ. Res. 2012, 110, 1159–1161. [Google Scholar] [CrossRef] [PubMed]
- Rajaram, R.D.; Dissard, R.; Faivre, A.; Ino, F.; Delitsikou, V.; Jaquet, V.; Cagarelli, T.; Lindenmeyer, M.; Jansen-Duerr, P.; Cohen, C.; et al. Tubular NOX4 expression decreases in chronic kidney disease but does not modify fibrosis evolution. Redox Biol. 2019, 26, 101234. [Google Scholar] [CrossRef] [PubMed]
- Yaribeygi, H.; Farrokhi, F.R.; Rezaee, R.; Sahebkar, A. Oxidative stress induces renal failure: A review of possible molecular pathways. J. Cell. Biochem. 2018, 119, 2990–2998. [Google Scholar] [CrossRef] [PubMed]
- Drummond, G.R.; Sobey, C.G. Endothelial NADPH oxidases: Which NOX to target in vascular disease? Trends Endocrinol. Metab. 2014, 25, 452–463. [Google Scholar] [CrossRef]
- Satoh, M.; Fujimoto, S.; Haruna, Y.; Arakawa, S.; Horike, H.; Komai, N.; Sasaki, T.; Tsujioka, K.; Makino, H.; Kashihara, N. NAD(P)H oxidase and uncoupled nitric oxide synthase are major sources of glomerular superoxide in rats with experimental diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2005, 288, F1144–F1152. [Google Scholar] [CrossRef]
- Diers, A.R.; Broniowska, K.A.; Hogg, N. Nitrosative stress and redox-cycling agents synergize to cause mitochondrial dysfunction and cell death in endothelial cells. Redox Biol. 2013, 1, 1–7. [Google Scholar] [CrossRef]
- Xia, N.; Förstermann, U.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef]
- Cai, S.; Khoo, J.; Channon, K.M. Augmented BH4 by gene transfer restores nitric oxide synthase function in hyperglycemic human endothelial cells. Cardiovasc. Res. 2005, 65, 823–831. [Google Scholar] [CrossRef]
- Novoa, U.; Soto, K.; Valdés, C.; Villaseñor, J.; Treuer, A.V.; González, D.R. Tetrahydrobiopterin (BH4) Supplementation Prevents the Cardiorenal Effects of Diabetes in Mice by Reducing Oxidative Stress, Inflammation and Fibrosis. Biomedicines 2022, 10, 2479. [Google Scholar] [CrossRef]
- Picot, D.; Loll, P.J.; Garavito, R.M. The X-ray crystal structure of the membrane protein prostaglandin H2 synthase-1. Nature 1994, 367, 243–249. [Google Scholar] [CrossRef]
- Liu, J.; Seibold, S.A.; Rieke, C.J.; Song, I.; Cukier, R.I.; Smith, W.L. Prostaglandin endoperoxide H synthases: Peroxidase hydroperoxide specificity and cyclooxygenase activation. J. Biol. Chem. 2007, 282, 18233–18244. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, N.; Gaw-Gonzalo, I.T.; Natarajan, R. Molecular Mechanisms of High Glucose-Induced Cyclooxygenase-2 Expression in Monocytes. Diabetes 2004, 53, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Francés, D.E.; AIngaramo, P.I.; Mayoral, R.; Través, P.; Casado, M.; Valverde, A.M.; Martín-Sanz, P.; Carnovale, C.E. Cyclooxygenase-2 over-expression inhibits liver apoptosis induced by hyperglycemia. J. Cell. Biochem. 2013, 114, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Xu, R.; Wu, G.; Yin, Z.; Zhang, H.; Li, H.; Chen, W. Aspirin Suppresses Hepatic Glucagon Signaling Through Decreasing Production of Thromboxane A2. Endocrinology 2023, 164, bqac217. [Google Scholar] [CrossRef]
- Gahalain, N.; Chaudhary, J.; Kumar, A.; Sharma, S.; Jain, A. Lipid peroxidation: An overview. Int. J. Pharm. Sci. Res. 2011, 2, 2757–2766. [Google Scholar]
- Kulkarni, A.; Nadler, J.L.; Mirmira, R.G.; Casimiro, I. Regulation of Tissue Inflammation by 12-Lipoxygenases. Biomolecules 2021, 11, 717. [Google Scholar] [CrossRef]
- Ibrahim, A.S.; Elshafey, S.; Sellak, H.; Hussein, K.A.; El-Sherbiny, M.; Abdelsaid, M.; Rizk, N.; Beasley, S.; Tawfik, A.M.; Smith, S.B.; et al. A lipidomic screen of hyperglycemia-treated HRECs links 12/15-Lipoxygenase to microvascular dysfunction during diabetic retinopathy via NADPH oxidase. J. Lipid Res. 2015, 56, 599–611. [Google Scholar] [CrossRef]
- Chen, S.; Zou, H. Key Role of 12-Lipoxygenase and Its Metabolite 12-Hydroxyeicosatetraenoic Acid (12-HETE) in Diabetic Retinopathy. Curr. Eye Res. 2022, 47, 329–335. [Google Scholar] [CrossRef]
- Østergaard, J.A.; Cooper, M.E.; Jandeleit-Dahm, K.A.M. Targeting oxidative stress and anti-oxidant defence in diabetic kidney disease. J. Nephrol. 2020, 33, 917–929. [Google Scholar] [CrossRef]
- Chung, H.Y.; Baek, B.S.; Song, S.H.; Kim, M.S.; Huh, J.I.; Shim, K.H.; Kim, K.W.; Lee, K.H. Xanthine dehydrogenase/Xanthine oxidase and oxidative stress. Age 1997, 20, 127–140. [Google Scholar] [CrossRef]
- Polito, L.; Bortolotti, M.; Battelli, M.G.; Bolognesi, A. Chronic kidney disease: Which role for xanthine oxidoreductase activity and products? Pharmacol. Res. 2022, 184, 106407. [Google Scholar] [CrossRef] [PubMed]
- Feillet-Coudray, C.; Fouret, G.; Bonafos, B.; Aoun, M.; Carillon, J.; Sutra, T.; Coudray, C. Xanthine oxidase is variably involved in nutritional and physio-pathologic oxidative stress situations. J. Physiobiochem. Metab. 2013, 2, 8. [Google Scholar] [CrossRef]
- Liu, J.; Wang, C.; Liu, F.; Lu, Y.; Cheng, J. Metabolomics revealed xanthine oxidase-induced oxidative stress and inflammation in the pathogenesis of diabetic nephropathy. Anal. Bioanal. Chem. 2015, 407, 2569–2579. [Google Scholar] [CrossRef] [PubMed]
- Desco, M.C.; Asensi, M.; Màrquez, R.; Martìnez-Valls, J.; Vento, M.; Pallardo, F.V.; Sastre, J.; Viña, J. Xanthine oxidase is involved in free radical production in type 1 diabetes. Protection by Allopurinol. Diabetes 2002, 51, 1118–1124. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.-J.; Choi, W.J.; Chang, Y.-K.; Park, C.W.; Kim, S.Y.; Hong, Y.A. Inhibition of Xanthine Oxidase Protects against Diabetic Kidney Disease through the Amelioration of Oxidative Stress via VEGF/VEGFR Axis and NOX-FoxO3a-eNOS Signaling Pathway. Int. J. Mol. Sci. 2023, 24, 3807. [Google Scholar] [CrossRef]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef]
- Ryter, S.W.; Tyrrell, R.M. The heme synthesis and degradation pathways: Role in oxidant sensitivity: Heme oxygenase has both pro- and antioxidant properties. Free Radic. Biol. Med. 2000, 28, 289–309. [Google Scholar] [CrossRef]
- Ndisang, J.F.; Lane, N.; Jadhav, A. Upregulation of the heme oxygenase system ameliorates postprandial and fasting hyperglycemia in type 2 diabetes. Endocrinol. Metab. 2009, 296, E1029–E1041. [Google Scholar] [CrossRef]
- de Souza Ferreira, C.; Araújo, T.H.; Ângelo, M.L.; Pennacchi, P.C.; Okada, S.S.; de Araújo, P.F.B.; Migliorini, S.; Rodrigues, M.R. Neutrophil dysfunction induced by hyperglycemia: Modulation of myeloperoxidase activity. Cell. Biochem. Funct. 2012, 7, 604–610. [Google Scholar] [CrossRef]
- Tian, R.; Ding, Y.; Peng, Y.Y.; Lu, N. Myeloperoxidase amplified high glucose-induced endothelial dysfunction in vasculature: Role of NADPH oxidase and hypochlorous acid. Biochem. Biophys. Res. Commun. 2017, 484, 572–578. [Google Scholar] [CrossRef]
- Lazarevic-Pasti, T.; Leskovac, A.; Vasic, V. Myeloperoxidase inhibitors as potential drugs. Curr. Drug. Metab. 2015, 16, 168–190. [Google Scholar] [CrossRef] [PubMed]
- Delporte, C.; Boudjeltia, K.Z.; Furtmüller, P.G.; Maki, R.A.; Dieu, M.; Noyon, C.; Soudi, M.; Dufour, D.; Coremans, C.; Nuyens, V.; et al. Myeloperoxidase-catalyzed oxidation of cyanide to cyanate: A potential carbamylation route involved in the formation of atherosclerotic plaques? J. Biol. Chem. 2018, 293, 6374–6386. [Google Scholar] [CrossRef] [PubMed]
- Bansal, S.; Liu, C.-P.; Sepuri, N.B.V.; Anandatheerthavarada, H.K.; Selvaraj, V.; Hoek, J.; Milne, G.L.; Guengerich, P.; Avadhani, N.G. Mitochondria-targeted cytochrome P450 2E1 induces oxidative damage and augments alcohol-mediated oxidative stress. J. Biol. Chem. 2010, 285, 24609–24619. [Google Scholar] [CrossRef] [PubMed]
- Mészáros, P.; Kovács, S.; Kulcsár, G.; Páskuj, M.; Almási, A. Investigation of Intestinal Absorption and Excretion of Paracetamol in Streptozotocin-Induced Hyperglycemia. Int. J. Mol. Sci. 2022, 23, 11913. [Google Scholar] [CrossRef]
- Papachristoforou, E.; Lambadiari, V.; Maratou, E.; Makrilakis, K. Association of glycemic indices (hyperglycemia, glucose variability, and hypoglycemia) with oxidative stress and diabetic complications. J. Diabetes Res. 2020, 2020, 7489795. [Google Scholar] [CrossRef] [PubMed]
- Darakjian, L.; Deodhar, M.; Turgeon, J.; Michaud, V. Chronic inflammatory status observed in patients with type 2 diabetes induces modulation of cytochrome P450 expression and activity. Int. J. Mol. Sci. 2021, 22, 4967. [Google Scholar] [CrossRef] [PubMed]
- Hybertson, B.M.; Gao, B.; Bose, S.K.; McCord, J.M. Oxidative stress in health and disease: The therapeutic potential of Nrf2 activation. Mol. Asp. Med. 2011, 8, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Liang, L.; Matsumoto, M.; Iwata, K.; Umemura, A.; He, F. Reactive Oxygen Species and NRF2 Signaling, Friends or Foes in Cancer? Biomolecules 2023, 13, 353. [Google Scholar] [CrossRef]
- Tossetta, G.; Marzioni, D. Natural and synthetic compounds in Ovarian Cancer: A focus on NRF2/KEAP1 pathway. Pharm. Res. 2022, 183, 106365. [Google Scholar] [CrossRef]
- Marzioni, D.; Mazzucchelli, R.; Fantone, S.; Tossetta, G. NRF2 modulation in TRAMP mice: An in vivo model of prostate cancer. Mol. Biol. Rep. 2023, 50, 873–881. [Google Scholar] [CrossRef]
- Tossetta, G.; Marzioni, D. Targeting the NRF2/KEAP1 pathway in cervical and endometrial cancers. Eur. J. Pharmacol. 2023, 941, 175503. [Google Scholar] [CrossRef] [PubMed]
- Baird, L. Yamamoto. M. The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol. Cel. Biol. 2020, 40, e00099-20. [Google Scholar] [CrossRef] [PubMed]
- Thimmulappa, R.K.; Mai, K.H.; Srisuma, S.; Kensler, T.W.; Yamamoto, M.; Biswal, S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002, 62, 5196–5203. [Google Scholar] [PubMed]
- Yu, J.; Zhao, Y.; Li, B.; Sun, L.; Huo, H. 17β-Estradiol regulates the expression of antioxidant enzymes in myocardial cells by increasing Nrf2 translocation. Inc. J. Biochem. Mol. Toxicol. 2012, 26, 264–269. [Google Scholar] [CrossRef]
- Liu, Y.Q.; Zhang, J.; Gao, L.N.; Wang, H.T. Effects of aerobic exercise on Nrf2-SOD pathway in the gastrocnemius of rats with high-glucose and high-fat diet. Chin. J. Appl. Physiol. 2020, 36, 481–485. [Google Scholar] [CrossRef]
- Obrenovich, M.; Li, Y.; Tayahi, M.; Reddy, V.P. Polyphenols and Small Phenolic Acids as Cellular Metabolic Regulators. Curr. Issues Mol. Biol. 2022, 44, 4152–4166. [Google Scholar] [CrossRef]
- Schnurr, K.; Hellwing, M.; Seidemann, B.; Jungblut, P.; Kuhn, P.H.; Rapaport, H.S.M.; Schewe, T. Oxygenation of biomembranes by mammalian lypoxygenases: The role of ubiquinone. Free Radic. Biol. Med. 1996, 20, 11–21. [Google Scholar] [CrossRef]
- Shahcheraghi, S.H.; Salemi, F.; Peirovi, N.; Ayatollahi, J.; Alam, W.; Khan, H.; Saso, L. Nrf2 Regulation by Curcumin: Molecular Aspects for Therapeutic Prospects. Molecules 2022, 27, 167. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Ahmadi, Z.; Mohammadinejad, R.; Farkhondeh, T.; Samarghandian, S. Curcumin activates the Nrf2 pathway and induces cellular protection against oxidative injury. Curr. Mol. Med. 2020, 20, 116–133. [Google Scholar] [CrossRef]
- Zhang, Q.; Deng, Q.; Zhang, J.; Ke, J.T.; Zhu, Y.; Wen, R.W.; Ye, Z.; Peng, H.; Su, Z.Z.; Wang, C.; et al. Activation of the Nrf2-ARE Pathway Ameliorates Hyperglycemia-Mediated Mitochondrial Dysfunction in Podocytes Partly Through Sirt1. Cell. Physiol. Biochem. 2018, 48, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.O.; Sanchez-Ramos, C.; Prieto-Arroyo, I.; Urbanek, P.; Steinbrenner, H.; Monsalve, M. Redox regulation of FoxO transcription factors. Redox Biol. 2015, 6, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Brunet, A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 2005, 24, 7410–7425. [Google Scholar] [CrossRef] [PubMed]
- Burk, R.F.; Hill, K.E.; Selenoprotein, P. An extracellular protein with unique physical characteristics and a role in selenium homeostasis. Annu. Rev. Nutr. 2005, 25, 215–235. [Google Scholar] [CrossRef]
- Lu, Q.; Zhai, Y.; Cheng, Q.; Liu, Y.; Gao, X.; Zhang, T.; Wei, Y.; Zhang, F.; Yin, X. The Akt-FoxO3a-manganese superoxide dismutase pathway is involved in the regulation of oxidative stress in diabetic nephropathy. Exp. Physiol. 2012, 98, 934–945. [Google Scholar] [CrossRef]
- Mortuza, R.; Chen, S.; Feng, B.; Sen, S.; Chakrabarti, S. High glucose induced alteration of SIRTs in endothelial cells causes rapid aging in ap300 and FOXO regulated pathway. PLoS ONE 2013, 8, e54514. [Google Scholar] [CrossRef]
- Kehrer, J.P.; Klotz, L.O. Free radicals and related reactive species as mediators of tissue injury and disease: Implications for Health. Crit. Rev. Toxicol. 2015, 45, 765–798. [Google Scholar] [CrossRef]
- Housley, M.P.; Udeshi, N.D.; Rodgers, J.T.; Shabanowitz, J.; Puigserver, P.; Hunt, D.F.; Hart, W.G. A PGC-1alpha-O-GlcNAc transferase complex regulates FoxO transcription factor activity in response to glucose. J. Biol. Chem. 2009, 284, 5148–5157. [Google Scholar] [CrossRef]
- Behl, Y.; Krothapalli, P.; Desta, T.; Roy, S.; Graves, D.T. FOXO1 plays an important role in enhanced microvascular cell apoptosis and microvascular cell loss in type 1 and type 2 diabetic rats. Diabetes 2009, 58, 917–925. [Google Scholar] [CrossRef]
- Nakae, J.; Biggs, W.H.; Kitamura, T.; Cavenee, W.K.; Wright, C.V.; Arden, K.C.; Accili, D. Regulation of insulin action and pancreatic beta-cell function by mutated alleles of the gene encoding fork head transcription factor Foxo1. Nat. Genet. 2002, 32, 245–253. [Google Scholar] [CrossRef]
- Farhan, M.; Silva, M.; Xingan, X.; Huang, Y.; Zheng, W. Role of FOXO transcription factors in cancer metabolism and angiogenesis. Cells 2020, 9, 1586. [Google Scholar] [CrossRef] [PubMed]
- Arcambal, A.; Taïlé, J.; Rondeau, P.; Viranaïcken, W.; Meilhac, O.; Gonthier, M.P. Free, Hyperglycemia modulates redox, inflammatory and vasoactive markers through specific signaling pathways in cerebral endothelial cells: Insights on insulin protective action. Free Radic. Biol. Med. 2019, 130, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Yaribeygi, H.; Noroozadeh, A.; Mohammadi, M.T.; Johnston, T.P.; Sahebkar, A. Crocin Improves Oxidative Stress by Potentiating Intrinsic Anti-Oxidant Defense Systems in Pancreatic Cells During Uncontrolled Hyperglycemia. J. Pharm. 2019, 22, 83–89. [Google Scholar] [CrossRef]
- Greer, E.L.; Oskoui, P.R.; Banko, M.R.; Maniar JMGygi MPGygi, S.P.; Brunet, A. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J. Biol. Chem. 2007, 282, 30107–30119. [Google Scholar] [CrossRef] [PubMed]
- Dromparis, P.; Michelakis, E.D. Mitochondria in vascular health and disease. Ann. Rev. Physiol. 2013, 75, 95–126. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Veenstra, A.; Palczewski, K.; Kern, T.S. Photoreceptor cells are major contributors to diabetes-induced oxidative stress and local inflammation in the retina. Proc. Natl. Acad. Sci. USA 2013, 110, 16586–16591. [Google Scholar] [CrossRef]
- Nguyen, N.H.; Tran, G.B.; Nguyen, C.T. Anti-oxidative effects of superoxide dismutase 3 on inflammatory diseases. J. Mol. Med. 2020, 98, 59–69. [Google Scholar] [CrossRef]
- Ikelle, L.; Naash, M.I.; Al-Ubaidi, M.R. Oxidative Stress, Diabetic Retinopathy, and Superoxide Dismutase 3. In Retinal Degenerative Diseases. Advances in Experimental Medicine and Biology; Bowes Rickman, C., Grimm, C., Anderson, R., Ash, J., LaVail, M., Hollyfield, J., Eds.; Springer: Cham, Switzerland, 2019; Volume 1185. [Google Scholar] [CrossRef]
- Hwang, I.; Lee, J.; Huh, J.Y.; Park, J.; Lee, H.B.; Ho, Y.S.; Ha, H. Catalase deficiency accelerates diabetic renal injury through peroxisomal dysfunction. Diabetes 2012, 61, 728–738. [Google Scholar] [CrossRef]
- Turkseven, S.; Kruger, A.; Mingone, C.J.; Kaminski, P.; Inaba, M.; Rodella, L.F.; Ikehara, S.; Wolin, M.S.; Abraham, N.G. Antioxidant mechanism of heme oxygenase-1 involves an increase in superoxide dismutase and catalase in experimental diabetes. Heart Circ. Physiol. 2005, 289, H701–H707. [Google Scholar] [CrossRef]
- Tang, X.; Luo, X.Y.; Chen, H.Z.; Liu, D.P. Mitochondria, endothelial cell function, and vascular diseases. Front. Physiol. 2014, 5, 175. [Google Scholar] [CrossRef]
- Oelze, M.; Kröller-Schön, S.; Steven, S.; Lubos, E.; Doppler, C.; Hausding, M.; Tobias, S.; Brochhausen, C.; Li, H.; Torzewski, M.; et al. Glutathione peroxidase-1 deficiency potentiates dysregulatory modifications of endothelial nitric oxide synthase and vascular dysfunction in aging. Hypertension 2014, 63, 390–396. [Google Scholar] [CrossRef]
- Altuhafi, A.; Altun, M.; Hadwan, M.H. The Correlation between Selenium-Dependent Glutathione Peroxidase Activity and Oxidant/Antioxidant Balance in Sera of Diabetic Patients with Nephropathy. Rep. Biochem. Mol. Biol. 2021, 10, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Perkins, A.; Nelson, K.J.; Parsonage, D.; Poole, L.B.; Karplus, P.A. Peroxiredoxins: Guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem. Sci. 2015, 40, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.; Chae, H.; Kim, K. Peroxiredoxins: A historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic. Biol. Med. 2005, 38, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Claiborne, A.; Yeh, J.; Mallett, T.; Luba, J.; Crane, E.; Charrier, V.; Parsonage, D. Protein-sulfenic acids: Diverse roles for an unlikely player in enzyme catalysis and redox regulation. Biochemistry 1999, 38, 15407–15416. [Google Scholar] [CrossRef]
- Monteiro, G.; Horta, B.B.; Pimenta, D.C.; Augusto, O.; Netto, L.E. Reduction of 1-Cys peroxiredoxins by ascorbate changes the thiol-specific antioxidant paradigm, revealing another function of vitamin C. Proc. Natl. Acad. Sci. USA 2007, 104, 4886–4891. [Google Scholar] [CrossRef] [PubMed]
- Arkat, S.; Umbarkar, P.; Singh, S.; Sitasawad, S.L. Mitochondrial Peroxiredoxin-3 protects against hyperglycemia induced myocardial damage in Diabetic cardiomyopathy. Free Radic. Biol. Med. 2016, 97, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Pacifici, F.; Arriga, R.; Sorice, G.P.; Capuani, B.; Scioli, M.G.; Pastore, D.; Giulia Donadel Bellia, A.; Caratelli, S.; Coppola, A.; Ferrelli, F.; et al. Peroxiredoxin 6, a novel player in the pathogenesis of diabetes. Diabetes 2014, 63, 3210–3220. [Google Scholar] [CrossRef]
- Haendeler, J.; Popp, R.; Goy, C.; Tischler, V.; Zeiher, A.M.; Dimmeler, S. Cathepsin D and H2O2 stimulate degradation of thioredoxin-1: Implication for endothelial cell apoptosis. J. Biol. Chem. 2005, 28, 42945–42951. [Google Scholar] [CrossRef]
- Tinkov, A.A.; Bjørklund, G.; Skalny, A.V.; Holmgren, A.; Skalnaya, M.G.; Chirumbolo, S.; Aaseth, J. The role of the thioredoxin/thioredoxin reductase system in the metabolic syndrome: Towards a possible prognostic marker? Cell. Mol. Life Sci. 2018, 75, 1567–1586. [Google Scholar] [CrossRef]
- Ng, C.J.; Shih, D.M.; Hama, S.Y.; Villa, N.; Navab, M.; Reddy, S.T. The paraoxonase gene family and atherosclerosis. Free Radic. Biol. Med. 2005, 38, 153–163. [Google Scholar] [CrossRef]
- Devarajan, A.; Bourquard, N.; Hama, S.; Navab, M.; Grijalva, V.R.; Morvardi, S.; Clarke, C.F.; Vergnes, L.; Reue, K.; Teiber, J.F.; et al. Paraoxonase 2 deficiency alters mitochondrial function and exacerbates the development of atherosclerosis. Antioxid. Redox Signal. 2011, 14, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Suehiro, T.; Inoue, M.; Nakauchi, Y.; Morita, T.; Arii, K.; Ito, H.; Kumon, Y.; Hashimoto, K. Serum Paraoxonase activity and its relationship to diabetic complications in patients with non-Insulin Dependent Diabetes Mellitus. Metabolism 1998, 47, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.; Reichert, C.O.; Bydlowski, S.P. Paraoxonases Activities and Polymorphisms in Elderly and Old-Age Diseases: An Overview. Antioxidants 2019, 8, 118. [Google Scholar] [CrossRef] [PubMed]
- Litvinov, D.; Mahini, H.; Garelnabi, M. Antioxidant and Anti-Inflammatory Role of Paraoxonase 1: Implication in Arteriosclerosis Diseases. N. Am. J. Med. Sci. 2012, 4, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Arab, Z.N.; Khayatan, D.; Razavi, S.M.; Zare, K.; Kheradkhah, E.; Momtaz, S.; Ferretti, G.; Bacchetti, T.; Sathyapalan, T.; Emami, S.A.; et al. Phytochemicals as Modulators of Paraoxonase-1 in Health and Diseases. Antioxidants 2022, 11, 1273. [Google Scholar] [CrossRef]
- Taler-Verčič, A.; Goličnik, M.; Bavec, A. The structure and function of paraoxonase-1 and its comparison to paraoxonase-2 and-3. Molecules 2020, 25, 5980. [Google Scholar] [CrossRef]
- Landry, J.; Sutton, A.; Tafrov, S.T.; Heller, R.C.; Stebbins, J.; Pillus, L.; Sternglanz, R. The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc. Natl. Acad. Sci. USA 2000, 97, 5807–5811. [Google Scholar] [CrossRef]
- Blander, G.; Guarente, L. The Sir2 family of protein deacetylases. Annu. Rev. Biochem. 2004, 73, 417–435. [Google Scholar] [CrossRef]
- Morris, B.J. Seven sirtuins for seven deadly diseases of aging. Free Radic. Biol. Med. 2013, 56, 133–171. [Google Scholar] [CrossRef]
- Guarente, L.; Imai, S.; Armstrong, C.M.; Kaeberlein, M. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000, 403, 795–800. [Google Scholar] [CrossRef]
- Morris, B.J. A forkhead in the road to longevity: The molecular basis of lifespan becomes clearer. J. Hypertens. 2005, 23, 1285–1309. [Google Scholar] [CrossRef] [PubMed]
- Ingram, D.K.; Zhu, M.; Mamczarz, J.; Zou, S.; Lane, M.A.; Roth, G.S.; de Cabo, R. Calorie restriction mimetics: An emerging research field. Aging Cell 2006, 5, 97–108. [Google Scholar] [CrossRef]
- Hall, J.A.; Dominy, J.E.; Lee, Y.; Puigserver, P. The sirtuin family’s role in aging and age-associated pathologies. J. Clin. Investig. 2013, 123, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Turkmen, K.; Karagoz, A.; Kucuk, A. Sirtuins as novel players in the pathogenesis of diabetes mellitus. World J. Diabetes 2014, 5, 894–900. [Google Scholar] [CrossRef] [PubMed]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Park, S.H.; Chang, H.C.; Shapiro, J.S.; Vassilopoulos, A.; Sawicki, K.T.; Chen, C.; Shang, M.; Burridge, P.W.; Epting, C.L.; et al. Sirtuin 2 regulates cellular iron homeostasis via deacetylation of transcription factor NRF2. J. Clin. Investig. 2017, 127, 1505–1516. [Google Scholar] [CrossRef]
- Patel, S.; Khan, H.; Majumdar, A. Crosstalk between Sirtuins and Nrf2: SIRT1 activators as emerging treatment for diabetic neuropathy. Metab. Brain Dis. 2022, 37, 2181–2195. [Google Scholar] [CrossRef]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Furukawa-Hibi, Y.; Chen, C.; Horio, Y.; Isobe, K.; Ikeda, K.; Motoyama, N. SIRT1 is critical regulator of FOXO-mediated transcription in response to oxidative stress. Int. J. Mol. Med. 2005, 16, 237–243. [Google Scholar] [CrossRef]
- Ogura, Y.; Kitada, M.; Koya, D. Sirtuins and Renal Oxidative Stress. Antioxidants 2021, 10, 1198. [Google Scholar] [CrossRef]
- Gounden, S.; Phulukdaree, A.; Moodley, D.; Chuturgoon, A. Increased SIRT3 Expression and Antioxidant Defense under Hyperglycemic Conditions in HepG2 Cells. Metab. Syndr. Relat. Disord. 2015, 13, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Sun, W.; Cheng, Y.; Xu, Z.; Cai, L. Management of diabetic nephropathy: The role of sirtuin-1. Future Med. Chem. 2019, 11, 2241–2245. [Google Scholar] [CrossRef] [PubMed]
- Nebbioso, M.; Lambiase, A.; Armentano, M.; Tucciarone, G.; Bonfiglio, V.; Plateroti, R.; Alisi, L. The Complex Relationship between Diabetic Retinopathy and High-Mobility Group Box: A Review of Molecular Pathways and Therapeutic Strategies. Antioxidants 2020, 9, 666. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
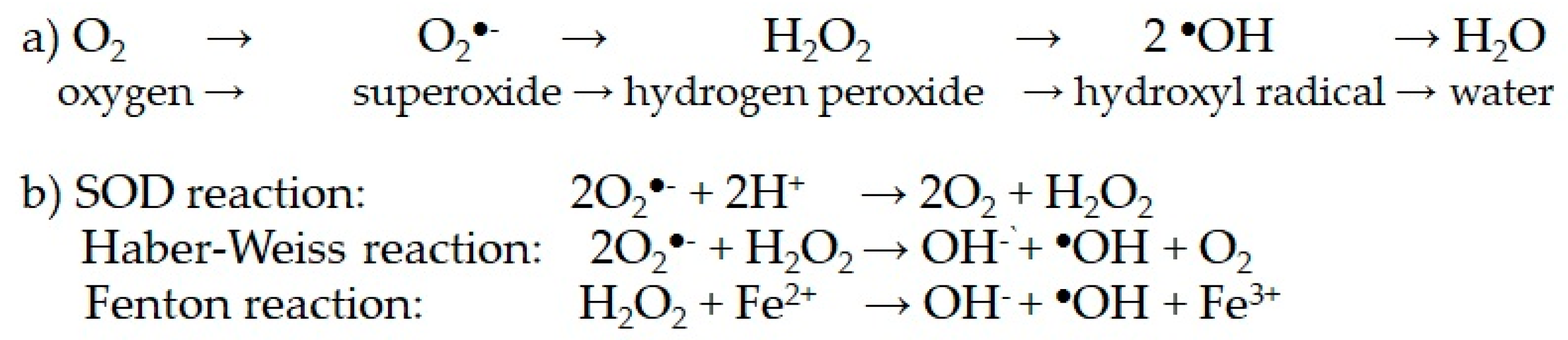{kind=link}
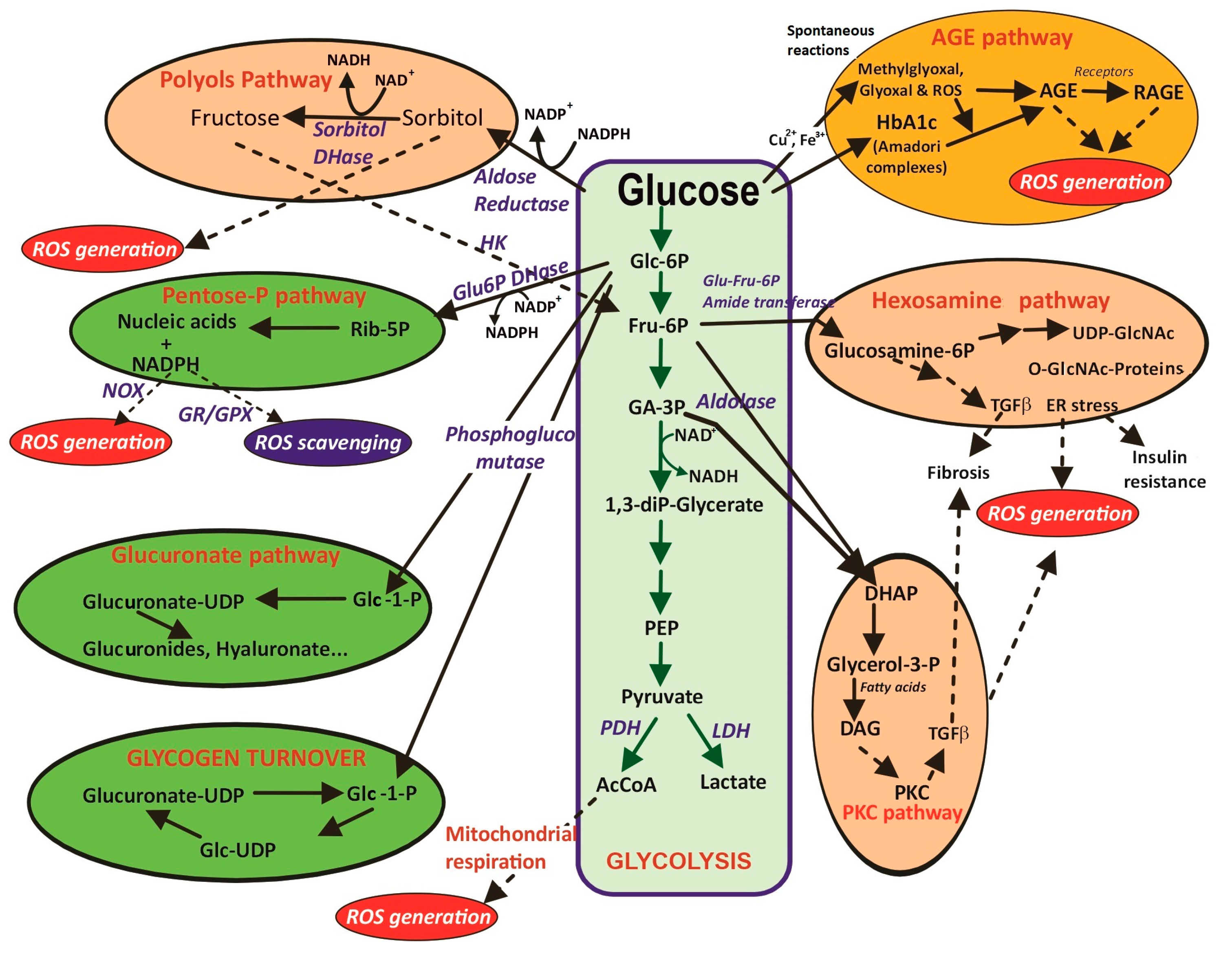
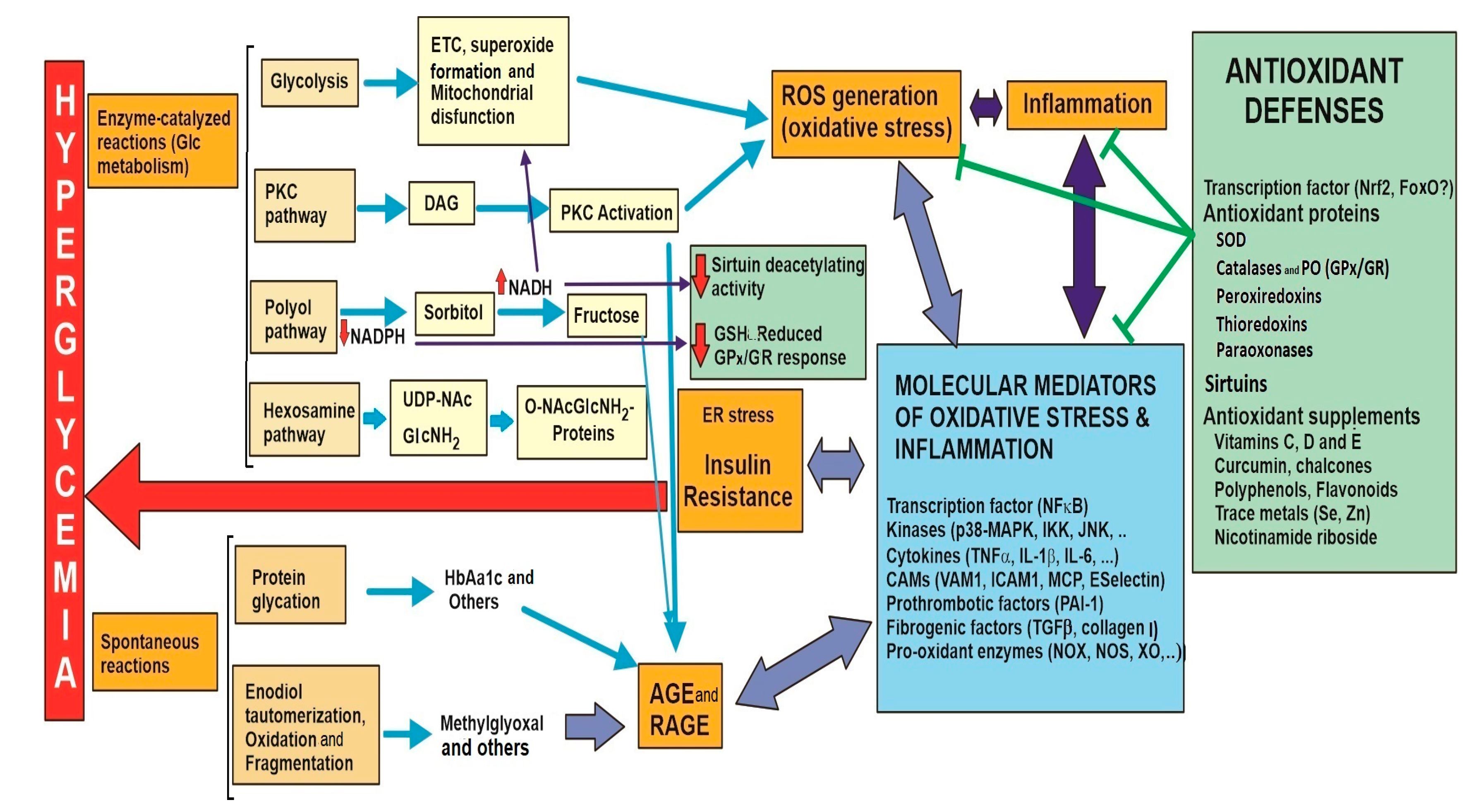
| 3.1. Glucose metabolism 3.1.1. Glycolysis to pyruvate, AcCoA, Krebs cycle and mitochondrial respiration through the electronic chain (ETC). Unpaired electrons and ROS formation. Lowered intramitochondrial antioxidant enzymes. Structural and functional damage of the inner membrane, mitochondrial complexes and mitochondrial DNA. | |
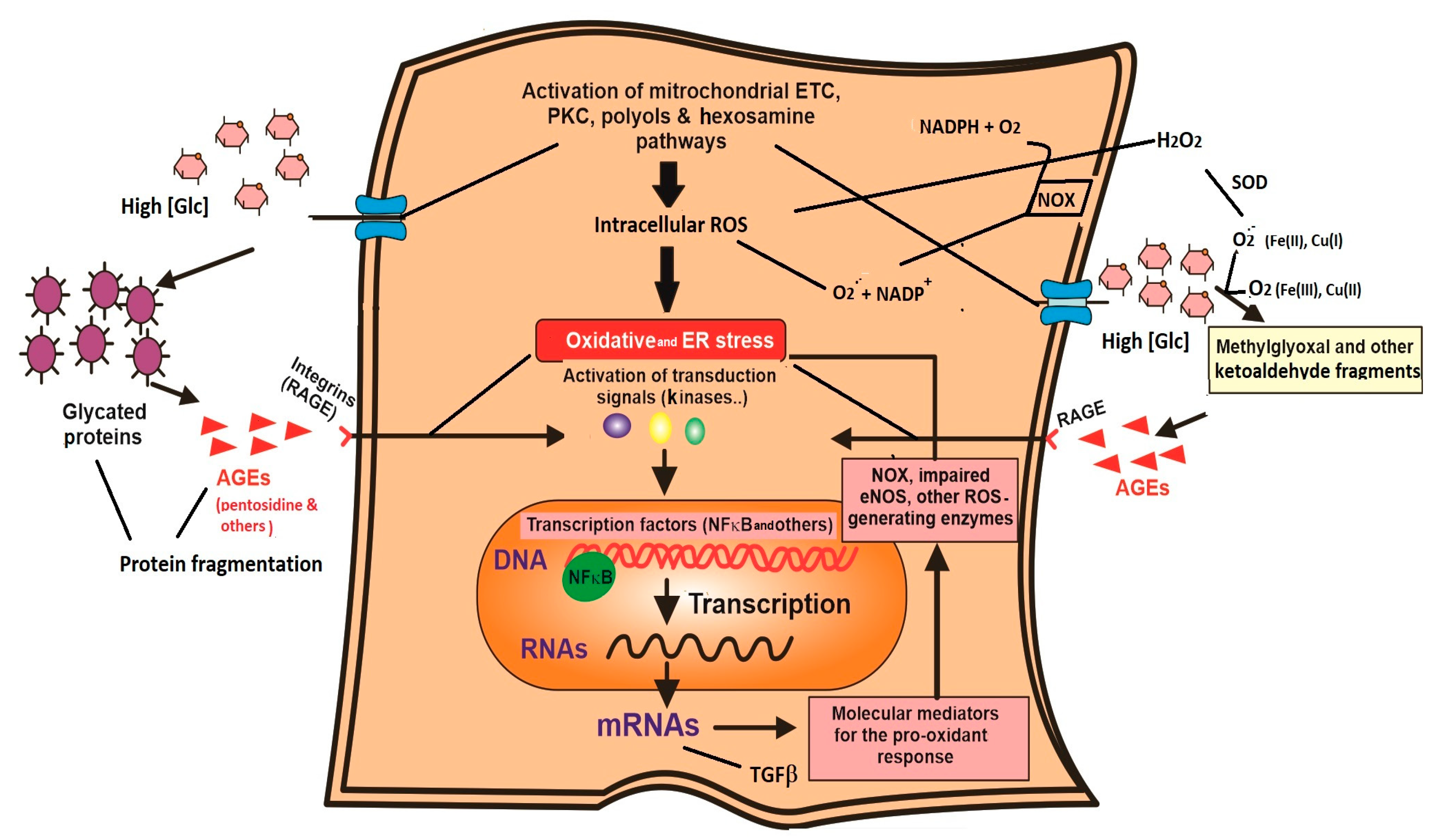
| 3.2. Glucose collateral metabolism 3.2.1. PKC pathway. Increase in DAG formation and activation of NOX isoenzymes. 3.2.2. Polyol pathway. Sorbitol and fructose formation, cofactor imbalance. 3.2.3. Hexosamine pathway. O-NAcGlucNH2 glycosylated proteins, ER stress and insulin resistance. | |
| 3.3. Spontaneous glucose reactions 3.3.1. Protein glycation. Generation of Amadori conjugates. Proteins alter stability with functional properties. 3.3.2. Glucose auto-oxidation. AGEs and AGE-RAGE. Dysfunctional molecules and altered transduction signals. | |
| 4. Main transcription factor involved in oxidative response: NFκB 4.1. Molecular mediators related to the pathological complications of hyperglycemia/oxidative stress. | |
| Type of mediator | Examples |
| Pro-inflammatory cytokines | TNFα, IL1β, IL6 |
| Vasoconstrictors | Angiotensin II, Endothelin1. |
| Cell adhesion molecules | ICAM-1, VCAM, MCP-1, E-Selectin |
| Pro-thrombotic molecules | PAI-1 |
| Fibrogenic molecules | TGFβ, Collagen type I |
| 4.2. Pro-oxidant enzymes that can alter the redox balance via ROS generation. | |
| Enzyme | Reaction catalyzed |
| NADPH Oxidases (NOX) | NADPH + 2O2 → NADP+ + O2●− |
| Coupled/Uncoupled Nitric Oxide Synthase (NOS) | Arginine + O2 → Citrulline + NO (coupled, neutral for ROS formation) |
| Arginine + O2 → Citrulline + NO + O2●− → ONOO●− (peroxynitrile) (uncoupled) | |
| PG G/H Synthase (PGHS) = Cyclooxygenase (COX) + Peroxidase | Arachidonate + 2O2 + AH2→ PGG2 + A (COX activity) |
| Arachidonate + 2O2 + AH2→ PGH2 + A+ H2O (complete PGHS activity) | |
| Lipoxygenases (LPO) | Unsaturated fatty acid + 2O2 → Hydroperoxide (R-OOH). (Lipid peroxidation of unsaturated fatty acids can break the carbon chain to yield MDA and other lipid peroxidation markers that generate ROS). |
| Xanthine Oxidase (XO) | Hypoxanthine (Xanthine) + O2 + H2O → Xanthine (Urate) + H2O2 + O2●− |
| Heme-oxygenase (HO1) | Heme + 3O2 + 3NADPH-reductase→ CO + Fe2+ + H+ + 3 H2O + 3NADP+-reductase + Biliverdin (bilirrubin). (Biliverdin could act as an antioxidant, but free Fe2+ could catalyze the Fenton reaction for ROS formation). |
| Myeloperoxidase (MPO) | Cl− + H2O2 → ClO− + H2O |
| Cytochrome P450 (CYP) | R-H + 2O2 → R-OH + H2O (H2O2, O2●− and ●OH are eventual products of some CYP isoenzymes) |
| 5.1. Superoxide dismutase (SOD) | 2 O2●− + 2H+ → H2O2 + O2 |
| 5.2.a Catalase (CAT) | 2 H2O2 → O2 + 2H2O |
| 5.2.b Glutathione peroxidase (GPx) Accessory Nrf2-induced enzymes Glutathione reductase (GR) Glutathione-S-transferase (GST) GC ligase (GCLC and GCLM) GSH synthase | H2O2 + 2 GSH → 2H2O + GSSG GSSG + NADPH→ 2 GSH + NAPD+ (GSH recycling) GSH + oxidized-Acceptor → GS-Acceptor conjugate + H2O Glu + Cys +ATP→ γ-Glutamyl-Cysteine (precursor GSH synthesis) +ADP+ Pi γ-Glutamyl-Cysteine + Gly + ATP → GSH + ADP+ Pi |
| 5.3. Peroxiredoxin (Prx) | H2O2 + GSH (Ascorb, Trx) → 2H2O + GSSG (Dehydroascorb, Trx) (GR or Trx reductase are needed for GSH and reduced Prx recycling respectively) |
| 5.4. Thioredoxin (Trx) Accessory enzyme: thioredoxin reductase (TrxR) | R-S-S-R + Reduced-Trx → 2 R-SH + Oxidized -Trx R-S-OH + Reduced-Trx → R-SH + Oxidized -Trx (protein thiol groups recycled from disulphide or sulfenic oxidations) Oxidized-Trx + NADPH→ Reduced-Trx + NAPD+ (Trx recycling) Alternatively, Trx is also regulated by the thioredoxin interacting protein (TxNIP) |
| 5.5. Paraoxonase (PON) | Oxidized Lipoproteins → Reduced Lipoproteins |
| 5.6. Sirtuin (Sirt) | Acetylated-Protein + NAD+ → Protein + ADP-Ribose + Nicotinamide + Acetate |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González, P.; Lozano, P.; Ros, G.; Solano, F. Hyperglycemia and Oxidative Stress: An Integral, Updated and Critical Overview of Their Metabolic Interconnections. Int. J. Mol. Sci. 2023, 24, 9352. https://doi.org/10.3390/ijms24119352
González P, Lozano P, Ros G, Solano F. Hyperglycemia and Oxidative Stress: An Integral, Updated and Critical Overview of Their Metabolic Interconnections. International Journal of Molecular Sciences. 2023; 24(11):9352. https://doi.org/10.3390/ijms24119352
Chicago/Turabian StyleGonzález, Patricia, Pedro Lozano, Gaspar Ros, and Francisco Solano. 2023. "Hyperglycemia and Oxidative Stress: An Integral, Updated and Critical Overview of Their Metabolic Interconnections" International Journal of Molecular Sciences 24, no. 11: 9352. https://doi.org/10.3390/ijms24119352
APA StyleGonzález, P., Lozano, P., Ros, G., & Solano, F. (2023). Hyperglycemia and Oxidative Stress: An Integral, Updated and Critical Overview of Their Metabolic Interconnections. International Journal of Molecular Sciences, 24(11), 9352. https://doi.org/10.3390/ijms24119352