_Putnam.png)
Roles of Virtual Screening and Molecular Dynamics Simulations in Discovering and Understanding Antimalarial Drugs

 , , and
, , and
Abstract
1. Introduction
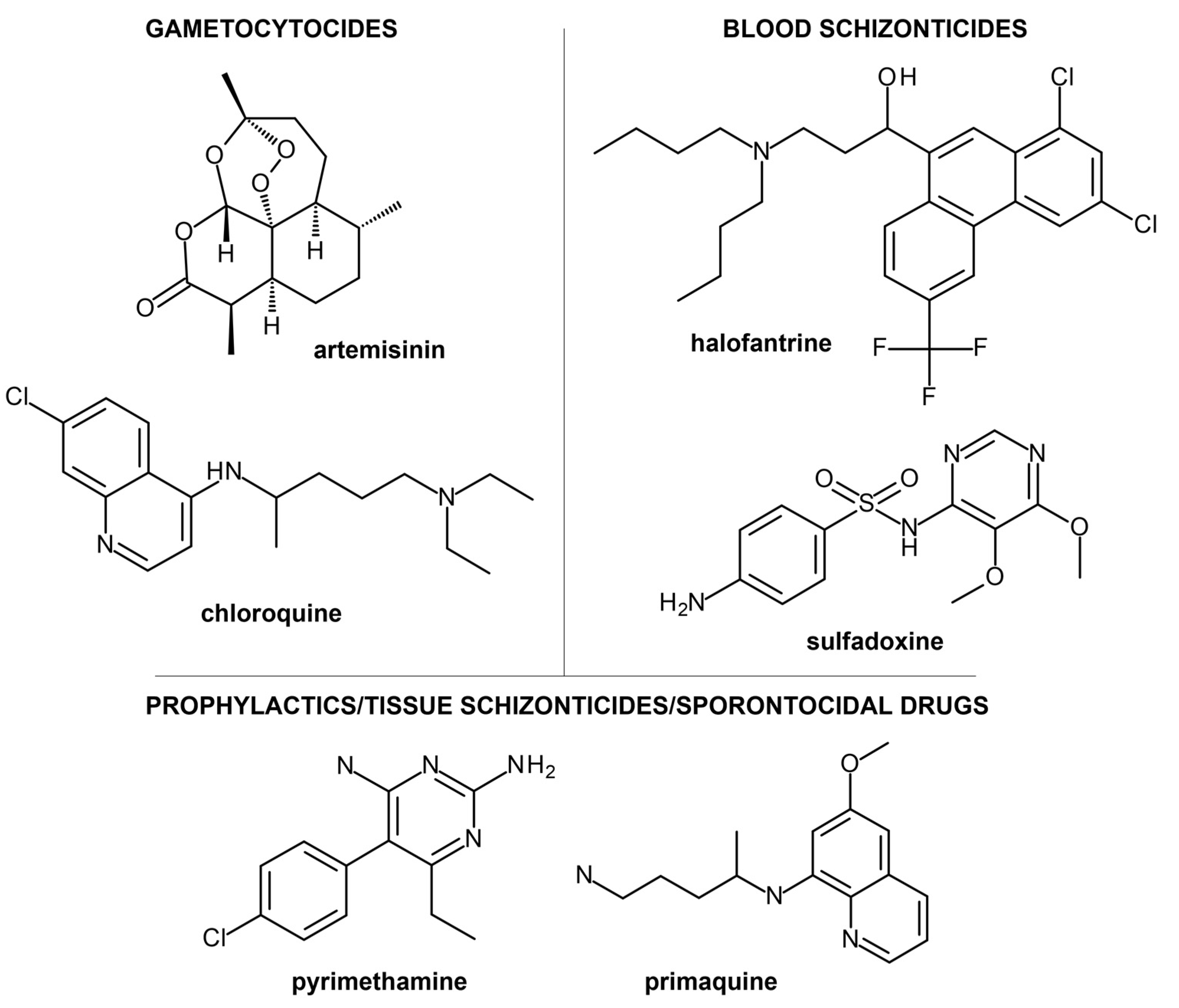
2. Current Treatment Strategies and Clinical Candidates Targeting Malaria
3. Computer-Aided Anti-Malarial Drug Discovery
3.1. Structure-Based Methods
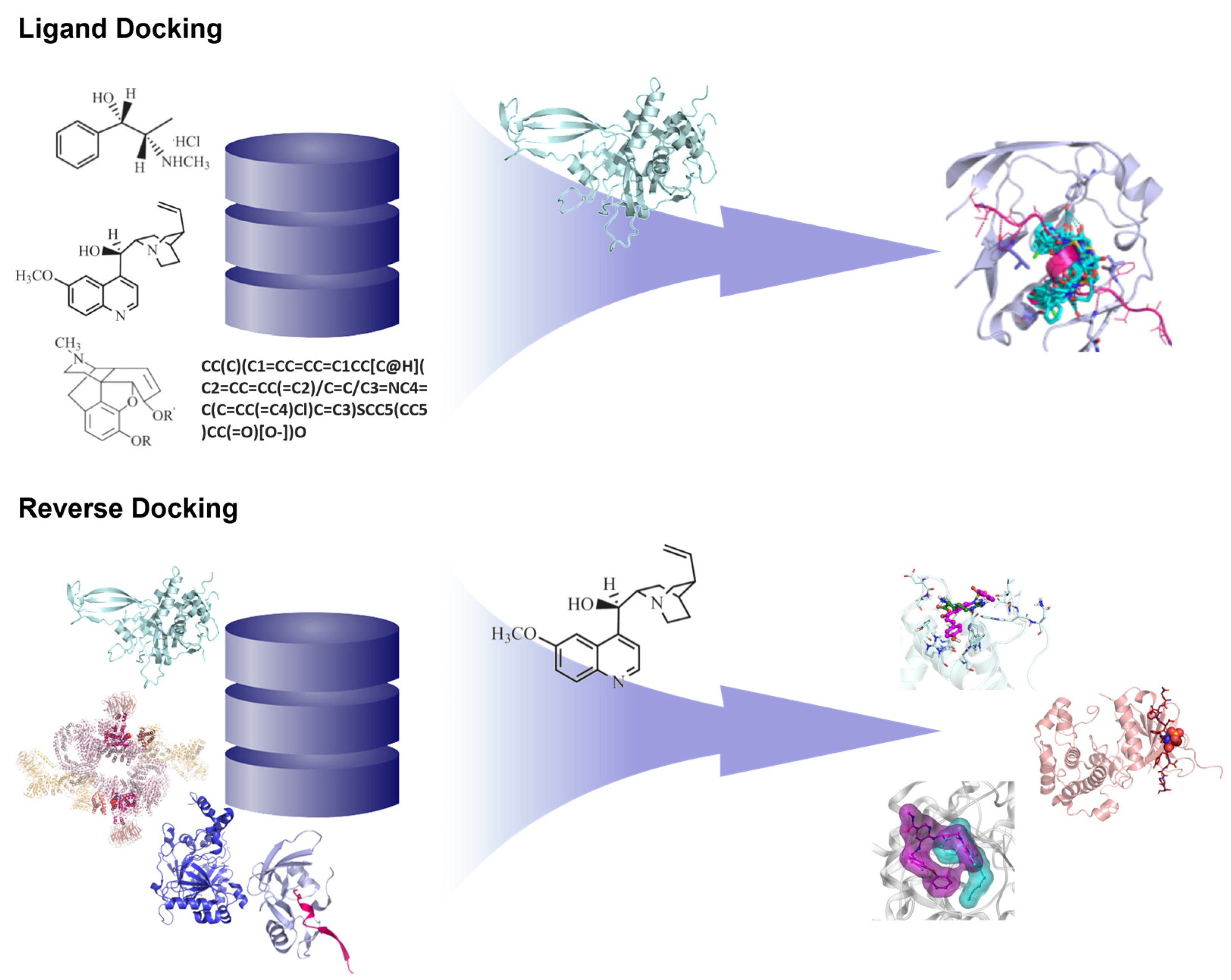
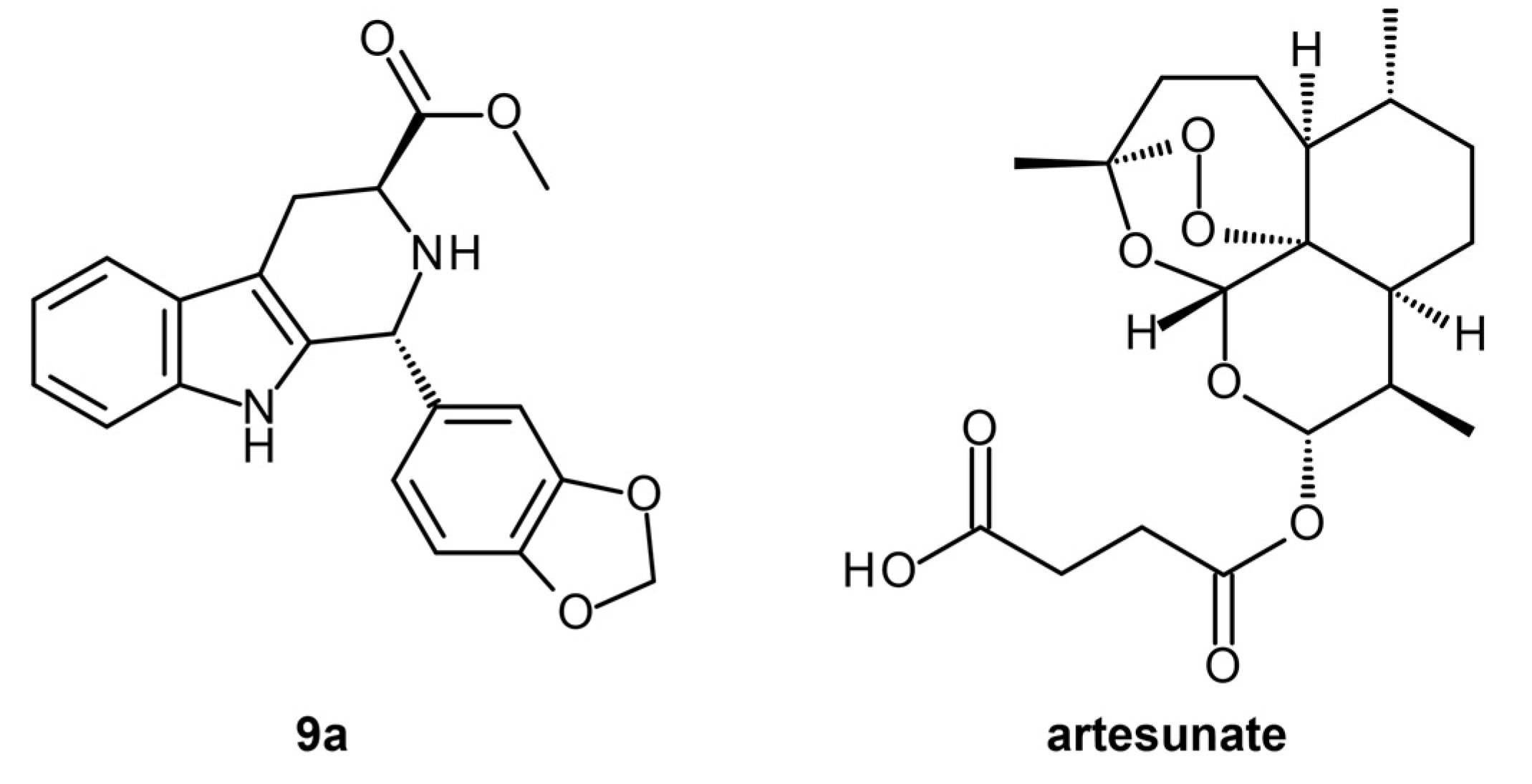
3.1.1. Reverse Docking of β-Carboline Derivatives
3.1.2. Anopheles gambiae Trehalase Ab Initio Modeling and Hit Identification
3.1.3. Drug Repurposing of FDA-Approved Drugs for Antimalarial Drug Discovery
3.2. Ligand-Based Methods
3.2.1. Machine Learning and Virtual Screening for P. falciparum Protein Kinases
3.2.2. Artificial Neural Network-Genetic Algorithm for Fusidic Acid Derivatives
4. Elucidation of Anti-Malarial Drugs Mechanism of Action
4.1. Artemisinin, Its Derivatives, and Combinatorial Therapy
4.2. Falcipains as Drug Targets
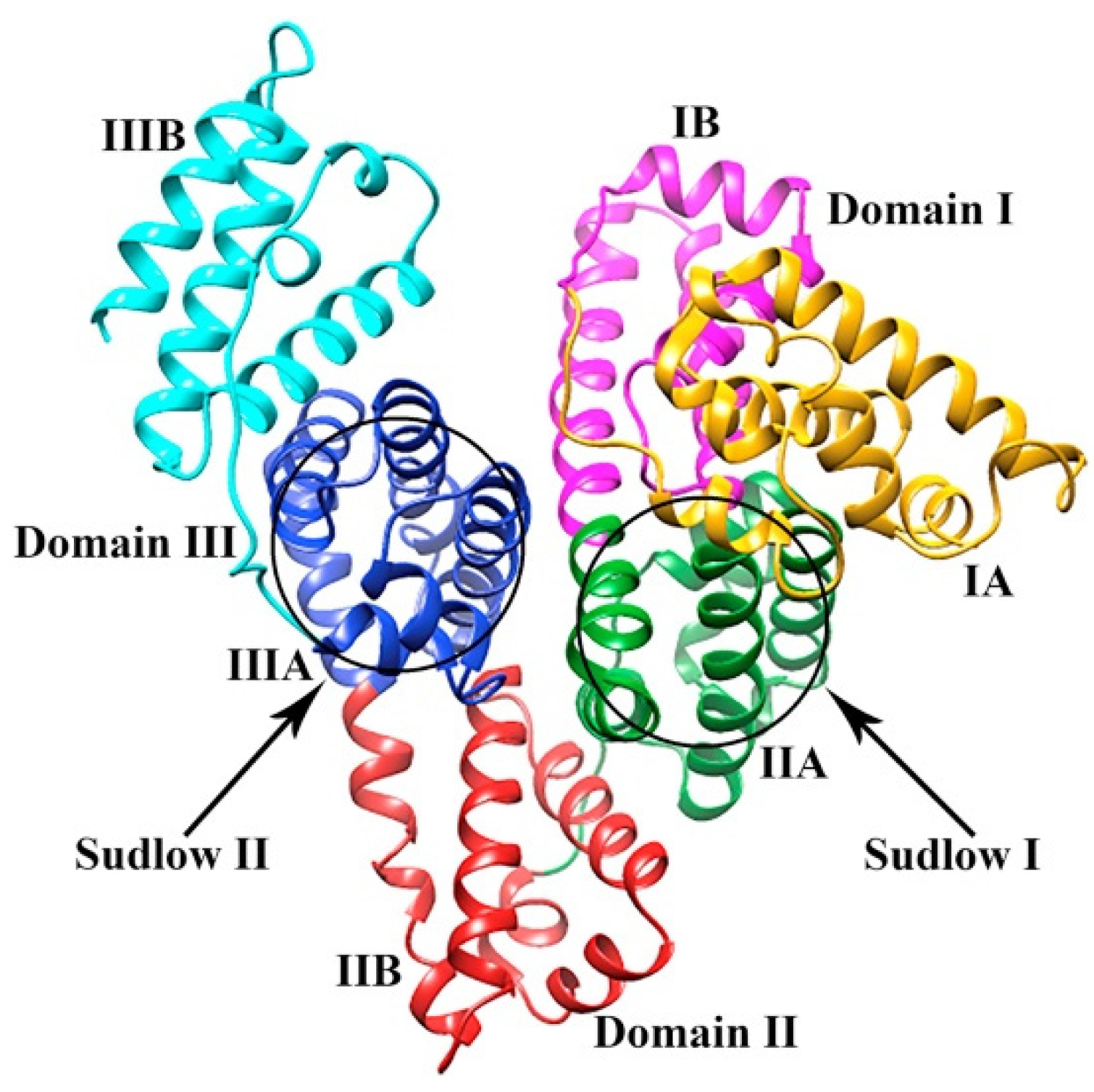
4.3. Interaction of Antimalarial Drugs with Serum Albumin
4.4. Molecular Mechanism of Anti-Malarial Drug Resistance
4.5. Other Potential Targets for Inhibition of Resistant Malarial Strains
5. MD Simulations of Peptide Immunogens for Vaccine Development
5.1. Circumsporozoite Protein
5.2. Ring-Infected Erythrocyte Surface Antigen
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. World Malaria Report 2022: Tracking Progress and Gaps in the Global Response to Malaria; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- Mawson, A.R. The pathogenesis of malaria: A new perspective. Pathog. Glob. Health 2013, 107, 122–129. [Google Scholar] [CrossRef]
- Miglianico, M.; Bolscher, J.M.; Vos, M.W.; Koolen, K.J.M.; de Bruijni, M.; Rajagopal, D.S.; Chen, E.; Kiczun, M.; Gray, D.; Campo, B.; et al. Assessment of the drugability of initial malaria infection through miniaturized sporozoite assays and high-throughput screening. Commun. Biol. 2023, 6, 216. [Google Scholar] [CrossRef]
- Herraiz, T.; Guillen, H.; Gonzalez-Pena, D.; Aran, V.J. Antimalarial Quinoline Drugs Inhibit beta-Hematin and Increase Free Hemin Catalyzing Peroxidative Reactions and Inhibition of Cysteine Proteases. Sci. Rep. 2019, 9, 15398. [Google Scholar] [CrossRef]
- Kumar, S.; Guha, M.; Choubey, V.; Maity, P.; Bandyopadhyay, U. Antimalarial drugs inhibiting hemozoin (beta-hematin) formation: A mechanistic update. Life Sci. 2007, 80, 813–828. [Google Scholar] [CrossRef]
- Egan, T.J. Recent advances in understanding the mechanism of hemozoin (malaria pigment) formation. J. Inorg. Biochem. 2008, 102, 1288–1299. [Google Scholar] [CrossRef]
- Tse, E.G.; Korsik, M.; Todd, M.H. The past, present and future of anti-malarial medicines. Malar. J. 2019, 18, 93. [Google Scholar] [CrossRef]
- Muller, I.B.; Hyde, J.E. Antimalarial drugs: Modes of action and mechanisms of parasite resistance. Future Microbiol. 2010, 5, 1857–1873. [Google Scholar] [CrossRef]
- Sevene, E.; Gonzalez, R.; Menendez, C. Current knowledge and challenges of antimalarial drugs for treatment and prevention in pregnancy. Expert. Opin. Pharmacother. 2010, 11, 1277–1293. [Google Scholar] [CrossRef]
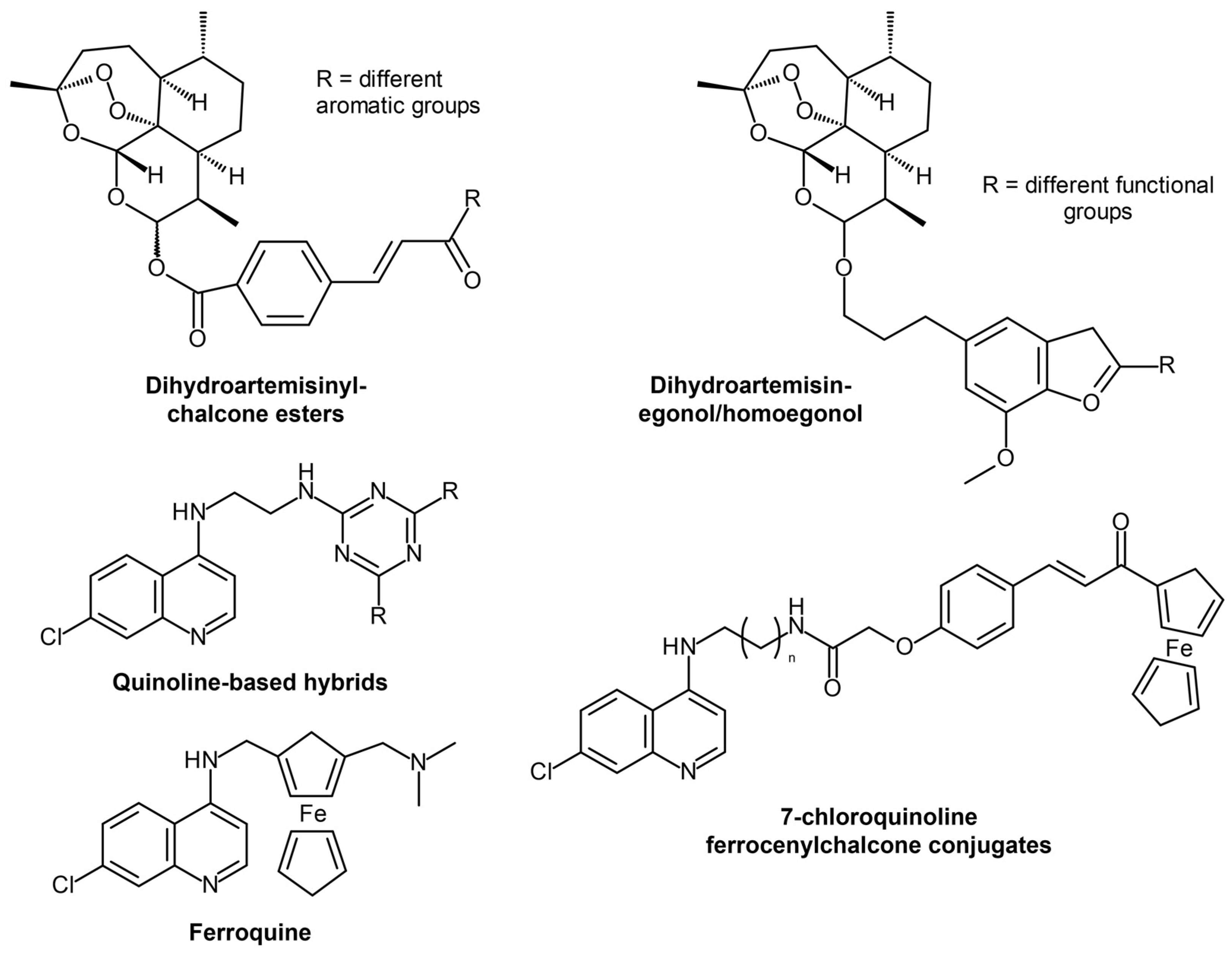
- Smit, F.J.; van Biljon, R.A.; Birkholtz, L.M.; N’Da, D.D. Synthesis and in vitro biological evaluation of dihydroartemisinyl-chalcone esters. Eur. J. Med. Chem. 2015, 90, 33–44. [Google Scholar] [CrossRef]
- Capci Karagoz, A.; Reiter, C.; Seo, E.J.; Gruber, L.; Hahn, F.; Leidenberger, M.; Klein, V.; Hampel, F.; Friedrich, O.; Marschall, M.; et al. Access to new highly potent antileukemia, antiviral and antimalarial agents via hybridization of natural products (homo)egonol, thymoquinone and artemisinin. Bioorg. Med. Chem. 2018, 26, 3610–3618. [Google Scholar] [CrossRef]
- Sahu, S.; Ghosh, S.K.; Kalita, J.; Dutta, M.; Bhat, H.R. Design, synthesis and antimalarial screening of some hybrid 4-aminoquinoline-triazine derivatives against pf-DHFR-TS. Exp. Parasitol. 2016, 163, 38–45. [Google Scholar] [CrossRef]
- Bhat, H.R.; Singh, U.P.; Thakur, A.; Kumar Ghosh, S.; Gogoi, K.; Prakash, A.; Singh, R.K. Synthesis, antimalarial activity and molecular docking of hybrid 4-aminoquinoline-1,3,5-triazine derivatives. Exp. Parasitol. 2015, 157, 59–67. [Google Scholar] [CrossRef]
- Bhat, H.R.; Singh, U.P.; Yadav, P.S.; Kumar, V.; Gahtori, P.; Das, A.; Chetia, D.; Prakash, A.; Mahanta, J. Synthesis, characterization and antimalarial activity of hybrid 4-aminoquinoline-1,3,5-triazine derivatives. Arab. J. Chem. 2016, 9, S625–S631. [Google Scholar] [CrossRef]
- Kumar, K.; Pradines, B.; Madamet, M.; Amalvict, R.; Kumar, V. 1H-1,2,3-triazole tethered mono- and bis-ferrocenylchalcone-beta-lactam conjugates: Synthesis and antimalarial evaluation. Eur. J. Med. Chem. 2014, 86, 113–121. [Google Scholar] [CrossRef]
- Raj, R.; Saini, A.; Gut, J.; Rosenthal, P.J.; Kumar, V. Synthesis and in vitro antiplasmodial evaluation of 7-chloroquinoline-chalcone and 7-chloroquinoline-ferrocenylchalcone conjugates. Eur. J. Med. Chem. 2015, 95, 230–239. [Google Scholar] [CrossRef]
- Garcia-Barrantes, P.M.; Lamoureux, G.V.; Perez, A.L.; Garcia-Sanchez, R.N.; Martinez, A.R.; San Feliciano, A. Synthesis and biological evaluation of novel ferrocene-naphthoquinones as antiplasmodial agents. Eur. J. Med. Chem. 2013, 70, 548–557. [Google Scholar] [CrossRef]
- Kondratskyi, A.; Kondratska, K.; Vanden Abeele, F.; Gordienko, D.; Dubois, C.; Toillon, R.A.; Slomianny, C.; Lemiere, S.; Delcourt, P.; Dewailly, E.; et al. Ferroquine, the next generation antimalarial drug, has antitumor activity. Sci. Rep. 2017, 7, 15896. [Google Scholar] [CrossRef]
- Ong, H.W.; Truong, A.; Kwarcinski, F.; de Silva, C.; Avalani, K.; Havener, T.M.; Chirgwin, M.; Galal, K.A.; Willis, C.; Kramer, A.; et al. Discovery of potent Plasmodium falciparum protein kinase 6 (PfPK6) inhibitors with a type II inhibitor pharmacophore. Eur. J. Med. Chem. 2023, 249, 115043. [Google Scholar] [CrossRef]
- Matassini, C.; Parmeggiani, C.; Cardona, F. New Frontiers on Human Safe Insecticides and Fungicides: An Opinion on Trehalase Inhibitors. Molecules 2020, 25, 3013. [Google Scholar] [CrossRef]
- Gogoi, N.; Chetia, D.; Gogoi, B.; Das, A. Multiple-targets Directed Screening of Flavonoid Compounds from Citrus Species to find out Antimalarial Lead with Predicted Mode of Action: An In Silico and Whole Cell-based In vitro Approach. Curr. Comput. Aided Drug Des. 2021, 17, 69–82. [Google Scholar] [CrossRef]
- Neves, B.J.; Braga, R.C.; Alves, V.M.; Lima, M.N.N.; Cassiano, G.C.; Muratov, E.N.; Costa, F.T.M.; Andrade, C.H. Deep Learning-driven research for drug discovery: Tackling Malaria. PLoS Comput. Biol. 2020, 16, e1007025. [Google Scholar] [CrossRef]
- Gorki, V.; Walter, N.S.; Singh, R.; Chauhan, M.; Dhingra, N.; Salunke, D.B.; Kaur, S. beta-Carboline Derivatives Tackling Malaria: Biological Evaluation and Docking Analysis. ACS Omega 2020, 5, 17993–18006. [Google Scholar] [CrossRef]
- Yadav, U.; Pandey, J. Molecular Docking Studies of Rifampicin-rpoB complex: Repurposing Drug Design Implications for against Plasmodium falciparum Malaria through a Computational Approach. Drug Res. (Stuttg) 2023, 73, 164–169. [Google Scholar] [CrossRef]
- Diallo, B.N.; Swart, T.; Hoppe, H.C.; Tastan Bishop, O.; Lobb, K. Potential repurposing of four FDA approved compounds with antiplasmodial activity identified through proteome scale computational drug discovery and in vitro assay. Sci. Rep. 2021, 11, 1413. [Google Scholar] [CrossRef]
- Ibrahim, Z.Y.; Uzairu, A.; Shallangwa, G.; Abechi, S. Theoretical design of novel antimalarial agents against P. falciparum strain, Dd(2) through the QSAR modeling of synthesized 2’-substituted triclosan derivatives. Heliyon 2020, 6, e05032. [Google Scholar] [CrossRef]
- Ya’u Ibrahim, Z.; Uzairu, A.; Shallangwa, G.; Abechi, S. Molecular docking studies, drug-likeness and in-silico ADMET prediction of some novel β-Amino alcohol grafted 1,4,5-trisubstituted 1,2,3-triazoles derivatives as elevators of p53 protein levels. Sci. Afr. 2020, 10, e00570. [Google Scholar] [CrossRef]
- Muhseen, Z.T.; Hameed, A.R.; Al-Bhadly, O.; Ahmad, S.; Li, G. Natural products for treatment of Plasmodium falciparum malaria: An integrated computational approach. Comput. Biol. Med. 2021, 134, 104415. [Google Scholar] [CrossRef]
- Adigun, R.A.; Malan, F.P.; Balogun, M.O.; October, N. Design, synthesis, and in silico-in vitro antimalarial evaluation of 1,2,3-triazole-linked dihydropyrimidinone quinoline hybrids. Struct. Chem. 2023. [Google Scholar] [CrossRef]
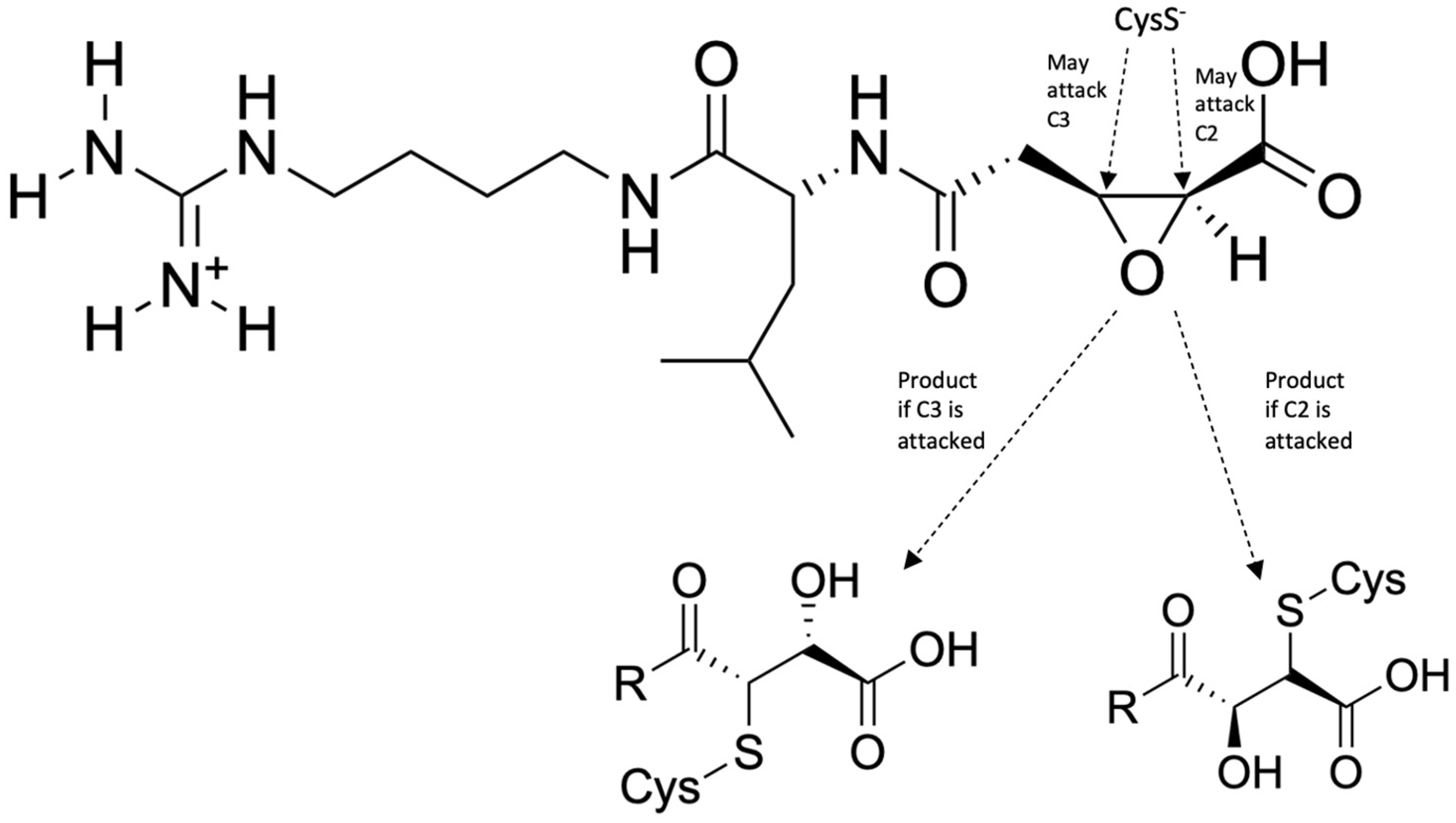
- Arafet, K.; Ferrer, S.; Marti, S.; Moliner, V. Quantum mechanics/molecular mechanics studies of the mechanism of falcipain-2 inhibition by the epoxysuccinate E64. Biochemistry 2014, 53, 3336–3346. [Google Scholar] [CrossRef]
- Danazumi, A.U.; Balogun, E.O. Microsecond-long simulation reveals the molecular mechanism for the dual inhibition of falcipain-2 and falcipain-3 by antimalarial lead compounds. Front. Mol. Biosci. 2022, 9, 1070080. [Google Scholar] [CrossRef]
- Lee, A.; Lee, K.; Kim, D. Using reverse docking for target identification and its applications for drug discovery. Expert. Opin. Drug Discov. 2016, 11, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Kuo, P.C.; Shi, L.S.; Damu, A.G.; Su, C.R.; Huang, C.H.; Ke, C.H.; Wu, J.B.; Lin, A.J.; Bastow, K.F.; Lee, K.H.; et al. Cytotoxic and antimalarial beta-carboline alkaloids from the roots of Eurycoma longifolia. J. Nat. Prod. 2003, 66, 1324–1327. [Google Scholar] [CrossRef] [PubMed]
- Franca, T.C. Homology modeling: An important tool for the drug discovery. J. Biomol. Struct. Dyn. 2015, 33, 1780–1793. [Google Scholar] [CrossRef] [PubMed]
- Ltd., V.S.T.P. VLifeMDS: Molecular Design Suite; Ltd., V.S.T.P.: Pune, India, 2010. [Google Scholar]
- Liu, K.; Dong, Y.; Huang, Y.; Rasgon, J.L.; Agre, P. Impact of trehalose transporter knockdown on Anopheles gambiae stress adaptation and susceptibility to Plasmodium falciparum infection. Proc. Natl. Acad. Sci. USA 2013, 110, 17504–17509. [Google Scholar] [CrossRef]
- Adedeji, E.O.; Oduselu, G.O.; Ogunlana, O.O.; Fatumo, S.; Koenig, R.; Adebiyi, E. Anopheles gambiae Trehalase Inhibitors for Malaria Vector Control: A Molecular Docking and Molecular Dynamics Study. Insects 2022, 13, 1070. [Google Scholar] [CrossRef]
- Hardin, C.; Pogorelov, T.V.; Luthey-Schulten, Z. Ab initio protein structure prediction. Curr. Opin. Struct. Biol. 2002, 12, 176–181. [Google Scholar] [CrossRef]
- Barbarin-Bocahu, I.; Graille, M. The X-ray crystallography phase problem solved thanks to AlphaFold and RoseTTAFold models: A case-study report. Acta Crystallogr. D Struct. Biol. 2022, 78, 517–531. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Zheng, W.; Zhang, C.; Li, Y.; Pearce, R.; Bell, E.W.; Zhang, Y. Folding non-homologous proteins by coupling deep-learning contact maps with I-TASSER assembly simulations. Cell. Rep. Methods 2021, 1, 100014. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef]
- Binkowski, T.A.; Naghibzadeh, S.; Liang, J. CASTp: Computed Atlas of Surface Topography of proteins. Nucleic Acids Res. 2003, 31, 3352–3355. [Google Scholar] [CrossRef] [PubMed]
- Dundas, J.; Ouyang, Z.; Tseng, J.; Binkowski, A.; Turpaz, Y.; Liang, J. CASTp: Computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006, 34, W116–W118. [Google Scholar] [CrossRef] [PubMed]
- Jendele, L.; Krivak, R.; Skoda, P.; Novotny, M.; Hoksza, D. PrankWeb: A web server for ligand binding site prediction and visualization. Nucleic Acids Res. 2019, 47, W345–W349. [Google Scholar] [CrossRef] [PubMed]
- Jakubec, D.; Skoda, P.; Krivak, R.; Novotny, M.; Hoksza, D. PrankWeb 3: Accelerated ligand-binding site predictions for experimental and modelled protein structures. Nucleic Acids Res. 2022, 50, W593–W597. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar] [CrossRef]
- Macalino, S.J.; Gosu, V.; Hong, S.; Choi, S. Role of computer-aided drug design in modern drug discovery. Arch. Pharm. Res. 2015, 38, 1686–1701. [Google Scholar] [CrossRef]
- Baell, J.B. Feeling Nature’s PAINS: Natural Products, Natural Product Drugs, and Pan Assay Interference Compounds (PAINS). J. Nat. Prod. 2016, 79, 616–628. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. admetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Sun, L.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. ADMETopt: A Web Server for ADMET Optimization in Drug Design via Scaffold Hopping. J. Chem. Inf. Model. 2018, 58, 2051–2056. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Henin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
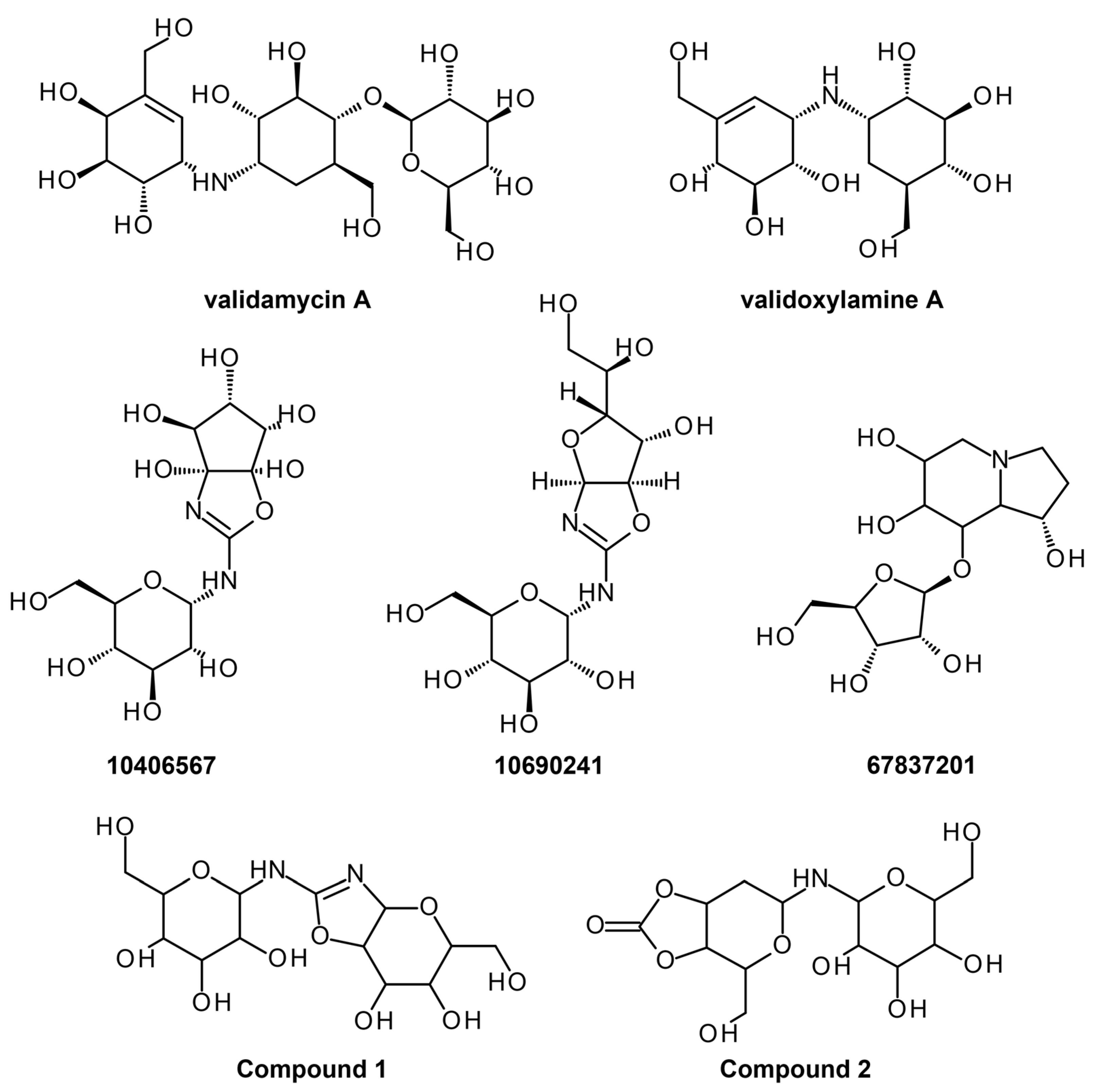
- Asano, N.; Takeuchi, M.; Kameda, Y.; Matsui, K.; Kono, Y. Trehalase inhibitors, validoxylamine A and related compounds as insecticides. J. Antibiot. (Tokyo) 1990, 43, 722–726. [Google Scholar] [CrossRef]
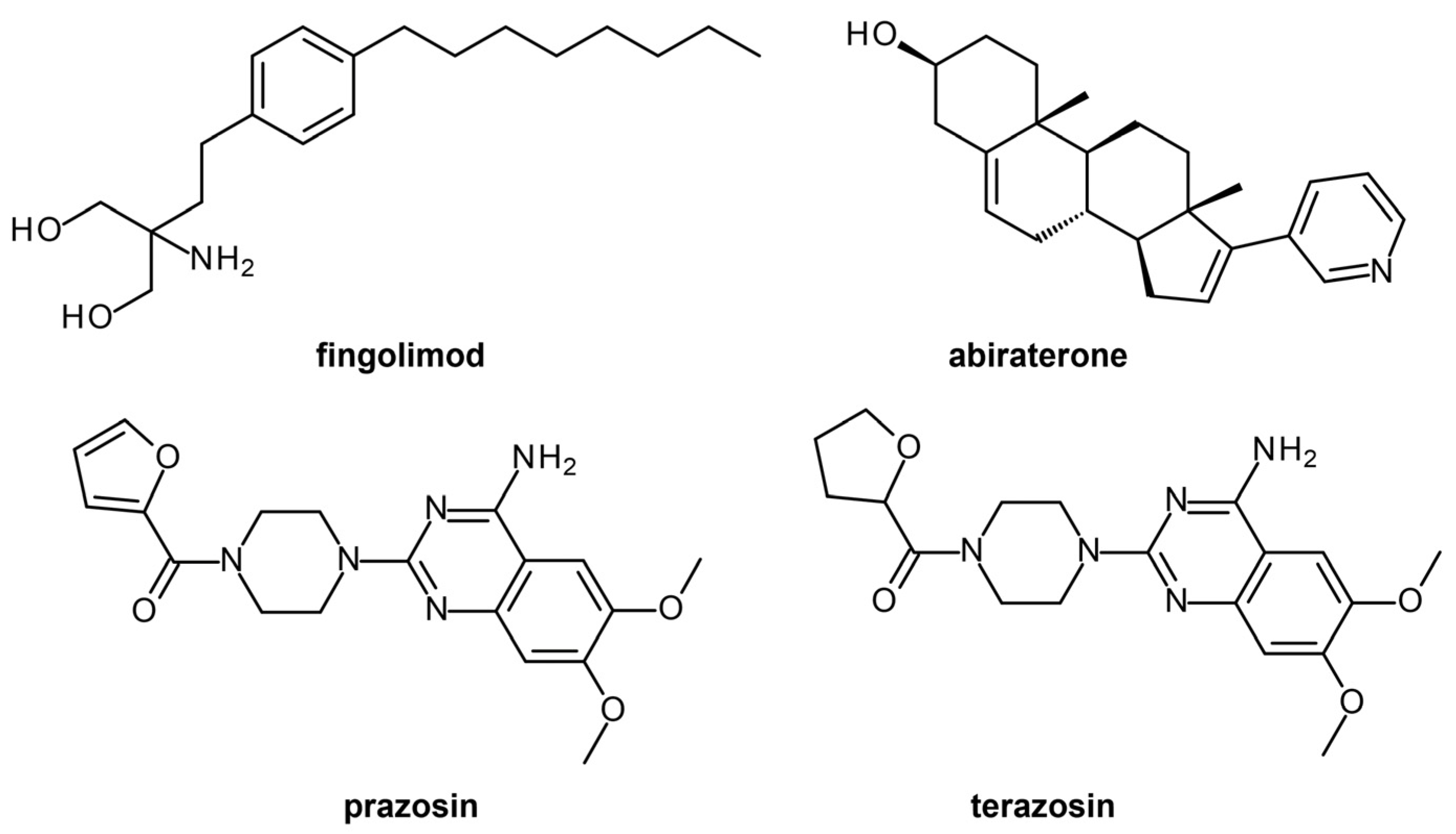
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Park, K. A review of computational drug repurposing. Transl. Clin. Pharmacol. 2019, 27, 59–63. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Cheng, D.; Shrivastava, S.; Tzur, D.; Gautam, B.; Hassanali, M. DrugBank: A knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008, 36, D901–D906. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Hassan, N.M.; Alhossary, A.A.; Mu, Y.; Kwoh, C.K. Protein-Ligand Blind Docking Using QuickVina-W With Inter-Process Spatio-Temporal Integration. Sci. Rep. 2017, 7, 15451. [Google Scholar] [CrossRef]
- Desaphy, J.; Raimbaud, E.; Ducrot, P.; Rognan, D. Encoding protein-ligand interaction patterns in fingerprints and graphs. J. Chem. Inf. Model. 2013, 53, 623–637. [Google Scholar] [CrossRef] [PubMed]
- Abad-Zapatero, C. Ligand efficiency indices for effective drug discovery: A unifying vector formulation. Expert. Opin. Drug Discov. 2021, 16, 763–775. [Google Scholar] [CrossRef] [PubMed]
- Pronk, S.; Pall, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Billker, O.; Dechamps, S.; Tewari, R.; Wenig, G.; Franke-Fayard, B.; Brinkmann, V. Calcium and a calcium-dependent protein kinase regulate gamete formation and mosquito transmission in a malaria parasite. Cell 2004, 117, 503–514. [Google Scholar] [CrossRef]
- Govindasamy, K.; Jebiwott, S.; Jaijyan, D.K.; Davidow, A.; Ojo, K.K.; Van Voorhis, W.C.; Brochet, M.; Billker, O.; Bhanot, P. Invasion of hepatocytes by Plasmodium sporozoites requires cGMP-dependent protein kinase and calcium dependent protein kinase 4. Mol. Microbiol. 2016, 102, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Lima, M.N.N.; Cassiano, G.C.; Tomaz, K.C.P.; Silva, A.C.; Sousa, B.K.P.; Ferreira, L.T.; Tavella, T.A.; Calit, J.; Bargieri, D.Y.; Neves, B.J.; et al. Integrative Multi-Kinase Approach for the Identification of Potent Antiplasmodial Hits. Front. Chem. 2019, 7, 773. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.C.; Rensi, S.E.; Torng, W.; Altman, R.B. Machine learning in chemoinformatics and drug discovery. Drug Discov. Today 2018, 23, 1538–1546. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.C.; Skillman, A.G.; Nicholls, A. Comparison of shape-matching and docking as virtual screening tools. J. Med. Chem. 2007, 50, 74–82. [Google Scholar] [CrossRef]
- Mendez-Lucio, O.; Medina-Franco, J.L. The many roles of molecular complexity in drug discovery. Drug Discov. Today 2017, 22, 120–126. [Google Scholar] [CrossRef]
- Cano, G.; Garcia-Rodriguez, J.; Garcia-Garcia, A.; Perez-Sanchez, H.; Benediktsson, J.A.; Thapa, A.; Barr, A. Automatic selection of molecular descriptors using random forest: Application to drug discovery. Expert. Syst. Appl. 2017, 72, 151–159. [Google Scholar] [CrossRef]
- Long, J.; Ji, W.; Zhang, D.; Zhu, Y.; Bi, Y. Bioactivities and Structure-Activity Relationships of Fusidic Acid Derivatives: A Review. Front. Pharmacol. 2021, 12, 759220. [Google Scholar] [CrossRef] [PubMed]
- Azmi, H.F.; Lhaksmana, K.M.; Kurniawan, I. QSAR Study of Fusidic Acid Derivative as Anti-Malaria Agents by using Artificial Neural Network-Genetic Algorithm. In Proceedings of the 2020 8th International Conference on Information and Communication Technology (ICoICT), Yogyakarta, Indonesia, 24–26 June 2020; pp. 1–4. [Google Scholar]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, H.; Tian, Y.S.; Kawashita, N.; Takagi, T. Mordred: A molecular descriptor calculator. J. Cheminform. 2018, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef] [PubMed]
- Mandlik, V.; Bejugam, P.R.; Singh, S. Chapter 6-Application of Artificial Neural Networks in Modern Drug Discovery. In Artificial Neural Network for Drug Design, Delivery and Disposition; Puri, M., Pathak, Y., Sutariya, V.K., Tipparaju, S., Moreno, W., Eds.; Academic Press: Boston, MA, USA, 2016; pp. 123–139. [Google Scholar]
- Jiménez-Luna, J.; Grisoni, F.; Schneider, G. Drug discovery with explainable artificial intelligence. Nat. Mach. Intell. 2020, 2, 573–584. [Google Scholar] [CrossRef]
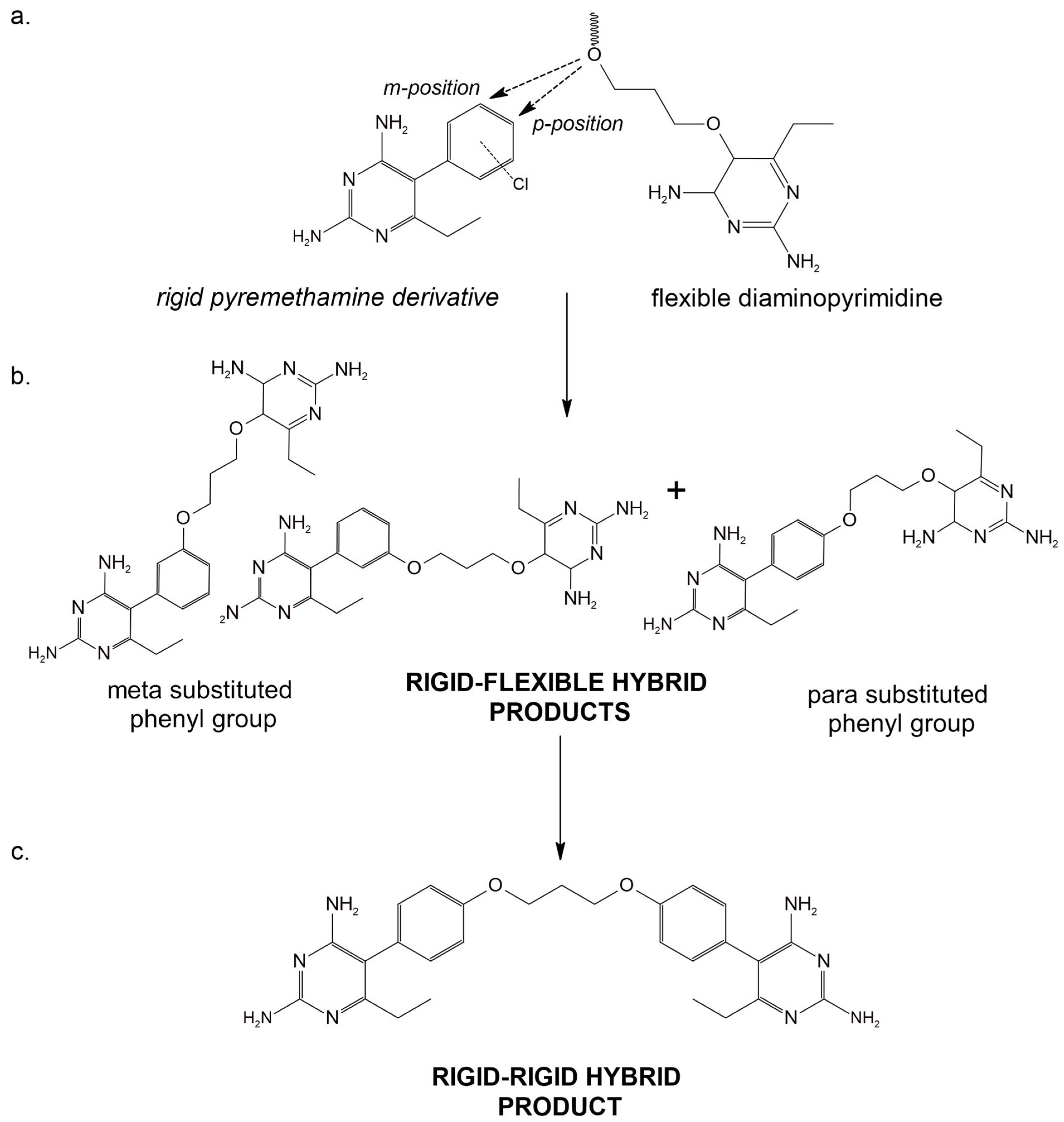
- Hecht, D.; Cheung, M.; Fogel, G.B. QSAR using evolved neural networks for the inhibition of mutant PfDHFR by pyrimethamine derivatives. Biosystems 2008, 92, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Scotti, L.; Ishiki, H.; Mendonca Junior, F.J.; da Silva, M.S.; Scotti, M.T. Artificial Neural Network Methods Applied to Drug Discovery for Neglected Diseases. Comb. Chem. High. Throughput Screen. 2015, 18, 819–829. [Google Scholar] [CrossRef]
- Meshnick, S.R.; Jefford, C.W.; Posner, G.H.; Avery, M.A.; Peters, W. Second-generation antimalarial endoperoxides. Parasitol. Today 1996, 12, 79–82. [Google Scholar] [CrossRef]
- Jefford, C.W.; Favarger, F.; Vicente, M.D.G.H.; Jacquier, Y. The Decomposition of cis-Fused Cyclopenteno-1,2,4-Trioxanes induced by Ferrous Salts and some oxophilic reagents. Helv. Chim. Acta 1995, 78, 452–458. [Google Scholar] [CrossRef]
- Wu, W.-M.; Wu, Y.; Wu, Y.-L.; Yao, Z.-J.; Zhou, C.-M.; Li, Y.; Shan, F. Unified Mechanistic Framework for the Fe(II)-Induced Cleavage of Qinghaosu and Derivatives/Analogues. The First Spin-Trapping Evidence for the Previously Postulated Secondary C-4 Radical. J. Am. Chem. Soc. 1998, 120, 3316–3325. [Google Scholar] [CrossRef]
- Arantes, C.; De Araujo, M.T.; Taranto, A.G.; De Carneiro, J.W.M. Relative stability of radicals derived from artemisinin: A semiempirical and DFT study. Int. J. Quantum Chem. 2005, 103, 749–762. [Google Scholar] [CrossRef]
- De Araujo, M.T.; De Carneiro, J.W.M.; Taranto, A.G. Solvent effects on the relative stability of radicals derived from artemisinin: DFT study using the PCM/COSMO approach. Int. J. Quantum Chem. 2006, 106, 2804–2810. [Google Scholar] [CrossRef]
- Taranto, A.G.; de Mesquita Carneiro, J.W.; de Araujo, M.T. DFT study of the reductive decomposition of artemisinin. Bioorg. Med. Chem. 2006, 14, 1546–1557. [Google Scholar] [CrossRef] [PubMed]
- Tonmunphean, S.; Parasuk, V.; Kokpol, S. Automated calculation of docking of artemisinin to heme. Mol. Model. Annu. 2001, 7, 26–33. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field. V. Extension of MMFF94 using experimental data, additional computational data, and empirical rules. J. Comput. Chem. 1996, 17, 616–641. [Google Scholar] [CrossRef]
- Mahapatra, R.K.; Behera, N.; Naik, P.K. Molecular modeling and evaluation of binding mode and affinity of artemisinin-quinine hybrid and its congeners with Fe-protoporphyrin-IX as a putative receptor. Bioinformation 2012, 8, 369–380. [Google Scholar] [CrossRef]
- Warhurst, D.C.; Craig, J.C.; Adagu, I.S.; Meyer, D.J.; Lee, S.Y. The relationship of physico-chemical properties and structure to the differential antiplasmodial activity of the cinchona alkaloids. Malar. J. 2003, 2, 26. [Google Scholar] [CrossRef]
- O’Brien, R.L.; Olenick, J.G.; Hahn, F.E. Reactions of quinine, chloroquine, and quinacrine with DNA and their effects on the DNA and RNA polymerase reactions. Proc. Natl. Acad. Sci. USA 1966, 55, 1511–1517. [Google Scholar] [CrossRef]
- Sijwali, P.S.; Koo, J.; Singh, N.; Rosenthal, P.J. Gene disruptions demonstrate independent roles for the four falcipain cysteine proteases of Plasmodium falciparum. Mol. Biochem. Parasitol. 2006, 150, 96–106. [Google Scholar] [CrossRef]
- Hanspal, M.; Dua, M.; Takakuwa, Y.; Chishti, A.H.; Mizuno, A. Plasmodium falciparum cysteine protease falcipain-2 cleaves erythrocyte membrane skeletal proteins at late stages of parasite development. Blood 2002, 100, 1048–1054. [Google Scholar] [CrossRef]
- Ibrahim, N.; Ibrahim, H.; Kim, S.; Nallet, J.P.; Nepveu, F. Interactions between antimalarial indolone-N-oxide derivatives and human serum albumin. Biomacromolecules 2010, 11, 3341–3351. [Google Scholar] [CrossRef] [PubMed]
- Bi, S.; Yan, L.; Sun, Y.; Zhang, H. Investigation of ketoprofen binding to human serum albumin by spectral methods. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2011, 78, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Bujacz, A.; Talaj, J.A.; Zielinski, K.; Pietrzyk-Brzezinska, A.J.; Neumann, P. Crystal structures of serum albumins from domesticated ruminants and their complexes with 3,5-diiodosalicylic acid. Acta Crystallogr. D Struct. Biol. 2017, 73, 896–909. [Google Scholar] [CrossRef] [PubMed]
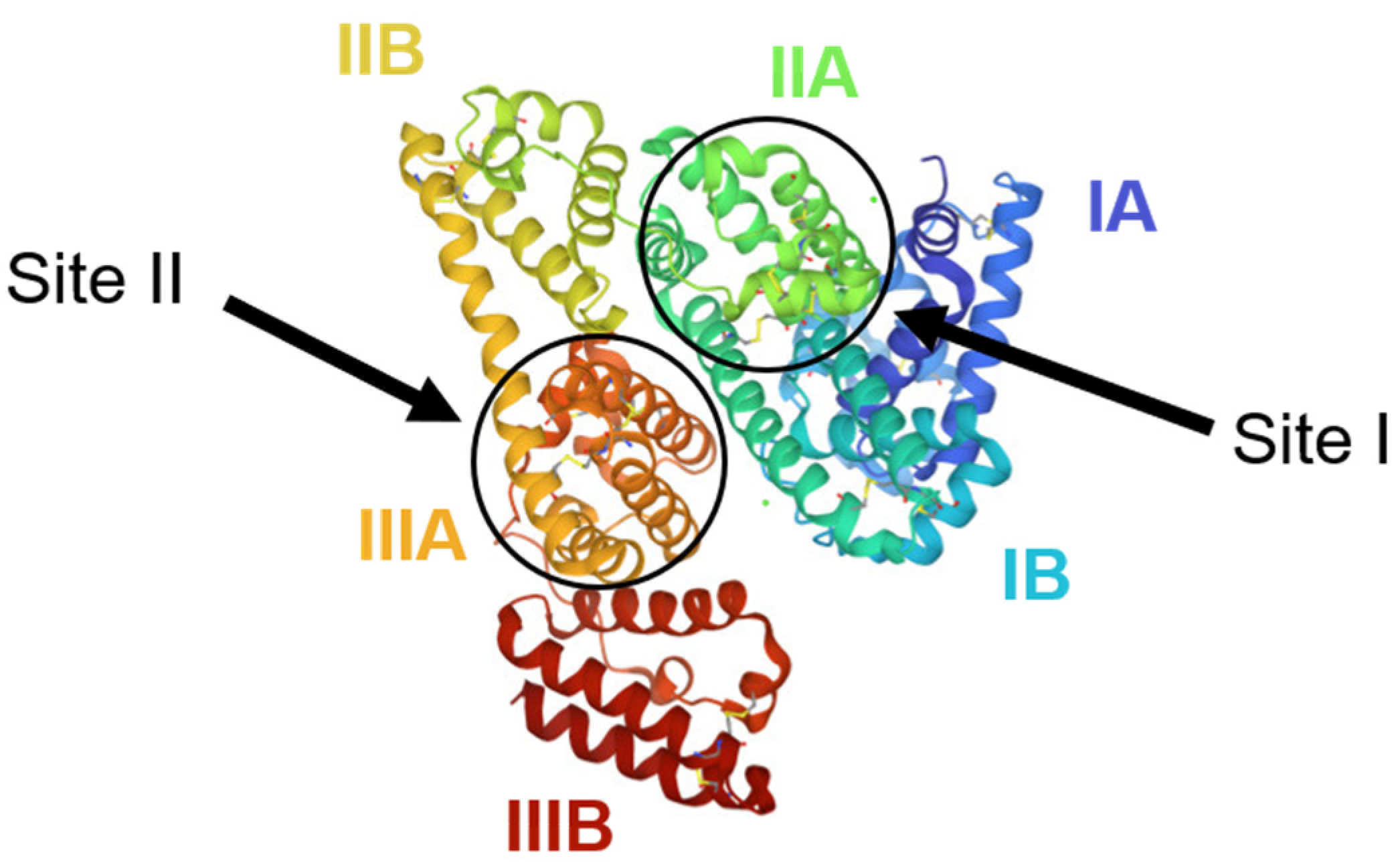
- Curry, S.; Mandelkow, H.; Brick, P.; Franks, N. Crystal structure of human serum albumin complexed with fatty acid reveals an asymmetric distribution of binding sites. Nat. Struct. Biol. 1998, 5, 827–835. [Google Scholar] [CrossRef]
- Sudlow, G.; Birkett, D.J.; Wade, D.N. The characterization of two specific drug binding sites on human serum albumin. Mol. Pharmacol. 1975, 11, 824–832. [Google Scholar]
- Mondal, M.; Krishna, R.; Sakthivel, N. Molecular interaction between human serum albumin (HSA) and phloroglucinol derivative that shows selective anti-proliferative potential. J. Lumin. 2017, 192, 990–998. [Google Scholar] [CrossRef]
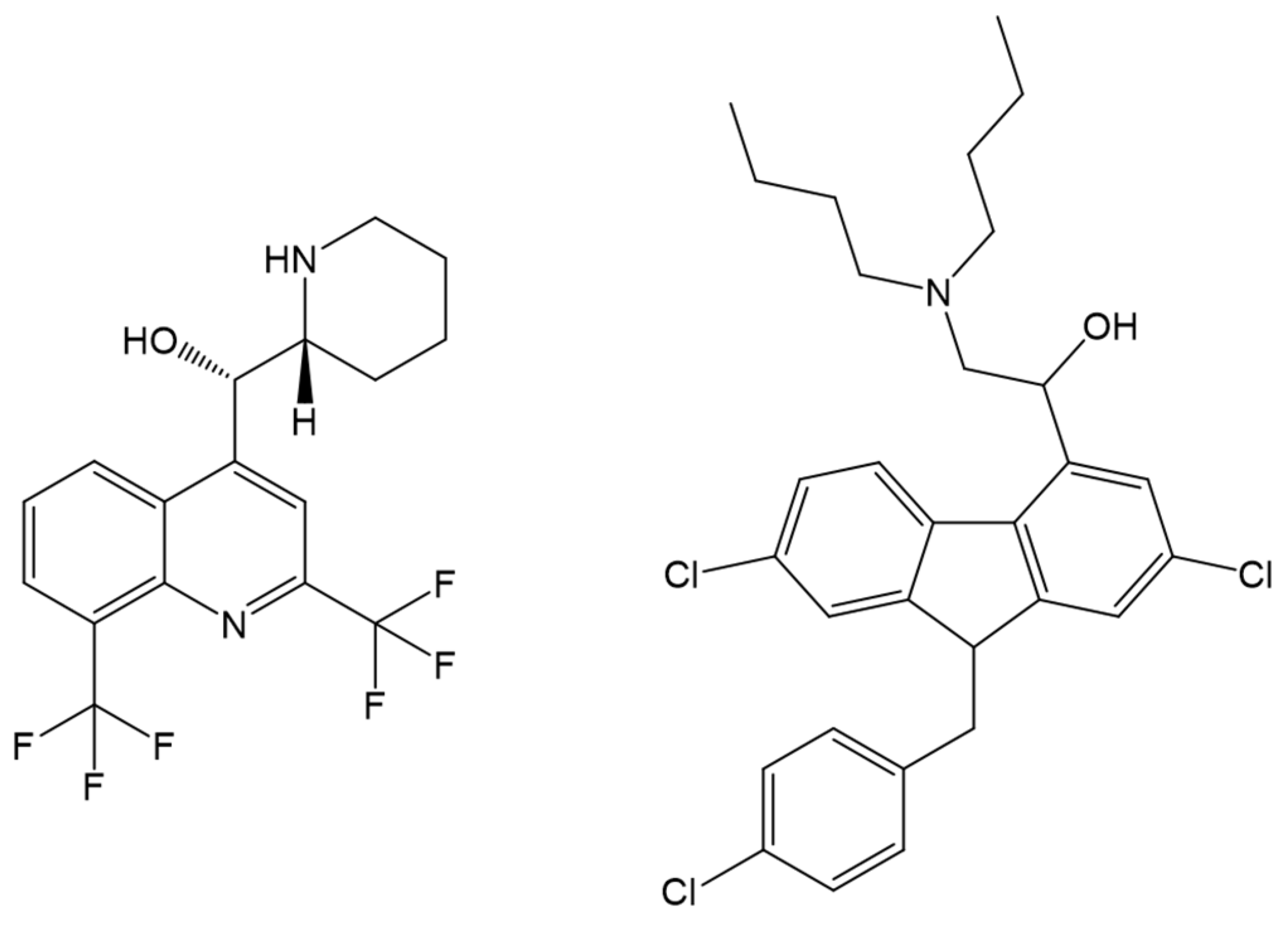
- Musa, K.A.; Ridzwan, N.F.W.; Mohamad, S.B.; Tayyab, S. Combination mode of antimalarial drug mefloquine and human serum albumin: Insights from spectroscopic and docking approaches. Biopolymers 2020, 111, e23337. [Google Scholar] [CrossRef]
- Musa, K.A.; Ridzwan, N.F.W.; Mohamad, S.B.; Tayyab, S. Exploring the combination characteristics of lumefantrine, an antimalarial drug and human serum albumin through spectroscopic and molecular docking studies. J. Biomol. Struct. Dyn. 2021, 39, 691–702. [Google Scholar] [CrossRef]
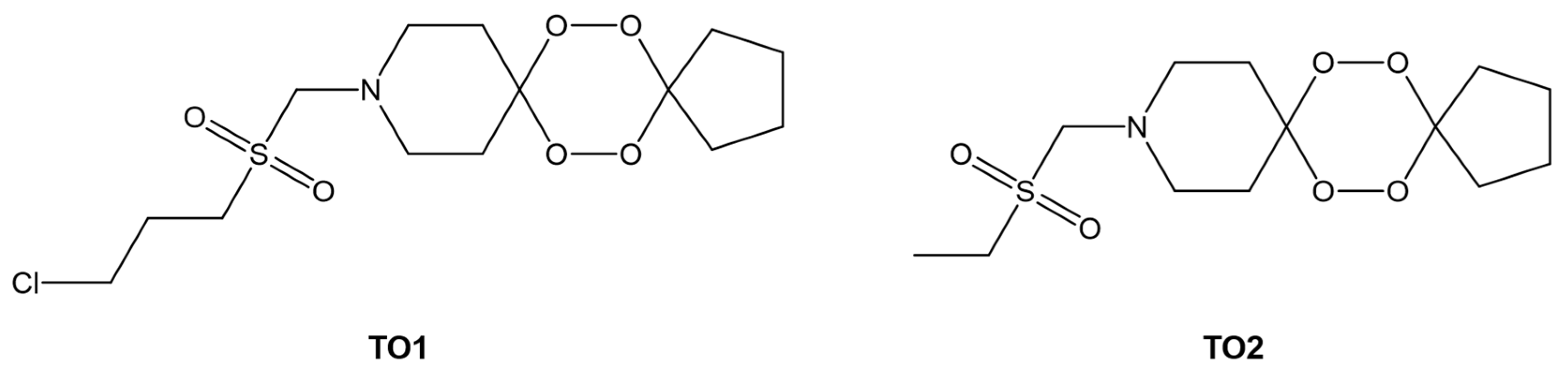
- Yadav, P.; Sharma, B.; Sharma, C.; Singh, P.; Awasthi, S.K. Interaction between the Antimalarial Drug Dispiro-Tetraoxanes and Human Serum Albumin: A Combined Study with Spectroscopic Methods and Computational Studies. ACS Omega 2020, 5, 6472–6480. [Google Scholar] [CrossRef]
- Ali, A.; Asif, M.; Alam, P.; Jane Alam, M.; Asif Sherwani, M.; Hasan Khan, R.; Ahmad, S.; Shamsuzzaman. DFT/B3LYP calculations, in vitro cytotoxicity and antioxidant activities of steroidal pyrimidines and their interaction with HSA using molecular docking and multispectroscopic techniques. Bioorg. Chem. 2017, 73, 83–99. [Google Scholar] [CrossRef]
- Singh, P.; Sharma, C.; Sharma, B.; Mishra, A.; Agarwal, D.; Kannan, D.; Held, J.; Singh, S.; Awasthi, S.K. N-sulfonylpiperidinedispiro-1,2,4,5-tetraoxanes exhibit potent in vitro antiplasmodial activity and in vivo efficacy in mice infected with P. berghei ANKA. Eur. J. Med. Chem. 2022, 244, 114774. [Google Scholar] [CrossRef] [PubMed]
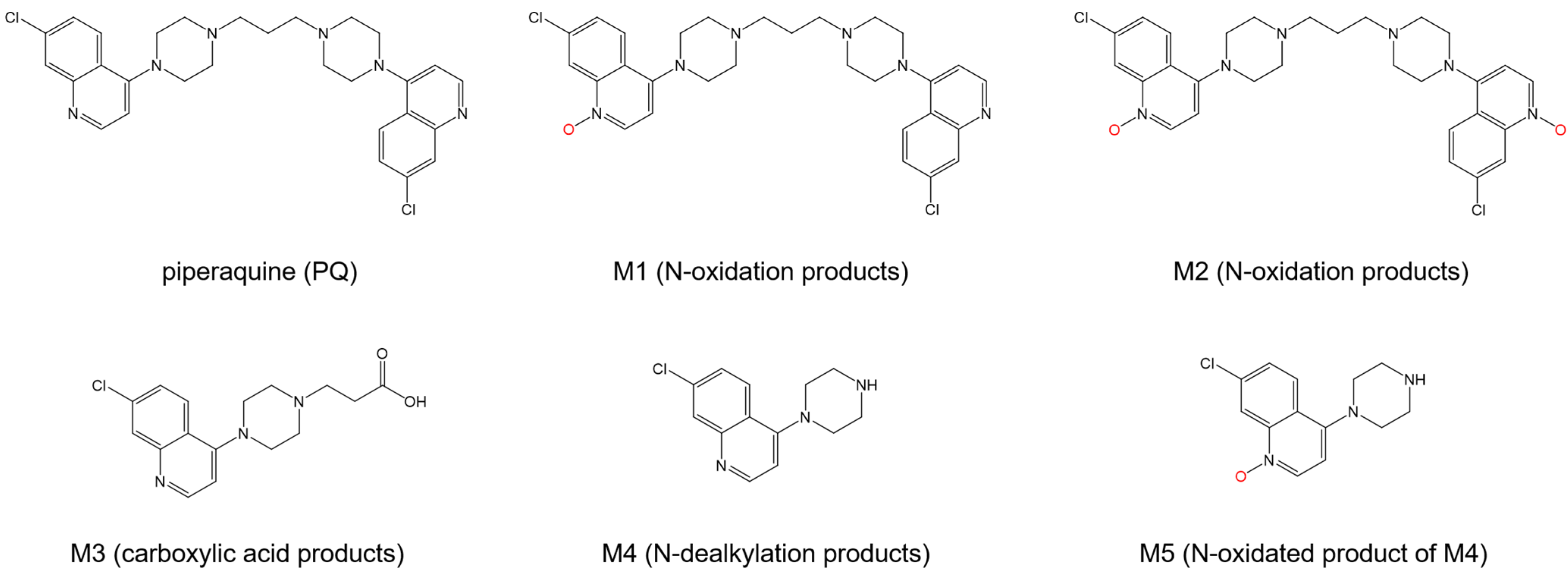
- Nguyen, D.V.; Nguyen, Q.P.; Nguyen, N.D.; Le, T.T.; Nguyen, T.D.; Dinh, D.N.; Nguyen, T.X.; Bui, D.; Chavchich, M.; Edstein, M.D. Pharmacokinetics and ex vivo pharmacodynamic antimalarial activity of dihydroartemisinin-piperaquine in patients with uncomplicated falciparum malaria in Vietnam. Antimicrob. Agents Chemother. 2009, 53, 3534–3537. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.H.; Huang, X.T.; Zhang, R.; Yang, L.; Huang, T.L.; Wang, N.S.; Mi, S.Q. Determination of CQP propionic acid in rat plasma and study of pharmacokinetics of CQP propionic acid in rats by liquid chromatography. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2008, 862, 189–195. [Google Scholar] [CrossRef]
- Ma, R.; Guo, D.X.; Li, H.F.; Liu, H.X.; Zhang, Y.R.; Ji, J.B.; Xing, J.; Wang, S.Q. Spectroscopic methodologies and molecular docking studies on the interaction of antimalarial drug piperaquine and its metabolites with human serum albumin. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2019, 222, 117158. [Google Scholar] [CrossRef] [PubMed]
- Majorek, K.A.; Porebski, P.J.; Dayal, A.; Zimmerman, M.D.; Jablonska, K.; Stewart, A.J.; Chruszcz, M.; Minor, W. Structural and immunologic characterization of bovine, horse, and rabbit serum albumins. Mol. Immunol. 2012, 52, 174–182. [Google Scholar] [CrossRef] [PubMed]
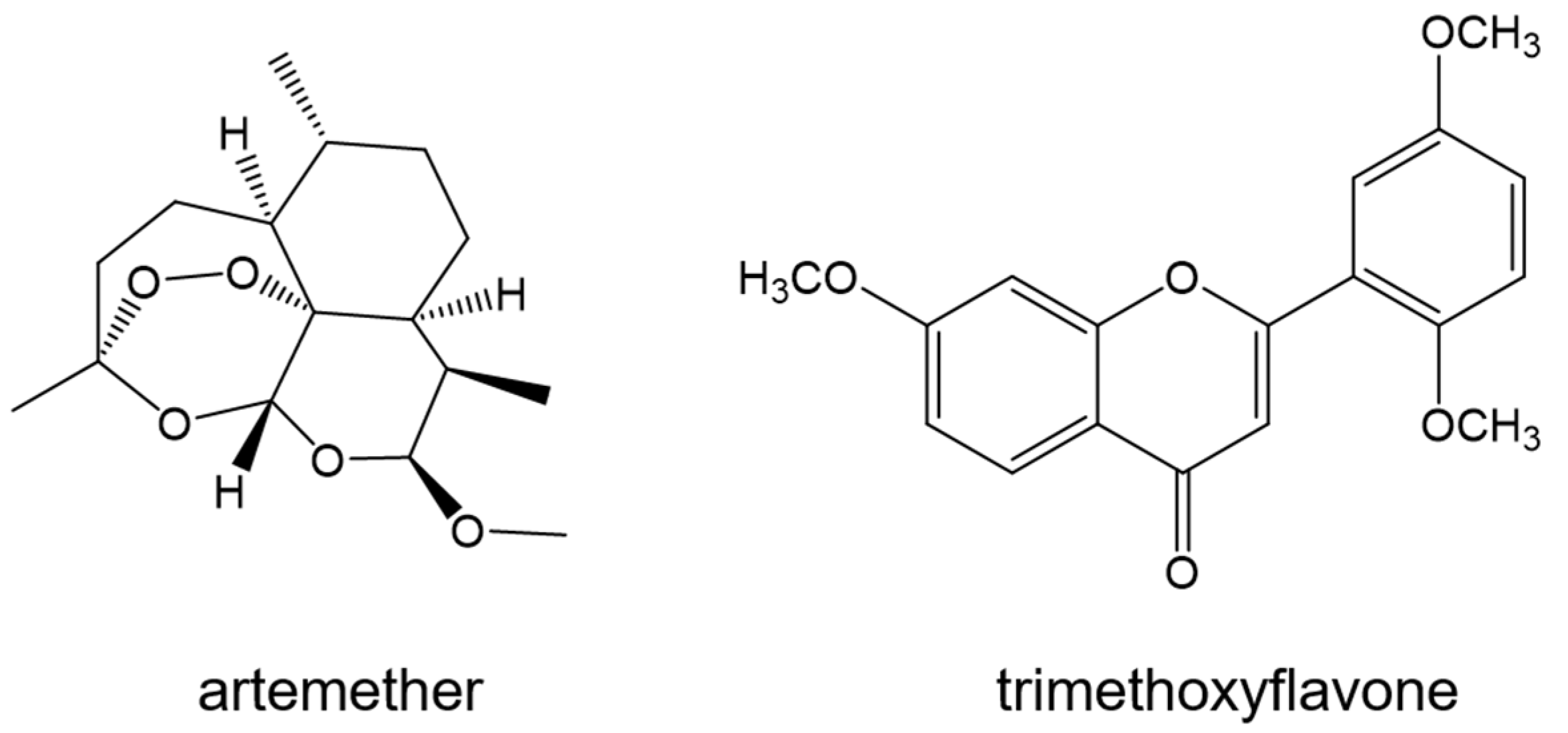
- Shi, J.H.; Pan, D.Q.; Wang, X.X.; Liu, T.T.; Jiang, M.; Wang, Q. Characterizing the binding interaction between antimalarial artemether (AMT) and bovine serum albumin (BSA): Spectroscopic and molecular docking methods. J. Photochem. Photobiol. B 2016, 162, 14–23. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C. Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J. Mol. Biol. 1995, 245, 43–53. [Google Scholar] [CrossRef]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins 2003, 52, 609–623. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Gokara, M.; Sudhamalla, B.; Amooru, D.G.; Subramanyam, R. Molecular interaction studies of trimethoxy flavone with human serum albumin. PLoS ONE 2010, 5, e8834. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Bharti, P.K.; Bansal, D.; Ali, N.A.; Raman, R.K.; Mohapatra, P.K.; Sehgal, R.; Mahanta, J.; Sultan, A.A.; Singh, N. Prevalence of mutations linked to antimalarial resistance in Plasmodium falciparum from Chhattisgarh, Central India: A malaria elimination point of view. Sci. Rep. 2017, 7, 16690. [Google Scholar] [CrossRef] [PubMed]
- Menard, D.; Dondorp, A. Antimalarial Drug Resistance: A Threat to Malaria Elimination. Cold Spring Harb. Perspect. Med. 2017, 7, a025619. [Google Scholar] [CrossRef] [PubMed]
- Patel, C.; Roy, D. A Computational Study of Molecular Mechanism of Chloroquine Resistance by Chloroquine Resistance Transporter Protein of Plasmodium falciparum via Molecular Modeling and Molecular Simulations. Physchem 2021, 1, 232–242. [Google Scholar] [CrossRef]
- Sullivan, D.J., Jr.; Gluzman, I.Y.; Russell, D.G.; Goldberg, D.E. On the molecular mechanism of chloroquine’s antimalarial action. Proc. Natl. Acad. Sci. USA 1996, 93, 11865–11870. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Singh, A.; Singh, M.; Sood, D.; Rathi, B.; Tomar, V.; Chandra, R. Modern Advancement in the Area of Antimalarial Drug Development. Indian J. Heterocycl. Chem. 2018, 28, 185. [Google Scholar]
- Yuthavong, Y.; Tarnchompoo, B.; Vilaivan, T.; Chitnumsub, P.; Kamchonwongpaisan, S.; Charman, S.A.; McLennan, D.N.; White, K.L.; Vivas, L.; Bongard, E.; et al. Malarial dihydrofolate reductase as a paradigm for drug development against a resistance-compromised target. Proc. Natl. Acad. Sci. USA 2012, 109, 16823–16828. [Google Scholar] [CrossRef]
- Amusengeri, A.; Tata, R.B.; Tastan Bishop, O. Understanding the Pyrimethamine Drug Resistance Mechanism via Combined Molecular Dynamics and Dynamic Residue Network Analysis. Molecules 2020, 25, 904. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Amusengeri, A.; Tastan Bishop, O. Discorhabdin N, a South African Natural Compound, for Hsp72 and Hsc70 Allosteric Modulation: Combined Study of Molecular Modeling and Dynamic Residue Network Analysis. Molecules 2019, 24, 188. [Google Scholar] [CrossRef]
- Doshi, U.; Holliday, M.J.; Eisenmesser, E.Z.; Hamelberg, D. Dynamical network of residue-residue contacts reveals coupled allosteric effects in recognition, catalysis, and mutation. Proc. Natl. Acad. Sci. USA 2016, 113, 4735–4740. [Google Scholar] [CrossRef] [PubMed]
- Yuvaniyama, J.; Chitnumsub, P.; Kamchonwongpaisan, S.; Vanichtanankul, J.; Sirawaraporn, W.; Taylor, P.; Walkinshaw, M.D.; Yuthavong, Y. Insights into antifolate resistance from malarial DHFR-TS structures. Nat. Struct. Biol. 2003, 10, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Abbat, S.; Jain, V.; Bharatam, P.V. Origins of the specificity of inhibitor P218 toward wild-type and mutant PfDHFR: A molecular dynamics analysis. J. Biomol. Struct. Dyn. 2015, 33, 1913–1928. [Google Scholar] [CrossRef] [PubMed]
- Tarnchompoo, B.; Chitnumsub, P.; Jaruwat, A.; Shaw, P.J.; Vanichtanankul, J.; Poen, S.; Rattanajak, R.; Wongsombat, C.; Tonsomboon, A.; Decharuangsilp, S.; et al. Hybrid Inhibitors of Malarial Dihydrofolate Reductase with Dual Binding Modes That Can Forestall Resistance. ACS Med. Chem. Lett. 2018, 9, 1235–1240. [Google Scholar] [CrossRef]
- Pastrana-Mena, R.; Dinglasan, R.R.; Franke-Fayard, B.; Vega-Rodriguez, J.; Fuentes-Caraballo, M.; Baerga-Ortiz, A.; Coppens, I.; Jacobs-Lorena, M.; Janse, C.J.; Serrano, A.E. Glutathione reductase-null malaria parasites have normal blood stage growth but arrest during development in the mosquito. J. Biol. Chem. 2010, 285, 27045–27056. [Google Scholar] [CrossRef]
- Kamaria, P.; Kawathekar, N. Ligand-based 3D-QSAR analysis and virtual screening in exploration of new scaffolds as Plasmodium falciparum glutathione reductase inhibitors. Med. Chem. Res. 2014, 23, 25–33. [Google Scholar] [CrossRef]
- Malonis, R.J.; Lai, J.R.; Vergnolle, O. Peptide-Based Vaccines: Current Progress and Future Challenges. Chem. Rev. 2020, 120, 3210–3229. [Google Scholar] [CrossRef]
- Caoili, S.E.C. Comprehending B-Cell Epitope Prediction to Develop Vaccines and Immunodiagnostics. Front. Immunol. 2022, 13, 908459. [Google Scholar] [CrossRef]
- Potocnakova, L.; Bhide, M.; Pulzova, L.B. An Introduction to B-Cell Epitope Mapping and In Silico Epitope Prediction. J. Immunol. Res. 2016, 2016, 6760830. [Google Scholar] [CrossRef]
- Julien, J.P.; Wardemann, H. Antibodies against Plasmodium falciparum malaria at the molecular level. Nat. Rev. Immunol. 2019, 19, 761–775. [Google Scholar] [CrossRef]
- Oyen, D.; Torres, J.L.; Aoto, P.C.; Flores-Garcia, Y.; Binter, S.; Pholcharee, T.; Carroll, S.; Reponen, S.; Wash, R.; Liang, Q.; et al. Structure and mechanism of monoclonal antibody binding to the junctional epitope of Plasmodium falciparum circumsporozoite protein. PLoS Pathog. 2020, 16, e1008373. [Google Scholar] [CrossRef] [PubMed]
- Perlmann, H.K.; Berzins, K.; Wahlin, B.; Udomsangpetch, R.; Ruangjirachuporn, W.; Wahlgren, M.; Perlmann, P.H. Absence of antigenic diversity in Pf155, a major parasite antigen in membranes of erythrocytes infected with Plasmodium falciparum. J. Clin. Microbiol. 1987, 25, 2347–2354. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, M.; Torii, M.; Sjölander, A.; Berzins, K.; Perlmann, P.; Miller, L.H. Pf155/RESA antigen is localized in dense granules of Plasmodium falciparum merozoites. Exp. Parasitol. 1990, 71, 326–329. [Google Scholar] [CrossRef] [PubMed]
- Rug, M.; Wickham, M.E.; Foley, M.; Cowman, A.F.; Tilley, L. Correct promoter control is needed for trafficking of the ring-infected erythrocyte surface antigen to the host cytosol in transfected malaria parasites. Infect. Immun. 2004, 72, 6095–6105. [Google Scholar] [CrossRef]
- Azimi-Resketi, M.; Heydaryan, S.; Kumar, N.; Takalou, A.; Dizaji, R.E.; Gorgani, B.N.; Shams, M. Computational Clues of Immunogenic Hotspots in Plasmodium falciparum Erythrocytic Stage Vaccine Candidate Antigens: In Silico Approach. Biomed. Res. Int. 2022, 2022, 5886687. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v.2--a server for in silico prediction of allergens. J. Mol. Model. 2014, 20, 2278. [Google Scholar] [CrossRef]
- Hebditch, M.; Carballo-Amador, M.A.; Charonis, S.; Curtis, R.; Warwicker, J. Protein-Sol: A web tool for predicting protein solubility from sequence. Bioinformatics 2017, 33, 3098–3100. [Google Scholar] [CrossRef]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Target | Computational Method | Hit or Lead Compound | Experimentally Validated | Drug Discovery Stage | Ref. |
|---|---|---|---|---|---|
| Essential kinase PfPK6 | Pharmacophore screening | Luceome | Yes | Lead identification | [19] |
| Trehalase | Homology modeling, Virtual screening, and absorption, distribution, metabolism, excretion, and toxicity (ADMET) screening | (4-(hydroxymethyl)-6-{[3,4,5-trihydroxy-6- (hydroxymethyl)oxan-2-yl]amino}-hexahydro-[1,3]dioxolo [4,5-c]pyran-2-one) Uniflorine | Yes | Lead optimization | [20] |
| Dihydroorotate dehydrogenase (PfDHODH), plasma membrane P-type cation translocating ATPase (PfATP4) | Structure-based pharmacophore, Density Functional Theory (DFT) study | Luteolin | Yes | Lead optimization | [21] |
| P. falciparum 3D7 and W2 strains | Deep learning, QSAR | 2-(4,6-diphenyl-1,2-dihydro-1,3,5-triazin-2-yl)phenol 4-{N-[3-(morpholin-4-yl)-1,4-dioxo-1,4-dihydronaphthalen-2-yl]acetamido}benzoic acid N2-(3-fluorophenyl)-N4-[(oxolan-2-yl)methyl]quinazoline-2,4-diamine | Yes | Lead identification | [22] |
| P. falciparum 3D7 and RKL-9 strains, P. berghei-infected erythrocytes | Homology modeling, Reverse docking, Drug likeness screening | (1R,3S)-methyl 1-(benzo[d][1,3]dioxol-5-yl)-2,3,4,9-tetrahydro-1H-pyrido [3,4-b]indole-3-carboxylate | Yes | Lead optimization | [23] |
| P. falciparum apicoplast-targeted proteins | Homology modeling, molecular docking | Rifampicin | No | Drug repurposing, Lead identification | [24] |
| 36 P. falciparum drug targets | Molecular docking, MD simulations | DrugBank Library | Yes | Drug repurposing, Lead identification | [25] |
| P. falciparum, strain Dd2, Enoyl-ACP (acyl carrier protein)-reductase (FabI) | QSAR | 2’-substituted triclosan derivatives | No | Lead identification | [26] |
| p53 | ADMET screening, Molecular docking | 1-(1-benzyl-5-phenyl-1H-1,2,3-triazol-4-yl)-1-(4-bromophenyl)-2-((3,4-dimethylphenyl)amino)ethanol | No | Lead identification | [27] |
| P. falciparum 1-deoxy-d-xylulose-5-phosphate reductoisomerase (PfDXR) | Pharmacophore modeling, Virtual screening, Molecular docking, MD simulations | Fosmidomycin Myricetin 3-rhamnoside, 7-O-Galloyltricetiflavan (25S)-5-beta-spirostan-3-beta-ol 3-O-beta-d-glucopyranosyl-(1->2)-beta-d-glucopyranoside Oleanolic acid 28-O-beta-d-glucopyranoside | No | Lead identification | [28] |
| P. falciparum glutathione reductase | Molecular docking, MD simulations | 1,2,3-triazole-linked dihydropyrimidinone quinoline hybrids | Yes | Optimization of drug structure | [29] |
| Falcipains | Quantum mechanics/molecular mechanics, MD simulations | N-[N-(1-hydroxycarboxyethylcarbonyl)leucylaminobutyl]guanidine (E64) | Yes | Lead optimization | [30] |
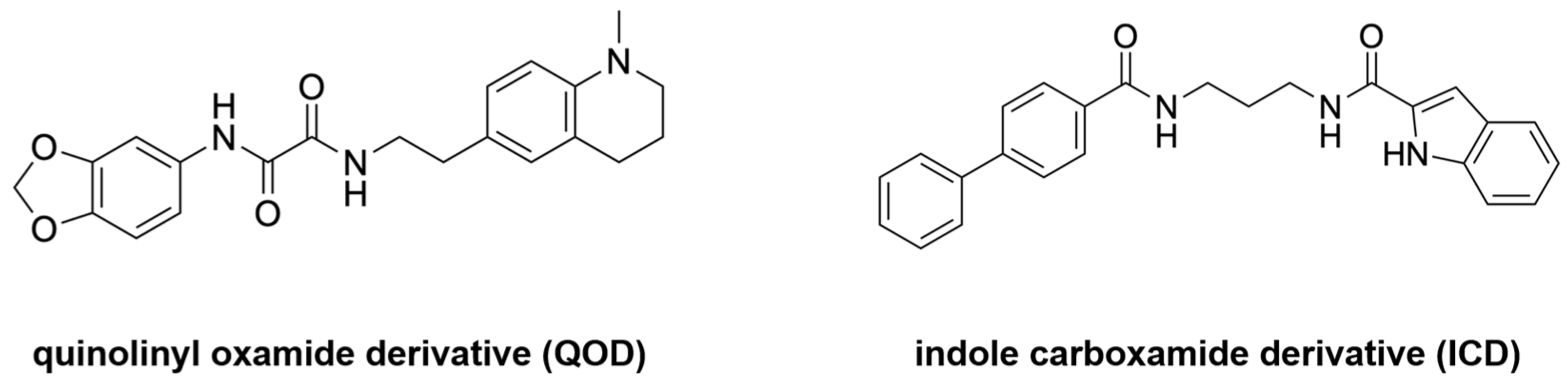
| MD simulations | N-(2H-1,3-benzodioxol-5-yl)-N’-[2(1-methyl-1,2,3,4-tetrahydroquinolin-6-yl) ethyl]ethanediamide N-{3-[(biphenyl-4yl carbonyl) amino]propyl}-1H-indole-2-carboxamide | Yes | Lead optimization | [31] |
| Drug Name | Binding Free Energy (kJ mol−1) a | Ref. | |
|---|---|---|---|
| Site I | Site II | ||
| Lumefantrine | −29.83 | −26.95 | [102] |
| TO1 b | −28.6 | - | [103] |
| TO2 b | −26.1 | - | |
| Piperaquine | −40 | - | [108] |
| M1 c | −28 | - | |
| M2 c | −29 | - | |
| M3 c | −32 | −33 | |
| M4 c | −41 | - | |
| M5 c | −42 | −36 | |
| Artemether | - | −26 | [110] |
| Tri-methoxy flavone | −28.01 | −32.40 | [115] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duay, S.S.; Yap, R.C.Y.; Gaitano, A.L., III; Santos, J.A.A.; Macalino, S.J.Y. Roles of Virtual Screening and Molecular Dynamics Simulations in Discovering and Understanding Antimalarial Drugs. Int. J. Mol. Sci. 2023, 24, 9289. https://doi.org/10.3390/ijms24119289
Duay SS, Yap RCY, Gaitano AL III, Santos JAA, Macalino SJY. Roles of Virtual Screening and Molecular Dynamics Simulations in Discovering and Understanding Antimalarial Drugs. International Journal of Molecular Sciences. 2023; 24(11):9289. https://doi.org/10.3390/ijms24119289
Chicago/Turabian StyleDuay, Searle S., Rianne Casey Y. Yap, Arturo L. Gaitano, III, June Alexis A. Santos, and Stephani Joy Y. Macalino. 2023. "Roles of Virtual Screening and Molecular Dynamics Simulations in Discovering and Understanding Antimalarial Drugs" International Journal of Molecular Sciences 24, no. 11: 9289. https://doi.org/10.3390/ijms24119289
APA StyleDuay, S. S., Yap, R. C. Y., Gaitano, A. L., III, Santos, J. A. A., & Macalino, S. J. Y. (2023). Roles of Virtual Screening and Molecular Dynamics Simulations in Discovering and Understanding Antimalarial Drugs. International Journal of Molecular Sciences, 24(11), 9289. https://doi.org/10.3390/ijms24119289