
Effects of Phosphorylation on the Activity, Inhibition and Stability of Carbonic Anhydrases

 and
and
Abstract
1. Introduction
2. Results and Discussion
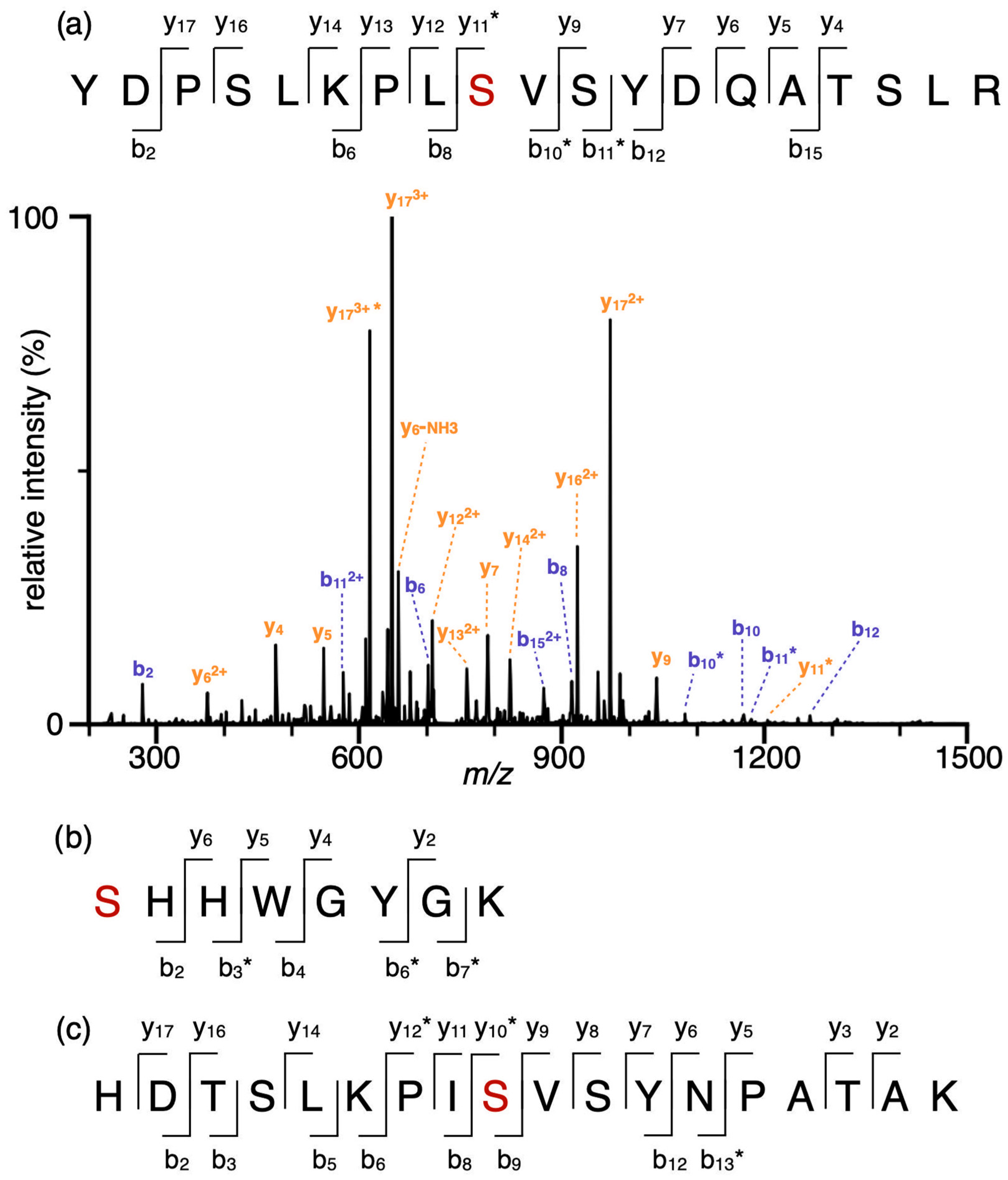
2.1. Phosphorylation Sites of Interest in hCAI and hCAII

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Entries for Phosphorylation (PhosphoSitePlus) [12] | Predicted Score a for Phosphorylation (PhosNet) [41] | Other Factors | ||
|---|---|---|---|---|
| hCAII | Ser2 | 2 | n/a | in vitro phosphorylation |
| Ser29 | 3 | Unsp. b (0.998) PKA (0.519) | only serine within 10 Å of the active site | |
| Ser48 | 1 | Unsp. b (0.728) | in vitro phosphorylation | |
| Ser50 | 2 | Unsp. b (0.565) PKC (0.515) | ||
| Ser99 | 3 | DNAPK (0.616) PKA (0.522) | ||
| Ser151 | 3 | Unsp. b (0.838) | ||
| Ser172 | 3 | Unsp. b (0.674) | ~12 Å to His64 (relevant to rate-limiting proton shuttle) | |
| hCAI | Ser30 | 1 | Unsp. b (0.918) | only serine within 10 Å of the active site |
| Ser49 | 1 | Unsp. b (0.887) | in vitro phosphorylation | |
| Ser51 | 1 | PKC (0.577) | conserved in hCAII | |
| Ser131 | 3 | Unsp. b (0.942) | ||
| Ser137 | 9 | Unsp. b (0.935) |
2.2. Screening Esterase Activity of CAs in Whole-Cell Lysates
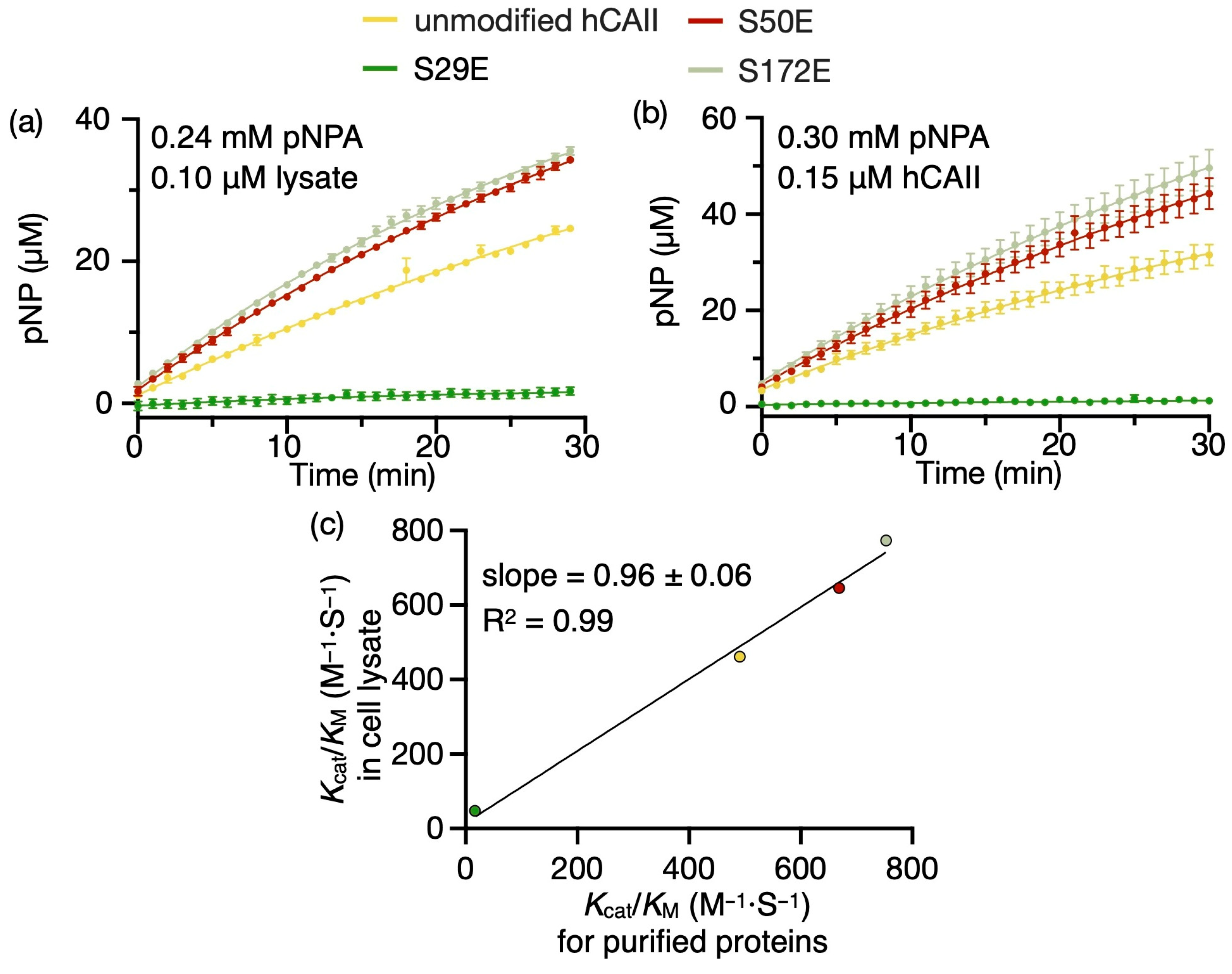
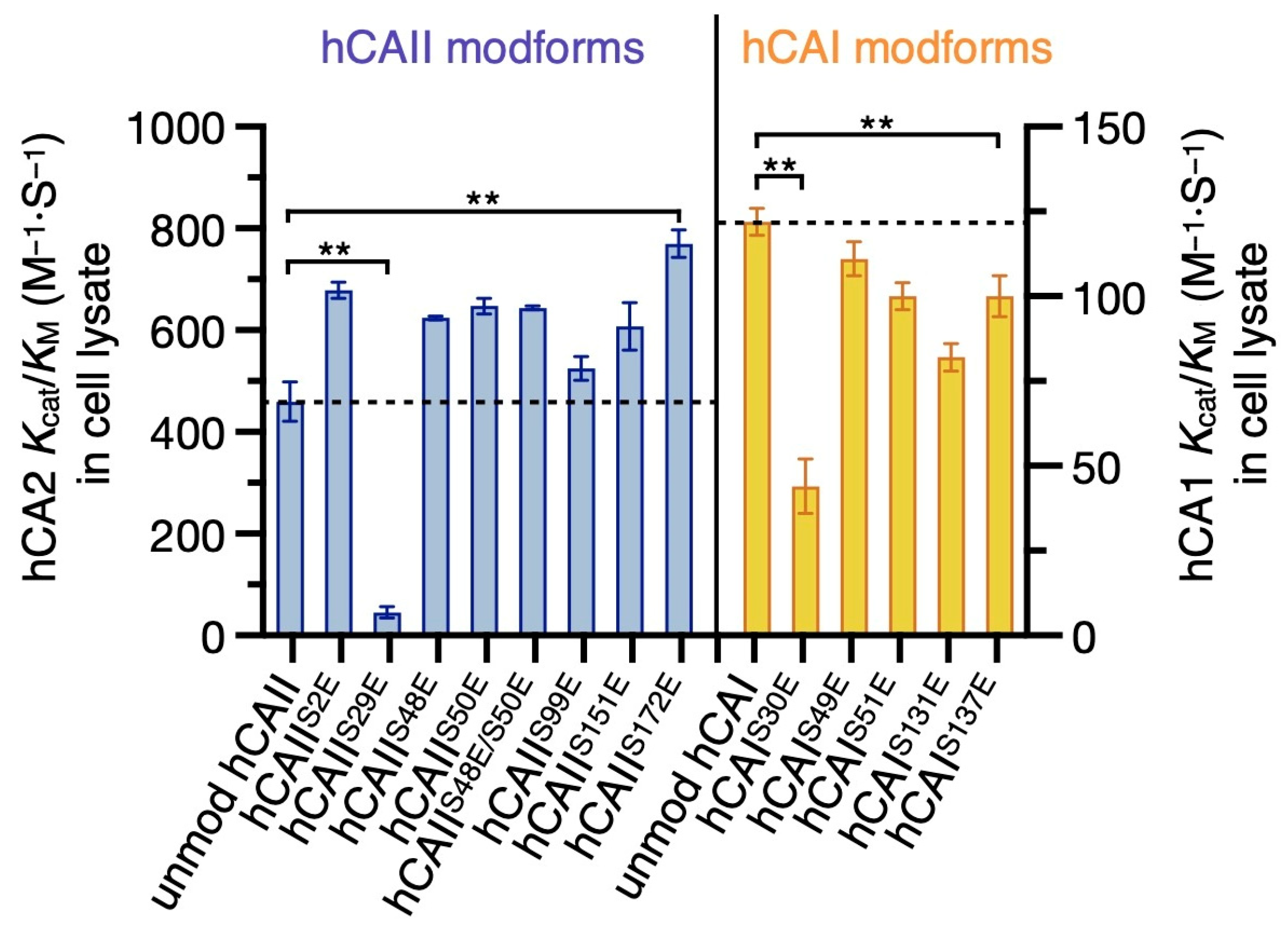
2.3. Phosphomimic Sites Can Strongly Affect the Esterase Activity of hCAs
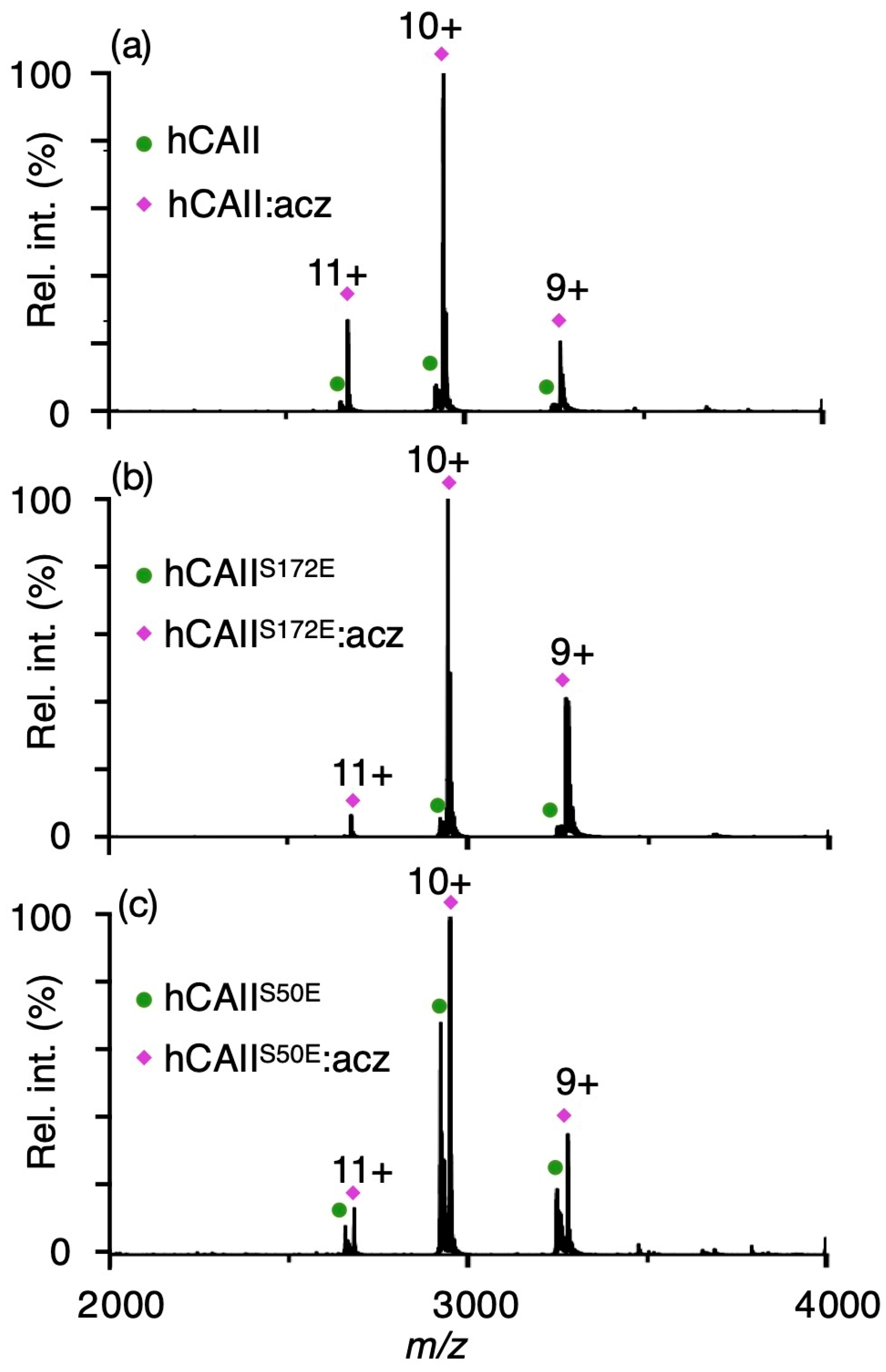
2.4. Effects of Phosphomimetic Sites on hCAII Interactions with Sulphonamide Inhibitors
3. Materials and Methods
3.1. Carbonic Anhydrase Recombinant Expression and Purification
3.2. In Vitro Phosphorylation
3.3. Esterase Activity Assay
3.4. Obtaining Catalytic Efficiencies from Enzyme Kinetics Measurements
3.5. Native Mass Spectrometry and Binding Affinity Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mboge, M.Y.; Mahon, B.P.; McKenna, R.; Frost, S.C. Carbonic Anhydrases: Role in pH Control and Cancer. Metabolites 2018, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.I.; Shajee, B.; Waheed, A.; Ahmad, F.; Sly, W.S. Structure, function and applications of carbonic anhydrase isozymes. Bioorg. Med. Chem. 2013, 21, 1570–1582. [Google Scholar] [CrossRef] [PubMed]
- Al-Samir, S.; Papadopoulos, S.; Scheibe, R.J.; Meißner, J.D.; Cartron, J.-P.; Sly, W.S.; Alper, S.L.; Gros, G.; Endeward, V. Activity and distribution of intracellular carbonic anhydrase II and their effects on the transport activity of anion exchanger AE1/SLC4A1. J. Physiol. 2013, 591, 4963–4982. [Google Scholar] [CrossRef]
- Li, X.; Liu, Y.; Alvarez, B.V.; Casey, J.R.; Fliegel, L. A Novel Carbonic Anhydrase II Binding Site Regulates NHE1 Activity. Biochemistry 2006, 45, 2414–2424. [Google Scholar] [CrossRef]
- Henry, R.P. Multiple Roles of Carbonic Anhydrase in Cellular Transport and Metabolism. Annu. Rev. Physiol. 1996, 58, 523–538. [Google Scholar] [CrossRef]
- Scozzafava, A.; Supuran, C.T. Glaucoma and the Applications of Carbonic Anhydrase Inhibitors. In Carbonic Anhydrase: Mechanism, Regulation, Links to Disease, and Industrial Applications; Frost, S.C., McKenna, R., Eds.; Springer: Dordrecht, The Netherlands, 2014; pp. 349–359. [Google Scholar]
- Ciccone, L.; Cerri, C.; Nencetti, S.; Orlandini, E. Carbonic Anhydrase Inhibitors and Epilepsy: State of the Art and Future Perspectives. Molecules 2021, 26, 6380. [Google Scholar] [CrossRef]
- Poggetti, V.; Salerno, S.; Baglini, E.; Barresi, E.; Da Settimo, F.; Taliani, S. Carbonic Anhydrase Activators for Neurodegeneration: An Overview. Molecules 2022, 27, 2544. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic anhydrases as drug targets—An overview. Curr. Top. Med. Chem. 2007, 7, 825–833. [Google Scholar] [CrossRef]
- Di Fiore, A.; Supuran, C.T.; Scaloni, A.; De Simone, G. Human carbonic anhydrases and post-translational modifications: A hidden world possibly affecting protein properties and functions. J. Enzyme Inhib. Med. Chem. 2020, 35, 1450–1461. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Zhang, Q.; Liu, Z.; Zhao, Q.; Zhang, X.; Wang, Y.; Wang, Z.-X.; Jin, Y.; Li, X.; Liu, Z.-X.; et al. qPhos: A database of protein phosphorylation dynamics in humans. Nucleic Acids Res. 2019, 47, D451–D458. [Google Scholar] [CrossRef] [PubMed]
- Ullah, S.; Lin, S.; Xu, Y.; Deng, W.; Ma, L.; Zhang, Y.; Liu, Z.; Xue, Y. dbPAF: An integrative database of protein phosphorylation in animals and fungi. Sci. Rep. 2016, 6, 23534. [Google Scholar] [CrossRef] [PubMed]
- Freemont, P.S. Ubiquitination: RING for destruction? Curr. Biol. 2000, 10, R84–R87. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, S.; Saraswathi, N.T. Non-enzymatic glycation mediated structure–function changes in proteins: Case of serum albumin. RSC Adv. 2016, 6, 90739–90753. [Google Scholar] [CrossRef]
- Gharib, R.; Khatibi, A.; Khodarahmi, R.; Haidari, M.; Husseinzadeh, S. Study of glycation process of human carbonic anhydrase II as well as investigation concerning inhibitory influence of 3-beta-hydroxybutyrate on it. Int. J. Biol. Macromol. 2020, 149, 443–449. [Google Scholar] [CrossRef]
- Decker, T.; Kovarik, P. Serine phosphorylation of STATs. Oncogene 2000, 19, 2628–2637. [Google Scholar] [CrossRef]
- Karin, M.; Hunter, T. Transcriptional control by protein phosphorylation: Signal transmission from the cell surface to the nucleus. Curr. Biol. 1995, 5, 747–757. [Google Scholar] [CrossRef]
- Carrie, D.; Gilmour, K.M. Phosphorylation increases the catalytic activity of rainbow trout gill cytosolic carbonic anhydrase. J. Comp. Physiol. B 2016, 186, 111–122. [Google Scholar] [CrossRef]
- Blanco-Rivero, A.; Shutova, T.; Román, M.J.; Villarejo, A.; Martinez, F. Phosphorylation Controls the Localization and Activation of the Lumenal Carbonic Anhydrase in Chlamydomonas reinhardtii. PLoS ONE 2012, 7, e49063. [Google Scholar] [CrossRef]
- Alterio, V.; Di Fiore, A.; D’Ambrosio, K.; Supuran, C.T.; De Simone, G. Multiple Binding Modes of Inhibitors to Carbonic Anhydrases: How to Design Specific Drugs Targeting 15 Different Isoforms? Chem. Rev. 2012, 112, 4421–4468. [Google Scholar] [CrossRef] [PubMed]
- Duan, G.; Walther, D. The roles of post-translational modifications in the context of protein interaction networks. PLoS Comput. Biol. 2015, 11, e1004049. [Google Scholar] [CrossRef] [PubMed]
- Harju, A.-K.; Bootorabi, F.; Kuuslahti, M.; Supuran, C.T.; Parkkila, S. Carbonic anhydrase III: A neglected isozyme is stepping into the limelight. J. Enzyme Inhib. Med. Chem. 2013, 28, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Engstrand, C.; Forsman, C.; Liang, Z.; Lindskog, S. Proton transfer roles of lysine 64 and glutamic acid 64 replacing histidine 64 in the active site of human carbonic anhydrase II. Biochim. Biophys. Acta 1992, 1122, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Innocenti, A.; Scozzafava, A.; Parkkila, S.; Puccetti, L.; De Simone, G.; Supuran, C.T. Investigations of the esterase, phosphatase, and sulfatase activities of the cytosolic mammalian carbonic anhydrase isoforms I, II, and XIII with 4-nitrophenyl esters as substrates. Bioorg. Med. Chem. Lett. 2008, 18, 2267–2271. [Google Scholar] [CrossRef]
- Uda, N.R.; Seibert, V.; Stenner-Liewen, F.; Müller, P.; Herzig, P.; Gondi, G.; Zeidler, R.; van Dijk, M.; Zippelius, A.; Renner, C. Esterase activity of carbonic anhydrases serves as surrogate for selecting antibodies blocking hydratase activity. J. Enzyme Inhib. Med. Chem. 2015, 30, 955–960. [Google Scholar] [CrossRef]
- Iyer, R.; Barrese, A.A.; Parakh, S.; Parker, C.N.; Tripp, B.C. Inhibition profiling of human carbonic anhydrase II by high-throughput screening of structurally diverse, biologically active compounds. J. Biomol. Screen. 2006, 11, 782–791. [Google Scholar] [CrossRef]
- Ekinci, D.; Beydemir, S.; Alim, Z. Some drugs inhibit in vitro hydratase and esterase activities of human carbonic anhydrase-I and II. Pharmacol. Rep. PR 2007, 59, 580–587. [Google Scholar]
- Jovcevski, B.; Kelly, M.A.; Rote, A.P.; Berg, T.; Gastall, H.Y.; Benesch JL, P.; Aquilina, J.A.; Ecroyd, H. Phosphomimics Destabilize Hsp27 Oligomeric Assemblies and Enhance Chaperone Activity. Chem. Biol. 2015, 22, 186–195. [Google Scholar] [CrossRef]
- Hackney, D.D.; Yeo, Y.; Lin, C.T. Huntington-Associated Phosphorylation of Kinesin-1 Enhances Autoinhibition in a Phosphomimic. Biophys. J. 2013, 104, 652a. [Google Scholar] [CrossRef]
- Gropengiesser, J.; Varadarajan, B.T.; Stephanowitz, H.; Krause, E. The relative influence of phosphorylation and methylation on responsiveness of peptides to MALDI and ESI mass spectrometry. J. Mass Spectrom. 2009, 44, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Duong-Ly, K.C.; Peterson, J.R. The Human Kinome and Kinase Inhibition as a therapeutic strategy. Curr. Protoc. Pharmacol. 2013, 2, Unit2.9. [Google Scholar]
- St-Denis, N.; Gingras, A.-C. Mass Spectrometric Tools for Systematic Analysis of Protein Phosphorylation. In Progress in Molecular Biology and Translational Science; Shenolikar, S., Ed.; Academic Press: Cambridge, MA, USA, 2012; pp. 3–32. [Google Scholar]
- Schlaepfer, D.D.; Hunter, T. Integrin signalling and tyrosine phosphorylation: Just the FAKs? Trends Cell Biol. 1998, 8, 151–157. [Google Scholar] [CrossRef]
- Håkansson, K.; Carlsson, M.; Svensson, L.A.; Liljas, A. Structure of native and apo carbonic anhydrase II and structure of some of its anion-ligand complexes. J. Mol. Biol. 1992, 227, 1192–1204. [Google Scholar] [CrossRef] [PubMed]
- Sitbon, E.; Pietrokovski, S. Occurrence of protein structure elements in conserved sequence regions. BMC Struct. Biol. 2007, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef]
- Lundby, A.; Secher, A.; Lage, K.; Nordsborg, N.B.; Dmytriyev, A.; Lundby, C.; Olsen, J.V. Quantitative maps of protein phosphorylation sites across 14 different rat organs and tissues. Nat. Commun. 2012, 3, 876. [Google Scholar] [CrossRef]
- Shiromizu, T.; Adachi, J.; Watanabe, S.; Murakami, T.; Kuga, T.; Muraoka, S.; Tomonaga, T. Identification of missing proteins in the neXtProt database and unregistered phosphopeptides in the PhosphoSitePlus database as part of the Chromosome-centric Human Proteome Project. J. Proteome Res. 2013, 12, 2414–2421. [Google Scholar] [CrossRef]
- Blom, N.; Gammeltoft, S.; Brunak, S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 1999, 294, 1351–1362. [Google Scholar] [CrossRef]
- Zhu, G.; Liu, Y.; Shaw, S. Protein kinase specificity. A strategic collaboration between kinase peptide specificity and substrate recruitment. Cell Cycle Georget. 2005, Tex 4, 52–56. [Google Scholar] [CrossRef]
- Kumar, V.; Kannan, K.K. Enzyme-substrate interactions. Structure of human carbonic anhydrase I complexed with bicarbonate. J. Mol. Biol. 1994, 241, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Vlastaridis, P.; Kyriakidou, P.; Chaliotis, A.; Van de Peer, Y.; Oliver, S.G.; Amoutzias, G.D. Estimating the total number of phosphoproteins and phosphorylation sites in eukaryotic proteomes. GigaScience 2017, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Konermann, L.; Douglas, D.J. Unfolding of proteins monitored by electrospray ionization mass spectrometry: A comparison of positive and negative ion modes. J. Am. Soc. Mass Spectrom. 1998, 9, 1248–1254. [Google Scholar] [CrossRef]
- Testa, L.; Brocca, S.; Santambrogio, C.; D’Urzo, A.; Habchi, J.; Longhi, S.; Uversky, V.N.; Grandori, R. Extracting structural information from charge-state distributions of intrinsically disordered proteins by non-denaturing electrospray-ionization mass spectrometry. Intrinsically Disord. Proteins 2013, 1, e25068. [Google Scholar] [CrossRef] [PubMed]
- Gaspari, R.; Rechlin, C.; Heine, A.; Bottegoni, G.; Rocchia, W.; Schwarz, D.; Bomke, J.; Gerber, H.-D.; Klebe, G.; Cavalli, A. Kinetic and Structural Insights into the Mechanism of Binding of Sulfonamides to Human Carbonic Anhydrase by Computational and Experimental Studies. J. Med. Chem. 2016, 59, 4245–4256. [Google Scholar] [CrossRef] [PubMed]
- Nguyen GT, H.; Tran, T.N.; Podgorski, M.N.; Bell, S.G.; Supuran, C.T.; Donald, W.A. Nanoscale Ion Emitters in Native Mass Spectrometry for Measuring Ligand–Protein Binding Affinities. ACS Cent. Sci. 2019, 5, 308–318. [Google Scholar] [CrossRef]
- Nguyen GT, H.; Nocentini, A.; Angeli, A.; Gratteri, P.; Supuran, C.T.; Donald, W.A. Perfluoroalkyl Substances of Significant Environmental Concern Can Strongly Inhibit Human Carbonic Anhydrase Isozymes. Anal. Chem. 2020, 92, 4614–4622. [Google Scholar] [CrossRef]
- Nocentini, A.; Angeli, A.; Carta, F.; Winum, J.-Y.; Zalubovskis, R.; Carradori, S.; Capasso, C.; Donald, W.A.; Supuran, C.T. Reconsidering anion inhibitors in the general context of drug design studies of modulators of activity of the classical enzyme carbonic anhydrase. J. Enzyme Inhib. Med. Chem. 2021, 36, 561–580. [Google Scholar] [CrossRef]
- Gibson, D.G.; Young, L.; Chuang, R.-Y.; Venter, J.C.; Hutchison, C.A.; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef]
- Cao, W.; De La Cruz, E.M. Quantitative full time course analysis of nonlinear enzyme cycling kinetics. Sci. Rep. 2013, 3, 2658. [Google Scholar] [CrossRef]
- Scozzafava, A.; Supuran, C.T. Hydroxyurea is a carbonic anhydrase inhibitor. Bioorg. Med. Chem. 2003, 11, 2241–2246. [Google Scholar] [CrossRef] [PubMed]




| Cell Lysate kcat/KM (M−1·S−1) | Purified Proteins kcat/KM (M−1·S−1) | ||
|---|---|---|---|
| hCAII | unmodified | 460 ± 38 | 490 ± 11 |
| S2E | 679 ± 16 | ||
| S29E | 45 ± 11 | 19 ± 13 | |
| S48E | 624 ± 4 | ||
| S50E | 648 ± 15 | 670 ± 11 | |
| S48E_S50E | 644 ± 4 | ||
| S99E | 525 ± 23 | ||
| S151E | 607 ± 47 | ||
| S172E | 770 ± 27 | 751 ± 8 | |
| hCAI | unmodified | 122 ± 4 | |
| S30E | 44 ± 8 | ||
| S49E | 111 ± 5 | ||
| S51E | 100 ± 4A1 | ||
| S131E | 82 ± 4 | ||
| S137E | 100 ± 6 | ||
| Unmodified hCAII | hCAIIS50E | hCAIIS172E | ||
|---|---|---|---|---|
| Kd (µM) | Kd (µM) | Kd (µM) | Ki (µM) [9] | |
| acetazolamide | <0.1 a ± 0.1 | 83.1 ± 2.4 | <0.2 a ± 0.1 | 0.012 |
| brinzolamide | <2.9 a ± 1.4 | 21.5 ± 6.2 | <4.2 a ± 2.5 | 0.009 |
| dichlorphenamide | <2.8 a ± 0.4 | 23.9 ± 0.3 | <4.2 a ± 1.8 | 0.038 |
| ethoxzolamide | <3.5 a ± 0.3 | 84.8 ± 7.5 | <2.5 a ± 0.5 | 0.008 |
| indapamide | 10.0 ± 2.0 | 102.3 ± 11.3 | 9.2 ± 2.5 | 2.52 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, X.; Winter, D.; Glover, D.J.; Supuran, C.T.; Donald, W.A. Effects of Phosphorylation on the Activity, Inhibition and Stability of Carbonic Anhydrases. Int. J. Mol. Sci. 2023, 24, 9275. https://doi.org/10.3390/ijms24119275
Huang X, Winter D, Glover DJ, Supuran CT, Donald WA. Effects of Phosphorylation on the Activity, Inhibition and Stability of Carbonic Anhydrases. International Journal of Molecular Sciences. 2023; 24(11):9275. https://doi.org/10.3390/ijms24119275
Chicago/Turabian StyleHuang, Xiaojing, Daniel Winter, Dominic J. Glover, Claudiu T. Supuran, and William A. Donald. 2023. "Effects of Phosphorylation on the Activity, Inhibition and Stability of Carbonic Anhydrases" International Journal of Molecular Sciences 24, no. 11: 9275. https://doi.org/10.3390/ijms24119275
APA StyleHuang, X., Winter, D., Glover, D. J., Supuran, C. T., & Donald, W. A. (2023). Effects of Phosphorylation on the Activity, Inhibition and Stability of Carbonic Anhydrases. International Journal of Molecular Sciences, 24(11), 9275. https://doi.org/10.3390/ijms24119275