1. Introduction
RNA interference (RNAi) is a natural defense mechanism against foreign RNA, triggered by the presence of double-stranded RNA (dsRNA) and resulting in the silencing of specific genes via mRNA degradation or block of translation. Since its discovery in 1998 in the nematode
Caenorhabditis elegans [
1], RNAi has been transformed into a molecular technique to selectively silence gene expression and it has been extensively used in many different organisms. Besides basic and widely used gene function studies via RNAi-mediated gene knockdown and assessment of the corresponding phenotype, RNAi is also being studied for its application in gene therapy [
2] or cancer therapy [
3,
4]. In addition, it can be used to screen lists of disease-causing gene candidates [
5], just to name a few examples.
In recent years, RNAi has been applied to insect pest control, as it offers the possibility to species-specifically kill pest species by silencing essential genes. For instance, ingestion of bacterial cells expressing dsRNA targeting the heat shock protein and the shibire genes lead to gene silencing and lethality at the larval stage of the invasive emerald ash borer (
Agrilus planipennis Fairmaire) [
6]. Another possible strategy for insect pest control by RNAi is to target genes responsible for the (metabolic) resistance against commonly used insecticides [
7]. Comprehensive reviews on this topic, including current achievements related to technical and insect-specific challenges, potential applications, regulatory considerations, and perspectives of RNAi for insect control have been recently published [
8,
9,
10,
11].
The potential of RNAi-based approaches for pest control has also gained attention in the industry. The first RNAi-based product for pest control to obtain approval from a regulatory agency was the maize “SmartStax
® Pro” (Bayer, Leverkusen, Germany) approved in 2017 in the US [
12]. According to its manufacturer, it expresses dsRNA that interferes in the natural production of a vital protein in the corn rootworm. Therefore, ingestion of this maize would be lethal for the insect. Other companies, such as Syngenta, also announced advances in the development of RNAi-based products (
https://www.syngenta.com/en/innovation-agriculture/research-and-development/rna-based-biocontrols, accessed on 24 April 2023).
The application of RNAi for selective gene silencing experiments in the laboratory requires the production of gene-specific dsRNA, which can be obtained through different approaches, including in vitro transcription or in vivo production using genetically modified bacteria or yeast strains [
13,
14,
15]. While the production of microgram to one digit milligram amounts of dsRNA via in vitro transcription is typically not a problem for the budget, the production of higher milligram or even gram amounts of clean dsRNA, for large scale laboratory experiments for insect pest control, can become financially challenging [
13,
16,
17]. Here, the production of dsRNA via genetically modified bacterial cells can be an alternative.
Since its genetic modification to silence expression of RNase III and the insertion of a T7- inducible system for production of dsRNA [
18,
19], the bacterial strain HT115 (DE3) has been widely used for dsRNA production [
6,
19,
20,
21,
22,
23]. Although the application of bacterial crude extracts containing the produced dsRNA has been investigated in gene silencing experiments [
19,
20,
22,
24,
25,
26], purified dsRNA, free of other bacterial nucleic acids and proteins, is preferable or required in many cases, as it not only avoids potential immune responses due to the presence of bacterial cells [
27], but also facilitates regulatory approvals [
10,
28].
Several methods have been described for dsRNA extraction from bacterial cells [
29,
30,
31,
32,
33,
34]. Phenol–guanidine-based protocols have been reported as efficient method for extraction of dsRNA at high yields (up to 30 µg/mL of bacterial cells) and good quality [
29,
34,
35]. Other alternatives rely on fixing bacterial cells in ethanol and phosphate-buffered saline solution for extraction of dsRNA [
32,
33] or performing a phenol/chloroform/isoamyl alcohol (P/C/I) extraction and further DNase and RNase digestions [
30,
31].
The purification based on commercially available phenol–guanidine solutions inflates dsRNA production costs for medium- to large-scale laboratory experiments. Scaling up published protocols to extract dsRNA for example from a 5 L-bacterial culture, e.g., following the protocol from Verdonckt et al.’s work [
34], would consume 500 mL of QIAzol
® (QIAGEN, Venlo, The Netherlands), corresponding to 771.75 EUR just for the organic reagent. Processing the same amount of cells with a protocol using TRIzol
TM (Invitrogen, Waltham, MA, USA) [
29] would cost nearly EUR 1700 (794 mL of the organic reagent needed). While the required amounts of dsRNA for effective gene silencing vary with the target species, it is predicted that 2 to 10 g of dsRNA per hectare for crop protection would be needed in the field [
36]. Based on the published yields, this would correspond to EUR 30,000 to 142,000 for TRIzol
TM, or EUR 38,000 to 192,000 for QIAzol
®. However, for medium- to large-scale laboratory experiments the costs would also quickly be in the thousands to tens of thousands of Euros.
While the price for dsRNA synthesis has dropped strongly nowadays, with predicted production costs as low as 1 USD/g for a proprietary cell-free methodology (
www.greenlightbiosciences.com, accessed on 24 April 2023), the versality of engineered HT115 (DE3) cells for dsRNA production allows scientists to readily adjust dsRNA sequences according to experimental requirements or for distinct applications. Having a reliable and cost-effective protocol for extraction of dsRNA from large HT115 (DE3) culture volumes would further broaden the usability of HT115 cultures for dsRNA production, e.g., for field trials or larger laboratory applications, such as high-throughput screening of potential targets for RNAi in different species. Therefore, we aimed to develop a low-cost protocol for dsRNA extraction with high yield and quality, but at a fraction of the costs from the best currently available alternatives.
3. Discussion
Several methods have been developed for extraction of bacterially produced dsRNA for application of RNAi in crop protection, vector control, or functional genomic analysis, among other research fields [
29,
30,
31,
32,
33,
34,
37]. In the initial experiments, we had tested most of these protocols for the dsRNA yield and purity and obtained the best results in yield with the protocol established by Ongvarrasopone et al., which is based on a TRIzol
TM extraction. TRIzol
TM (Invitrogen), TRI Reagent
® (Merck, Darmstadt, Germany) and QIAzol
® (QIAGEN) are examples of commercially available phenol–guanidine-based reagents that have been used for extraction of bacterially produced dsRNA for RNAi application [
29,
34]. They offer an all-in-one solution for cell lysis, protein denaturation and deactivation of nucleases by the chaotropic agent guanidinium thiocyanate in solution with phenol [
35]. However, these commercially available monophasic solutions of phenol–guanidine are expensive. For example, in a protocol using QIAzol
® the costs for the reagent contribute to up to 85% of the total costs of dsRNA extraction [
34]. This becomes especially relevant when upscaling dsRNA purification from several liters of culture. Therefore, we exchanged the TRIzol
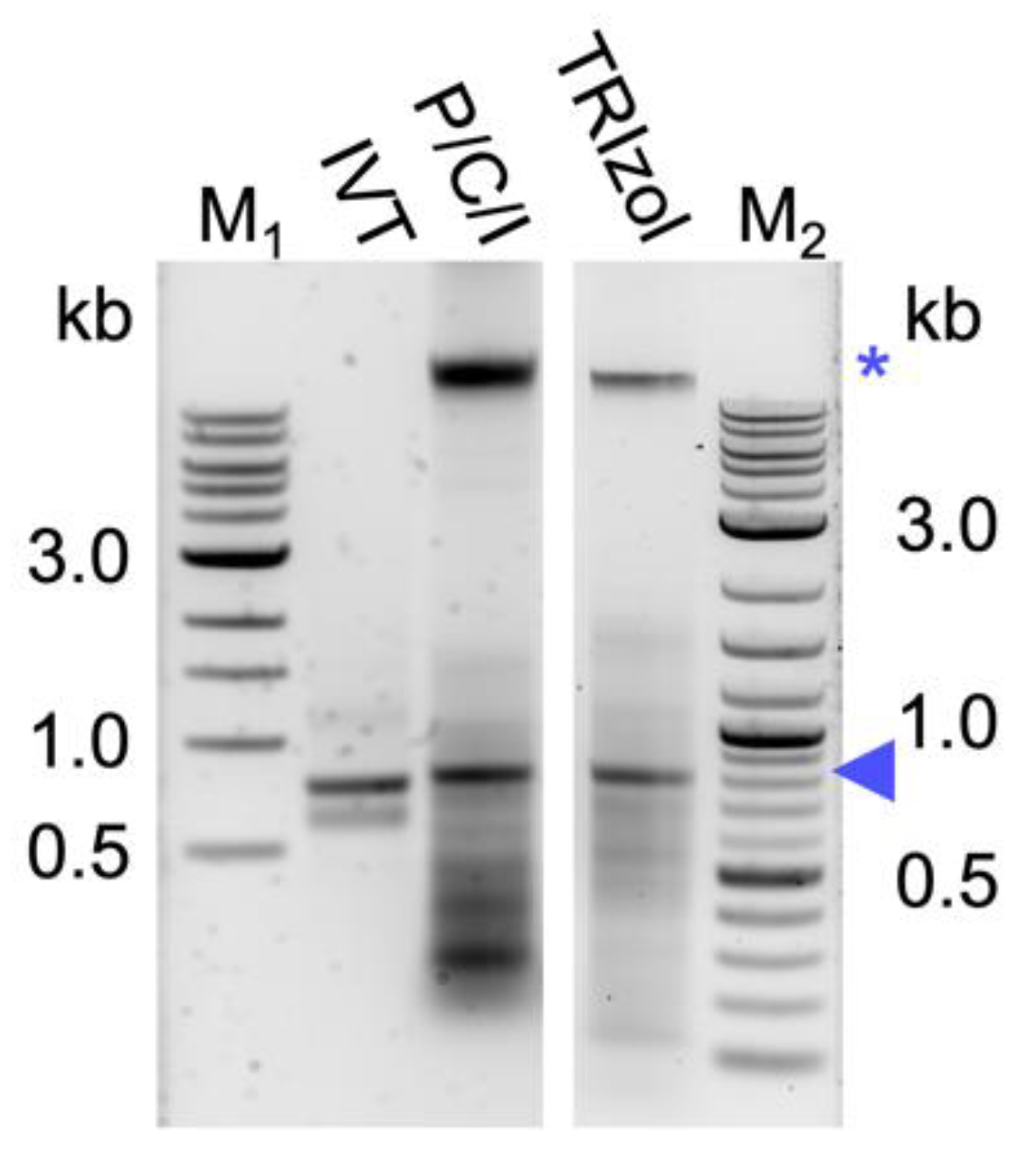
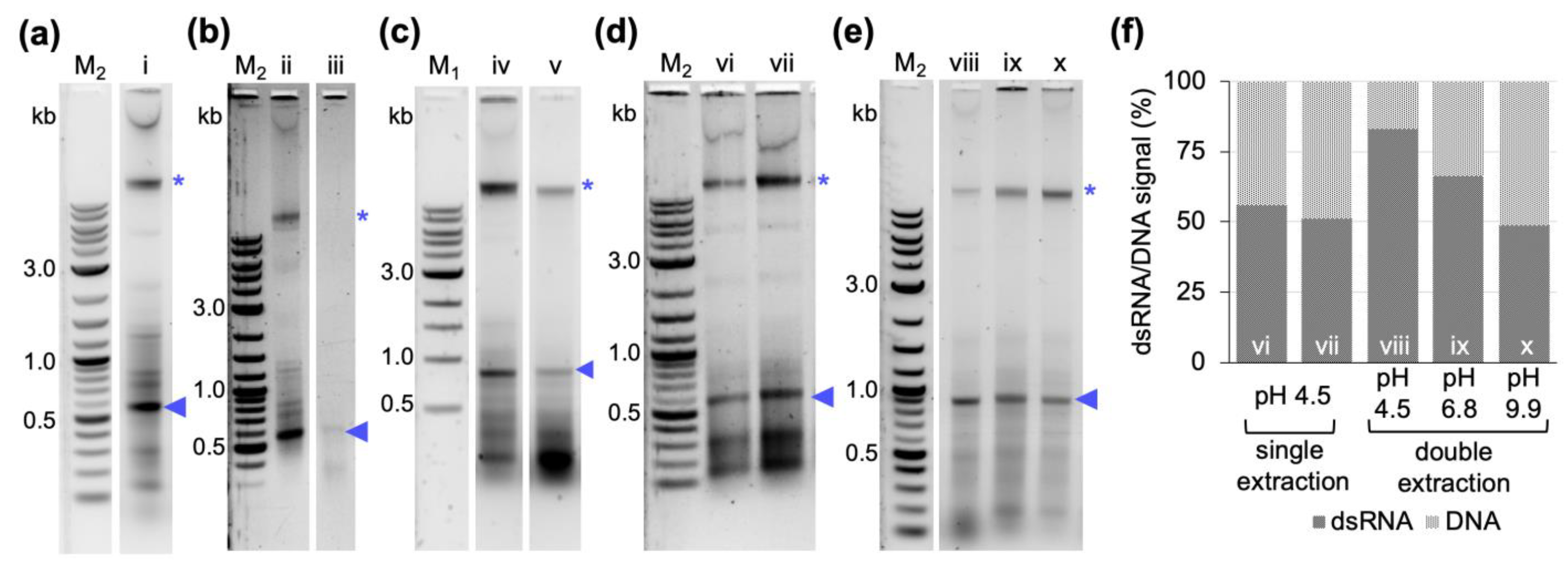
TM for the about 10-fold cheaper chemical P/C/I (25:24:1), pH 4.5–5. This resulted in a higher background of copurified bacterial RNAs and also (genomic) DNA (
Figure 1). The latter was unexpected, as the acidic conditions of the P/C/I extraction should partition the DNA into the interphase and organic phase [
35]. However, the acidity of the P/C/I itself was not sufficient for this effect, and also acidifying the reaction before the P/C/I addition did not improve the partitioning. In a systematic optimization process we found that adding a 2
nd acidic P/C/I extraction step reduces the copurified nucleic acids to a similar level as in the original TRIzol
TM-based protocol. Serial extractions to improve the quality of the dsRNA have previously been performed not only with phenol/chloroform-based protocols [
30,
31] but also with phenol–guanidine-based extraction methods [
34]. Altogether, our findings suggest that the pH of the used P/C/I might not always be sufficient to result in effective separation of RNA and DNA in phenol-based extractions, and that stabilization of the acidic conditions in the sample by using a low pH-buffer might be needed to obtain better results. Moreover, the presence of certain chemicals in the aqueous phase might interfere with the proper separation of the nucleic acids in the respective phase during the P/C/I extraction, as in our case the additional acidification of the reaction before adding P/C/I did not improve the result. Only when the already one-step P/C/I-purified dsRNA dissolved in pure water was acidified, followed by a 2
nd P/C/I extraction, could we achieve a better DNA and RNA separation. We suspect the SDS of the lysis buffer to be the interfering substance.
With regard to reducing the overall dsRNA production costs, we looked into possibilities to further increase the yield per culture volume and reduce extraction volumes to save on the most expensive component of the protocol, the organic reagent.
Cell lysis is a critical step in the extraction of bacterially produced dsRNA, as dsRNA would be lost with unlysed cells. Pre-treatment of cells, including sonication, heating, and enzymatic digestions, has been shown to increase the yield of dsRNA extraction [
31,
34]. However, quality analysis via gel electrophoresis has also shown that dsRNA extracted from sonicated samples yields less strong bands and smearing in the background, suggesting degradation of dsRNA [
31,
34]. We could show here that boiling in a reduced volume of the published buffer [
29] efficiently lysed the bacterial cells while at the same time preserving the dsRNA quality (
Figure 1 and
Figure 2), thereby abolishing the need for further treatments for cell lysis. Obtaining dsRNA free of bacterial cells is not only important to avoid possible unwanted immune responses in in vivo applications [
27], but is also highly relevant for regulatory aspects. In Europe, for instance, if no genetically modified organism (GMO) has been used or it is proven to be inactivated, RNAi-based products intended for agricultural pest control would be regulated following the same regulations applied for classical synthetic chemical pesticides [
28], which would apply to the dsRNA produced with our protocol.
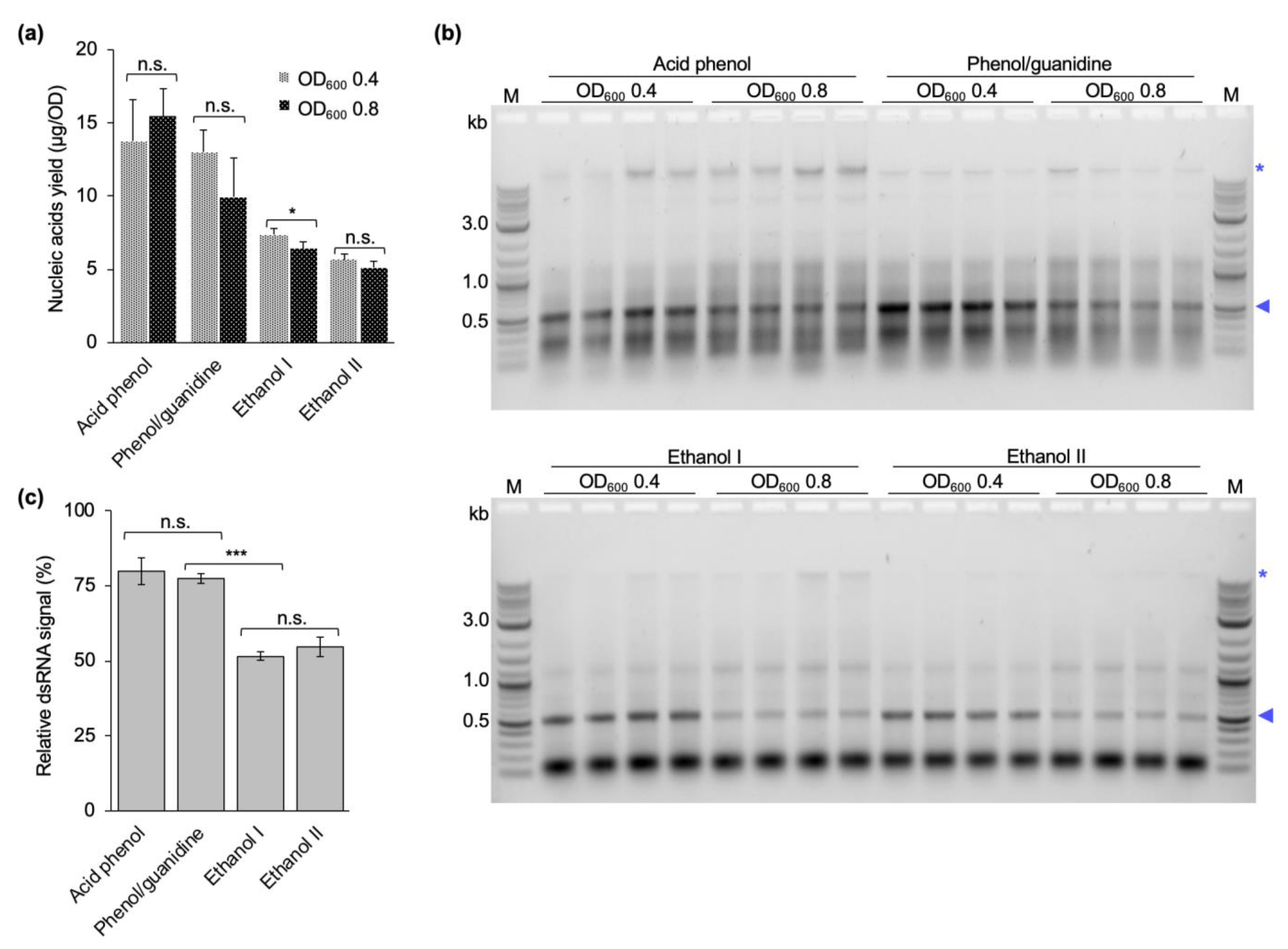
Different studies in the literature have induced dsRNA production at cell densities ranging from 0.4 to 0.8 OD
600, and harvested cells after 4 to 5 h of dsRNA production or when the OD
600 reached 1.0 [
29,
30,
31,
32,
33,
34]. Following the rationale that more cells per culture volume should yield more dsRNA per culture volume, we initially induced dsRNA production at a late exponential phase (OD
600 = 0.8). To prove this point, we additionally performed dsRNA extractions from cultures induced closer to the mid-exponential phase (OD
600 of 0.4). Regardless of the induction timepoint, the cells were allowed to produce dsRNA for 4 h before harvesting. In contrast to our expectations, the induction at the higher OD did not yield more dsRNA. On the contrary, the amount of dsRNA produced per cell was lower. In addition, a stronger background of copurified nucleic acids was observed. This was independent of the purification protocol used (
Figure 5). We assume that at a late timepoint in the exponential phase the bacterial cells are less transcriptionally active due to nutrients starting to become limiting, and maybe also become less responsive to IPTG.
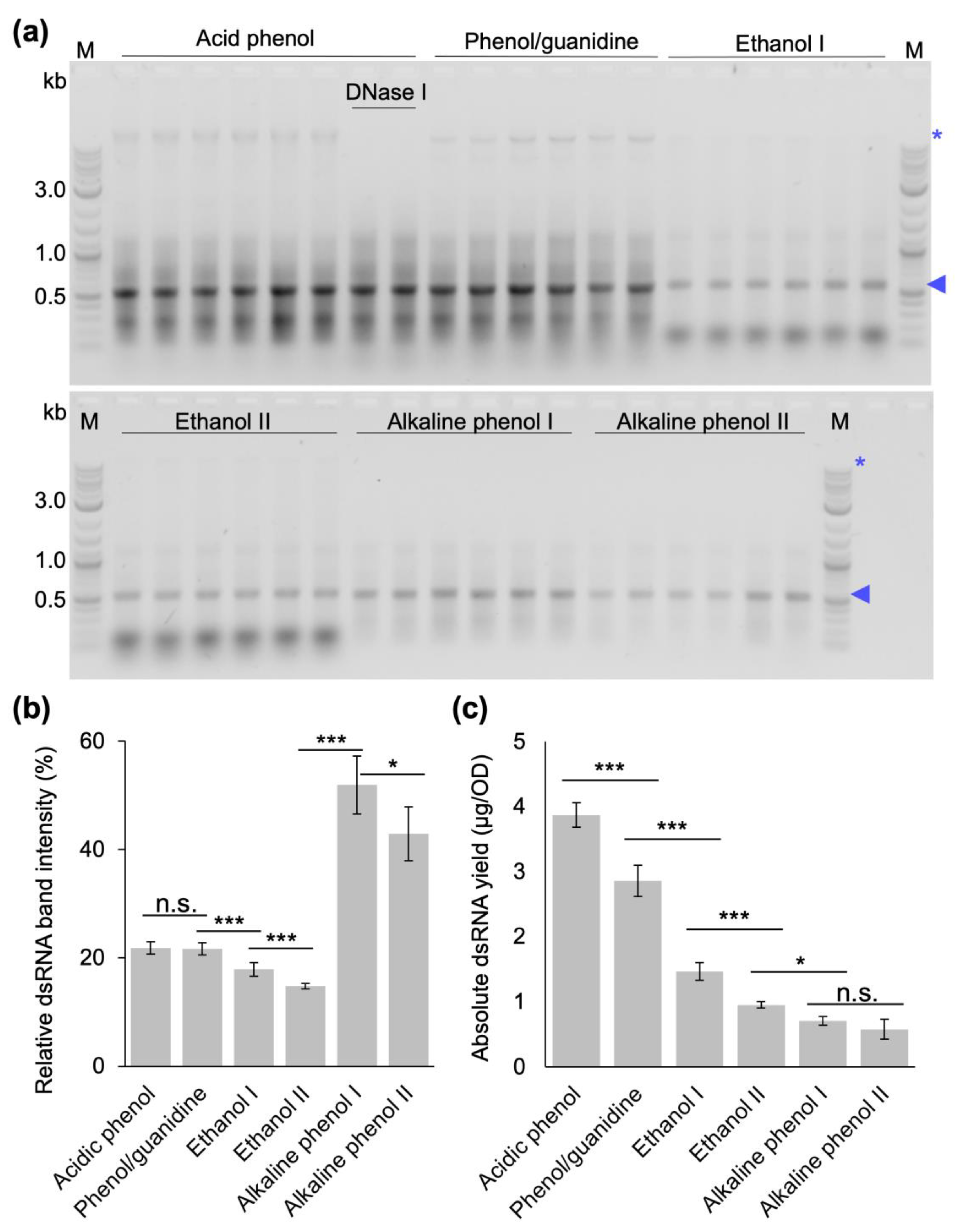
Finally, we compared the yield and quality of dsRNA extracted from bacterial cells induced at OD
600 of 0.4 with our optimized protocol, the original protocol established by Ongvarrasopone et al. and the published extraction protocols that do not require expensive monophasic solutions of phenol–guanidine [
29,
30,
31,
32,
33] in extensive side-by-side extractions. This experiment showed that our optimized protocol produces the highest absolute amount of dsRNA (
Figure 6), and had the highest nucleic acid concentrations. The gel electrophoretic analysis also showed that the different protocols resulted in very different dsRNA qualities. For our protocol and the phenol–guanidine-based protocol, there are still remnants of bacterial RNAs and DNA (the latter can be removed via additional DNase digestion after the first P/C/I extraction if required for specific applications). The ethanol-based protocols showed a strong contamination with small bacterial RNAs in addition to the rather moderate dsRNA yield, while, in our hands, the alkaline phenol-based protocol performed best with regard to purity, but at the lowest quantitative yields (
Figure 6a,b).
Interestingly, despite following the protocols as closely as the details reported in the respective methods sections allowed, we could not reproduce the reported dsRNA yields published in the literature. Possible reasons for the differences could be the different expression systems (strains and plasmids), culture media and types (batch or fed-batch), how the yields were determined, and the dsRNA constructs themselves. For instance, it has been shown that the dsRNA construct has a significative impact on the yield of the extracted dsRNA using a phenol–guanidine-based protocol [
34]. Regarding the different plasmids for expression of dsRNA, the plasmid L4440 has been used in our experiments, while pET3a and pET17b were used in some of the other studies [
29,
32]. Moreover, the nutrient content of the medium used to grow the cells might influence the dsRNA yield. We have preliminary data confirming that the amount of extracted dsRNA (by absorption measurement) doubles when cells are grown in the nutrient-rich terrific broth (TB) medium compared to LB medium. A very similar observation was made by Thammasorn et al. [
38]. Ongvarrasopone et al., report obtaining 30 µg dsRNA per 1 OD of cells grown in 2xYT medium, while we obtained about 13 µg per OD from cells grown in LB medium with their protocol (both numbers based on absorption measurement at 260 nm, i.e., measuring the total nucleic acid content). Potentially, the combination of different expression plasmid and nutrient supply is responsible for the observed difference. Likewise, Ahn et al. obtained approximately 4.85 µg per OD from cell grown in 2xYT medium, while we obtained about 1.4 µg/OD in LB. Besides the nutrient content, the high salt present in some media, such as TB, could contribute to regulate the pH of the cultures, thereby optimizing cell growth and dsRNA production [
38].
Taking into account only the costs for the organic reagent per microgram of produced dsRNA, our protocol is comparable to the two ethanol-based protocols (
Table 1). All three are 15- to 75-fold cheaper than the other tested methods. When users have to decide between the three most cost-effective protocols, aspects to consider will be the use of ethanol versus P/C/I, the strong contamination with small bacterial RNAs in the ethanol-based protocols, and, last but not least, that for ethanol-based protocols about 4 times larger culture volumes are required to produce the same absolute amount of dsRNA as with our protocol. This might become a deciding factor especially for large-scale dsRNA productions.
In summary, based on the results reported in the literature and our findings with the parallel purifications, we suggest that for a true comparison of the efficiency of a purification protocol, exactly the same dsRNA production system has to be used. Moreover, besides the extraction method, also the expression system, the culture conditions, and the induction time point for dsRNA production should be taken into account to further optimize the dsRNA yields from bacterial cultures. Finally, the combination of obtained dsRNA yields, dsRNA purity, and estimated production costs per milligram of purified dsRNA (based only on the organic reagent costs) suggests that when high purity dsRNA is required for the planned experiments, the alkaline phenol-based protocols perform best, but at considerable costs if high amounts are needed. In contrast, our optimized protocol has the best combined yield to cost and yield to culture volume ratios at only a moderate copurification of bacterial nucleic acids. This makes it the ideal protocol for dsRNA production for large scale laboratory or even field trial experiments not requiring the highest purity of dsRNA, meeting also potential regulatory demands for applications in the field.
4. Materials and Methods
4.1. In Vitro Production of dsRNA
Templates for in vitro transcription (IVT) of dsRNA were produced via polymerase chain reaction (PCR) using a fragment of the gene AAEL002851 from Aedes aegypti cloned into pCR4 TOPO vector and primers containing at T7 promoter overhang (marked in bold). PCR reactions contained 32.6 µL of nuclease-free water, 10 µL of 5x Q5 buffer, 5 µL of 2 mM dNTP mix, 0.5 µL of forward primer P884 (CCCTTTAATACGACTCACTATAGGGAGAAGGAAATCATCTCCGACGAAC) at 10 µM, 0.5 µL of reverse primer P885 (CCCTTTAATACGACTCACTATAGGGAGAACACGGTACTGTTGCGATCC) at 10 µM, 1 ng of DNA template and 0.5 µL of Q5 Taq polymerase (New England Biolabs, Inc., Ipswich, MA, USA). The PCR reaction was performed as follows: 30 s at 98 °C; 30 cycles of 98 °C for 10 s, 65 °C for 20 s and 72 °C for 1 min; and final elongation at 72 °C for 2 min. The PCR product was analyzed and purified via gel electrophoresis using Zymo Research Gel DNA recovery kit (Zymo Research Europe GmbH, Freiburg, Germany) following the manufacturer’s instructions. The purified template (200 ng) was used to generate dsRNA via in vitro transcription using the MegaScript Kit followed by purification of the in vitro transcription product using the MEGAclearTM kit according to the manufacturer’s instructions (Ambion/Life Technologies, Thermo Fischer Scientific Inc., Waltham, MA, USA).
4.2. In Vivo Production of dsRNA
For in vivo production of dsRNAs, 275 mL of lysogeny broth (LB-Lennox) medium (Carl Roth GmbH + Co. KG, Karlsruhe, Germany) supplemented with 100 µg/mL ampicillin (Carl Roth GmbH + Co. KG, Karlsruhe, Germany) and 12.5 µg/mL tetracycline (Fisher BioReagents, Geel, Belgium) were inoculated with 2.2 mL of an overnight culture of HT115 DE3 cells (F-, mcrA, mcrB, IN(rrnD-rrnE)1, rnc14::Tn10(DE3 lysogen: lavUV5 promoter -T7 polymerase, from the Caenorhabditis Genetics Center) transformed with L4440 plasmid encoding either a dsRNA against eGFP (480 bp) or Aae beta-tubulin (AAEL002851) (800 bp). Cells were grown at 37 °C and 180 rpm until the optical density measured at 600 nm (OD600) reached 0.4 or 0.8. dsRNA production was then induced via addition of isopropyl-β-D-thiogalactopyranoside (IPTG) (Carl Roth GmbH + Co. KG, Karlsruhe, Germany) to a final concentration of 0.4 mM and cells grown for another 4 h. Cells were harvested at 4 °C for 15 min at 3214 rcf, resuspended in clean LB-Lennox medium, aliquoted in batches of 10 OD or 2 OD in 2 mL-tubes, collected via centrifugation and stored at −80 °C until further use.
4.3. dsRNA Extraction from HT115 DE3 Cells via Phenol/Chloroform/Isoamyl Alcohol Extraction
Extraction of dsRNA using P/C/I was performed with the following modifications from the published phenol–guanidine-based protocol [
29]: bacterial cells were resuspended in 12.5 µL of 0.1% sodium dodecyl sulfate (SDS) per one OD of cells and incubated for 2 min at 95 °C. Then, 16.25 µL of neutral (pH 7.5) solution of 300 mM sodium acetate, 10 mM Tris-HCl, 5 mM EDTA, 1 µg RNase A per 1 OD cells were added to the lysed cells and incubated at 37 °C for 15 min. One volume of Roti
® Aqua phenol/chloroform/isoamyl alcohol (P/C/I, 25:24:1, pH 4.5–5) (Carl Roth GmbH + Co. KG, Karlsruhe, Germany) was added to the mixture and vortexed at full speed for 15 s. After incubating at 65 °C for 15 min, the tubes were centrifugated at room temperature for 15 min at 17,949 rcf. The aqueous upper phase was transferred to a fresh 2 mL-tube without disturbing the protein interphase or the organic lower phase. Ethanol precipitation was performed by adding 1/10 volume of 3 M sodium acetate, pH 5.2, and 2.5 volumes of 100% ethanol to the aqueous phase, followed by vortexing for 5 s. After precipitation for 1 h at −20 °C, dsRNA was pelleted at 17,949 rcf for 30 min at 4 °C. The dsRNA pellet was washed twice with 70% (
v/
v) cold ethanol, air dried, dissolved in 100 µL of nuclease-free water, incubated at 55 °C for 15 min and immediately placed on ice. For comparison with the original phenol–guanidine-based protocol, bacterial cells were also processed using TRIzol
TM (Invitrogen, Thermo Fischer Scientific, Waltham, MA, USA) as published in the literature [
29]. Concentration and quality of purified dsRNA was assessed via absorbance measurement and gel electrophoresis, respectively.
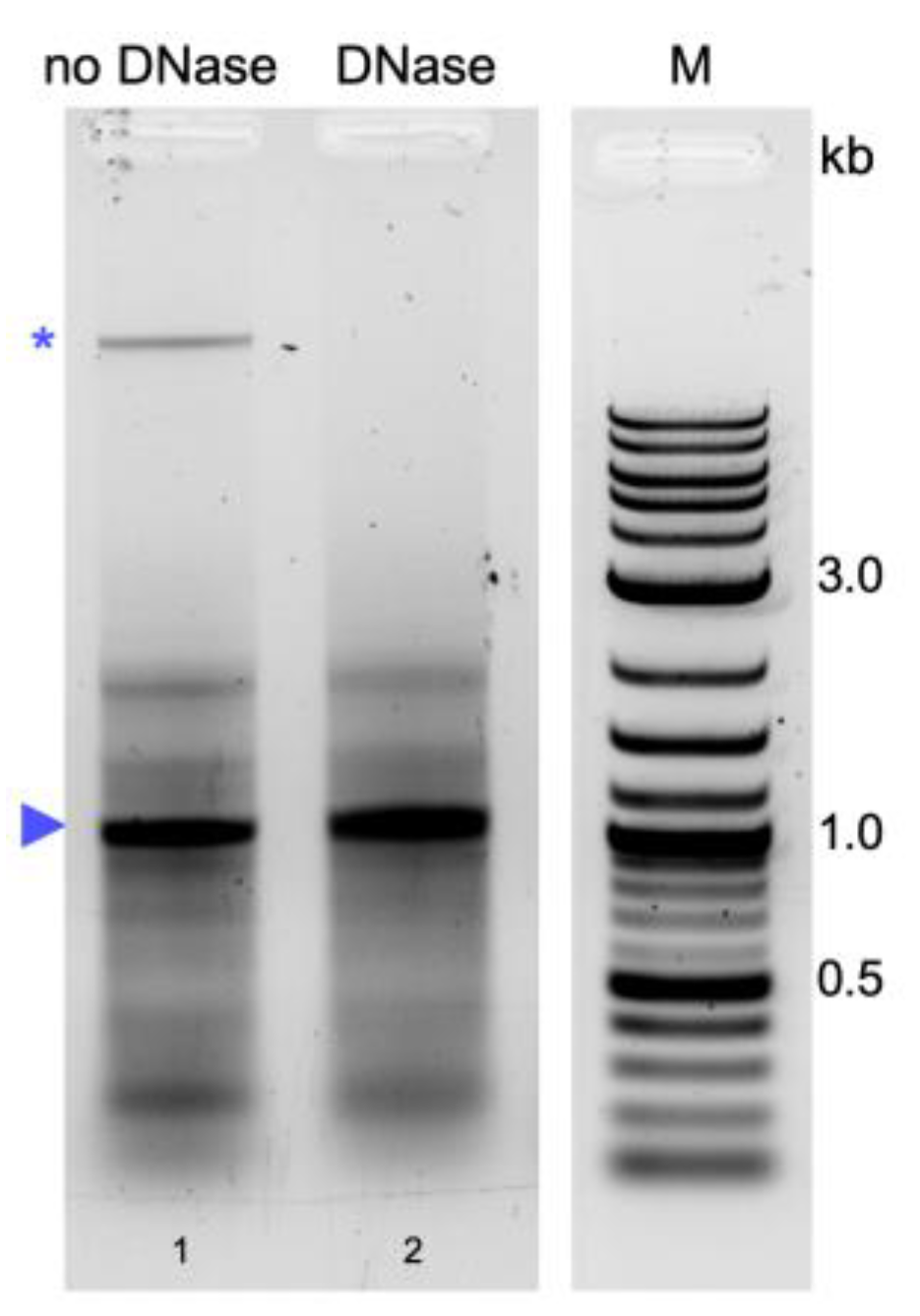
4.4. Removal of High-Molecular-Weight Nucleic Acid Contamination by DNase Digestion
Removal of high-molecular-weight nucleic acid contamination from dsRNA obtained via the phenol–guanidine-based extraction with TRIzol
TM [
29] was evaluated via DNase digestion. The final product from TRIzol
TM extraction was resuspended in nuclease-free water and DNase digestion was performed at room temperature for 15 min. A mock digestion was performed for comparison. Following DNase digestion, the nucleic acids were extracted with P/C/I (25:24:1, pH 7.5–8) and precipitated with ethanol. Concentration measurement and quality assessment via gel electrophoresis were performed as described above.
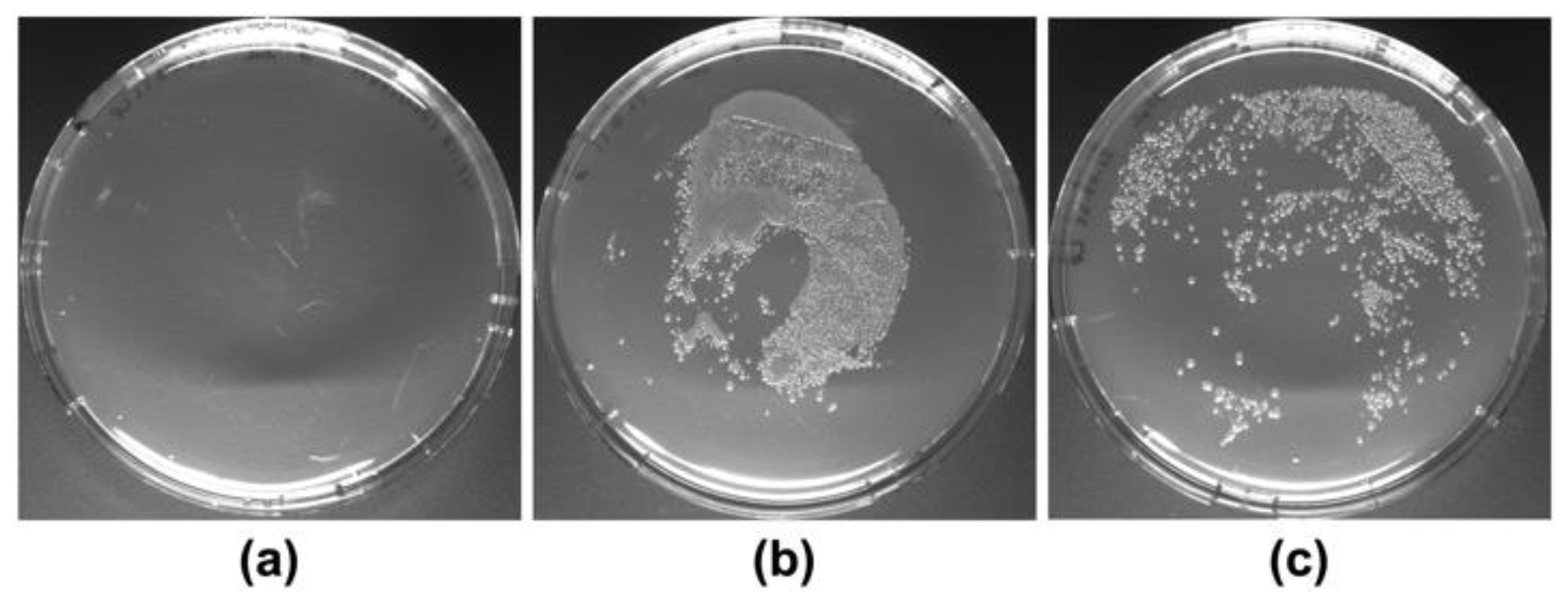
4.5. Cell Lysis Efficiency Testing
Efficiency of cell lysis by boiling HT115 (DE3) cells at 95 °C for two minutes after resuspension in 12.5 µL of SDS 0.1% (w/v) per OD600 of cells was evaluated through a cell viability assay. After boiling, cells were homogenized and spread into LB-agar 1.5% (w/v) plates containing ampicillin (100 µg/mL). As control, cells were resuspended in the same volume of sterile LB-medium 2% (w/v) and incubated at room temperature for two minutes. Then, cells were homogenized and aliquots of the undiluted cells suspension and of 1:100 dilution were spread into LB-agar plates. All plates were incubated overnight at 37 °C before evaluation of colony development. The experiments were performed in biological duplicates.
4.6. Assessment of pH and Serial P/C/I Extractions on dsRNA Purity
The protocol described in
Section 4.3 was tested in four different modifications to further improve dsRNA purity: (1) cell lysis was performed at 95 °C for 2 min in 0.1% (
w/
v) SDS, 0.1 M sodium acetate, pH 4.5. The rest of the protocol remained unchanged. (2) the RNase A digest was performed in 300 mM sodium acetate, 10 mM Tris-HCl, 5 mM EDTA, pH 4.5. The rest of the protocol remained unchanged. (3) The reaction was acidified by addition of 2.5 volumes of 0.1 M sodium acetate, pH 4.5 before the addition of one-volume equivalent (1000 µL) of P/C/I (25:24:1, pH 4.5–5.0). The rest of the protocol remained unchanged. (4) The product from the first P/C/I extraction, obtained as described in 4.3 and resuspended in 100 µL of nuclease-free water, was mixed with 300 µL of either 0.1 M sodium acetate at pH 4.5 or 0.2 M sodium phosphate dibasic at pH 6.8 or 0.1 M sodium bicarbonate/sodium carbonate at pH 9.9. Then, a second extraction was performed using 400 µL of P/C/I (25:24:1, pH 4.5–5). The downstream steps of dsRNA ethanol precipitation, washing, and resuspension in 100 µL of nuclease-free water, were performed as described in
Section 4.3. Concentration measurement and quality assessment via gel electrophoresis of the obtained dsRNA from each experiment were performed as described above.
4.7. Optimization of the Induction OD600
dsRNA was extracted from bacterial cells induced to produce dsRNA at OD
600 of 0.4 or 0.8 using a phenol–guanidine-based protocol [
29], two ethanol-based extraction protocols published in the literature [
32,
33], and the final version of the protocol developed in this study (two-step acidic P/C/I extraction, see
Section 4.6). All extractions were performed as published with the exception of the phenol–guanidine-based protocol, where the RNase digestion was performed for 15 min instead of 5 min as originally published. Concentration measurement and quality assessment via gel electrophoresis were performed as described above. Extraction yields and dsRNA purity after induction at different cell densities was compared between all four protocols.
4.8. Systematic Comparison of dsRNA Extraction Protocol Efficiencies
To compare the yield and purity of phenol–guanidine-based [
29], ethanol-based [
32,
33], and alkaline phenol-based [
30,
31] extraction protocols with the protocol developed in this study, dsRNA production in HT115 DE3 cells was induced at an OD
600 of 0.4. Experiments were performed in three biological replicates and two technical replicates each. Importantly, the quantitative yields were calculated considering the initial amount of cells and volume of nuclease-free water in the final resuspension step of each protocol. The method by Ongavarrasopone et al. [
29] was followed as published, except for an RNase A digestion time of 15 min instead of 5 min. The methods established by Posiri et al. [
32] and Papic et al. [
33] were followed in their entirety as published. The protocol established by Ahn et al. [
31] was adapted by opening the cells via homogenization in tubes containing ceramic beads in a tissue homogenizer (Precellys
®, Bertin Instruments, Montigny-le-Bretonneux, France) for two runs at 6000 rpm for 20 s with an intermediate incubation on ice for 1 min. The method established by Solis et al. [
30] was followed in its entirety, with exception of using Monarch DNase I (1 U/µL) and Monarch DNase I reaction buffer (New England Biolabs, Inc., Ipswich, MA, USA) for DNase digestion instead of Turbo DNase and DNase buffer (Ambion/Life Technologies, Thermo Fischer Scientific Inc., Waltham, MA, USA). These results were compared to the final version of the protocol developed in this study: Bacterial cells were resuspended in 12.5 µL of 0.1 % (
v/
v) SDS per OD of cells and incubated at 95 °C for 2 min. Then, 16.25 µL of 300 mM sodium acetate, 10 mM Tris-Cl, 5 mM EDTA, pH 7.5, 1 µg of PureLink RNase A per OD of cells were added and incubated at 37 °C for 15 min. The equivalent volume of P/C/I (25:24:1, pH 4.5–5) was added, thoroughly mixed for 15 s and incubated at 65 °C for 15 min. After centrifugation at 17,949 rcf for 15 min at 20 °C, the aqueous phase containing dsRNA was carefully transferred to a new tube without disturbing the protein interphase and the organic lower phase. Ethanol precipitation was performed by adding 1/10 volume of 3 M sodium acetate, pH 5.2, and 2.5 volumes of 100% ethanol to the aqueous phase, followed by vortexing for 5 s and precipitation for 1 h at −20 °C. The dsRNA was pelleted at 17,949 rcf for 30 min at 4 °C and washed once with 70% (
v/
v) cold ethanol. The air-dried dsRNA pellet was dissolved in 100 µL of nuclease-free water and incubated at 55 °C for 10 min. Optional DNase digestion was performed with 25 µL DNase I mix and incubated at 30 °C for 15 min. Then, 375 µL of 0.1 M acetate buffer at pH 4.5 was added before repeating the P/C/I extraction and ethanol precipitation as described above, this time applying an additional ethanol wash step. Finally, the dsRNA pellet was dissolved in 100 µL of nuclease-free water, incubated at 55 °C for 10 min, and immediately placed on ice. DsRNA yields were calculated as total amount of nucleic acid quantified via spectrophotometry at 260 nm divided by the total OD of bacterial cells used for extraction to result in µg dsRNA/1 OD of cells. The quality of extracted dsRNA was inspected via gel electrophoresis analyzing the same volume of extracted dsRNA from each technical and biological replicates from each extraction method.
4.9. Analysis of Band Intensity from Gel Electrophoresis
Images acquired from the gel documentation system were analyzed using the software Adobe Photoshop 2023 (Adobe Inc., 2023, San José, CA, USA). In general, the bands or the region of interest or the complete lane were identified and their integrated density was quantified.
4.10. Statistical Analysis
Data analysis was carried out using the software SigmaPlot (Version 14.0, Systat Software Inc., San José, CA, USA) and MiniTab
® (Minitab, LLC., State College, CA, USA). Yields of nucleic acids extracted from bacterial cells induced to produce dsRNA at OD
600 of 0.4 or 0.8, and yield of extracted dsRNA using different published methods and the optimized version of our protocol were analyzed via one-way analysis of variance (ANOVA), with Shapiro–Wilk test for normality and Brown–Forsythe test for equal variance. Holm–Sidak method was used for multiple comparison of the data from each condition, when the differences in the mean values among the groups were greater than would be expected by chance (α = 0.05). Data of the relative dsRNA band intensity, which failed Shapiro–Wilk test for normality, underwent a Box Cox transformation [
39] prior to conducting the ANOVA. For the comparative analysis of the amounts of nucleic acids extracted using different published methods and the optimized version of our protocol, the yields of nucleic acid per OD
600 for each extraction method were analyzed via Welch’s test, which does not assume equal variances for the analysis. The means were compared with the Games–Howell pairwise comparison at 95% confidence level.

 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}