

Epigenetic Histone Methylation of PPARγ and CPT1A Signaling Contributes to Betahistine Preventing Olanzapine-Induced Dyslipidemia

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
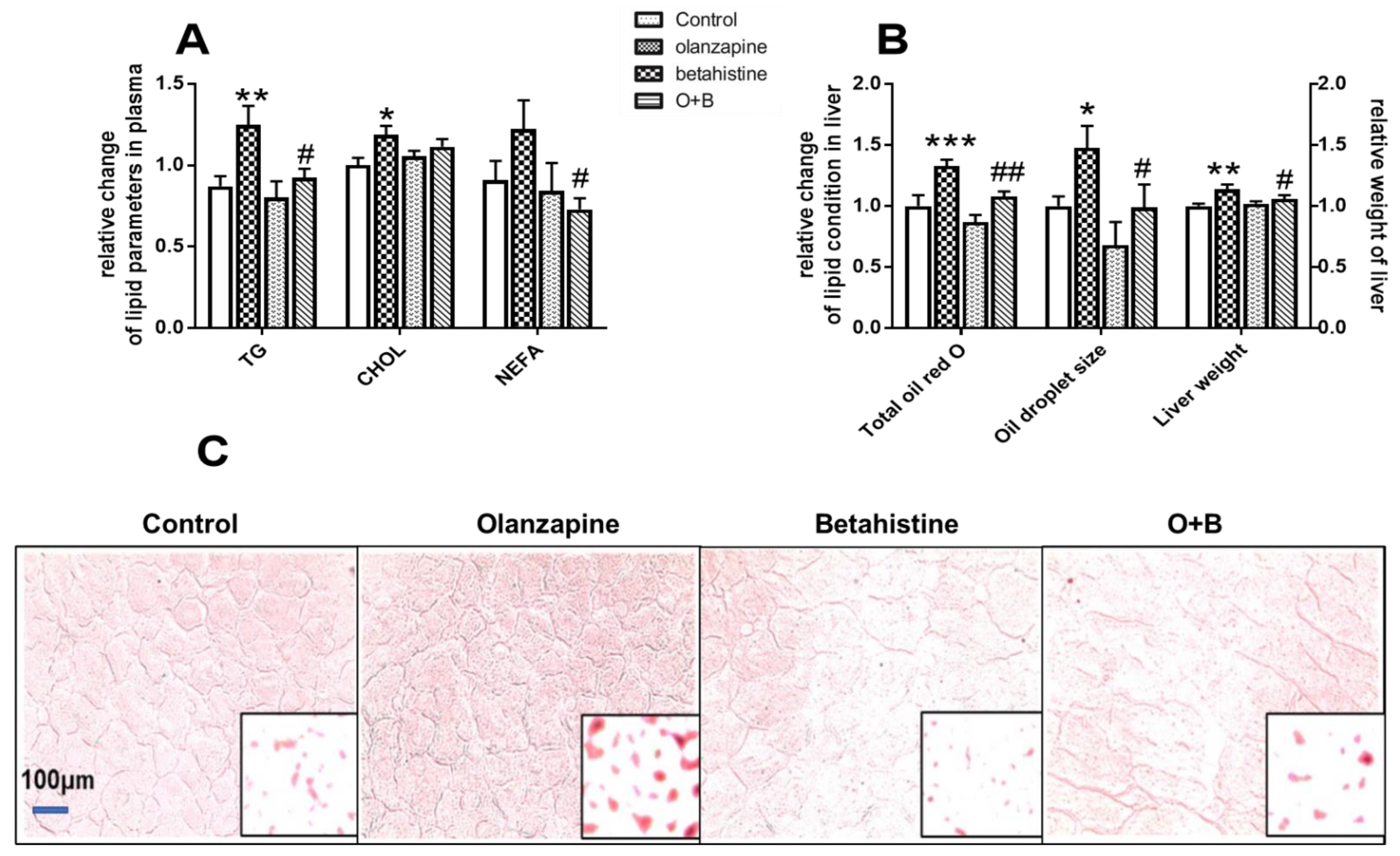
2.1. Betahistine Co-Treatment Ameliorated Dyslipidemia Caused by Olanzapine
2.2. Betahistine Co-Treatment Partly Alleviated Activation of the PPARγ/C/EBPα Pathway Induced by Olanzapine
2.3. Betahistine Co-Treatment Reversed the CPT1A Inhibition Induced by Olanzapine
2.4. Global Profile of H3K4me, H3K9me, and H3K27me in the Hepatic Tissue
2.5. Betahistine Co-Treatment Reversed the Activation of Kdm1a and Kdm4 Induced by Olanzapine
2.6. Betahistine Co-Treatment Increased the Enrichment of H3k9me3 Binding on the Promoter Region Pparg2, and H3K4me2 Binding on the Promoter Region Cpt1a
3. Discussion
4. Materials and Methods
4.1. Animal Treatment
4.2. Plasma and Liver Lipid Assays
4.3. Examining Hepatic Gene Expression Using Quantitative Reverse Transcription PCR (qRT-PCR)
4.4. Analyses of Global Histone Methylation Using Western Blot
4.5. Analysis of DNA Fragment Binding on Histone Methylation Marks Using ChIP-qPCR
4.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AMPK | activating protein kinase; |
| C/EBPs | CCAAT/enhancer binding proteins; |
| ChIP | chromatin immunoprecipitation; |
| CPT1A | carnitine palmitoyltransferase 1A; |
| FAS | fatty acid synthase; |
| H3K4me | histone H3 methylations at 4 lysine residue; |
| H3K9me | histone H3 methylations at 9 lysine residue; |
| KDM1A | lysine (K)-specific demethylase 1A; |
| KDM4B | lysine demethylase 4B; |
| NAFLD | non-alcoholic fatty liver disease; |
| NEFA | non-esterified fatty acid; |
| NPY | neuropeptide Y; |
| PPAR | peroxisome proliferator-activated receptor; |
| PHF2 | PHD finger protein 2; |
| qRT-PCR | quantitative reverse transcription PCR; |
| SGAs | second-generation antipsychotics; |
| TC | cholesterol; |
| TG | total triglycerides. |
References
- Nagai, N.; Watanabe, K. Olanzapine. Nihon Rinsho 2013, 71, 666–672. [Google Scholar] [PubMed]
- Hirsch, L.; Yang, J.; Bresee, L.; Jette, N.; Patten, S.; Pringsheim, T. Second-Generation Antipsychotics and Metabolic Side Effects: A Systematic Review of Population-Based Studies. Drug Saf. 2017, 40, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Deng, C. Effects of antipsychotic medications on appetite, weight, and insulin resistance. Endocrinol. Metab. Clin. N. Am. 2013, 42, 545–563. [Google Scholar] [CrossRef] [PubMed]
- Lian, J.; Huang, X.-F.; Pai, N.; Deng, C. Ameliorating antipsychotic-induced weight gain by betahistine: Mechanisms and clinical implications. Pharmacol. Res. 2016, 106, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Kaar, S.J.; Natesan, S.; McCutcheon, R.; Howes, O.D. Antipsychotics: Mechanisms underlying clinical response and side-effects and novel treatment approaches based on pathophysiology. Neuropharmacology 2020, 172, 107704. [Google Scholar] [CrossRef]
- Deng, C.; Weston-Green, K.; Huang, X.-F. The role of histaminergic H1 and H3 receptors in food intake: A mechanism for atypical antipsychotic-induced weight gain? Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2010, 34, 1–4. [Google Scholar] [CrossRef]
- Kroeze, W.K.; Hufeisen, S.J.; Popadak, B.A.; Renock, S.M.; Steinberg, S.; Ernsberger, P.; Jayathilake, K.; Meltzer, H.Y.; Roth, B.L. H1-histamine receptor affinity predicts short-term weight gain for typical and atypical antipsychotic drugs. Neuropsychopharmacology 2003, 28, 519–526. [Google Scholar] [CrossRef]
- He, M.; Zhang, Q.; Deng, C.; Wang, H.; Lian, J.; Huang, X.-F. Hypothalamic histamine H1 receptor-AMPK signaling time-dependently mediates olanzapine-induced hyperphagia and weight gain in female rats. Psychoneuroendocrinology 2014, 42, 153–164. [Google Scholar] [CrossRef]
- Fossati, A.; Barone, D.; Benvenuti, C. Binding affinity profile of betahistine and its metabolites for central histamine receptors of rodents. Pharmacol. Res. 2001, 43, 389–392. [Google Scholar] [CrossRef]
- Barak, N.; Beck, Y.; Albeck, J.H. Betahistine decreases olanzapine-induced weight gain and somnolence in humans. J. Psychopharmacol. 2016, 30, 237–241. [Google Scholar] [CrossRef]
- Naguy, A.; AlShalabi, S.R.; AlKhadhari, S. Betahistine-Associated Weight Loss and Improved Cognitive and Negative Symptoms: Domain in Early-Onset Schizophrenia. Am. J. Ther. 2019, 26, e790–e792. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Lian, J.; Pai, N.; Huang, X.-F. Reducing olanzapine-induced weight gain side effect by using betahistine: A study in the rat model. J. Psychopharmacol. 2012, 26, 1271–1279. [Google Scholar] [CrossRef] [PubMed]
- Lian, J.; Huang, X.-F.; Pai, N.; Deng, C. Preventing olanzapine-induced weight gain using betahistine: A study in a rat model with chronic olanzapine treatment. PLoS ONE 2014, 9, e104160. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.C.; Maayan, L.; Wu, R.; Youssef, M.; Jing, Z.; Sershen, H.; Szabo, V.; Meyers, J.; Jin, H.; Zhao, J.; et al. Betahistine effects on weight-related measures in patients treated with antipsychotic medications: A double-blind placebo-controlled study. Psychopharmacology 2018, 235, 3545–3558. [Google Scholar] [CrossRef]
- Lian, J.; Huang, X.-F.; Pai, N.; Deng, C. Betahistine ameliorates olanzapine-induced weight gain through modulation of histaminergic, NPY and AMPK pathways. Psychoneuroendocrinology 2014, 48, 77–86. [Google Scholar] [CrossRef]
- Liu, X.; Lian, J.; Hu, C.-H.; Deng, C. Betahistine co-treatment ameliorates dyslipidemia induced by chronic olanzapine treatment in rats through modulation of hepatic AMPKα-SREBP-1 and PPARα-dependent pathways. Pharmacol. Res. 2015, 100, 36–46. [Google Scholar] [CrossRef]
- Subramani, P.; Khor, M.-Z.; Urmila, B.; Parayil, V.-C. Ameliorative Effect of Curcumin on Olanzapine-induced Obesity in Sprague-Dawley Rats. Pharmacognosy. Res. 2017, 9, 247–252. [Google Scholar]
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications—A review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef]
- Silva, A.K.S.; Peixoto, C.A. Role of peroxisome proliferator-activated receptors in non-alcoholic fatty liver disease inflammation. Cell. Mol. Life Sci. 2018, 75, 2951–2961. [Google Scholar] [CrossRef]
- Zhang, F.; Kong, D.; Lu, Y.; Zheng, S. Peroxisome proliferator-activated receptor-γ as a therapeutic target for hepatic fibrosis: From bench to bedside. Cell. Mol. Life Sci. 2013, 70, 259–276. [Google Scholar] [CrossRef]
- Yang, M.D.; Chiang, Y.-M.; Higashiyama, R.; Asahina, K.; Mann, D.A.; Mann, J.; Wang, C.C.; Tsukamoto, H. Rosmarinic acid and baicalin epigenetically derepress peroxisomal proliferator-activated receptor γ in hepatic stellate cells for their antifibrotic effect. Hepatology 2012, 55, 1271–1281. [Google Scholar] [CrossRef]
- Xu, X.; Hu, J.; McGrath, B.C.; Cavener, D.R. GCN2 in the brain programs PPARγ2 and triglyceride storage in the liver during perinatal development in response to maternal dietary fat. PLoS ONE 2013, 8, e75917. [Google Scholar] [CrossRef]
- Yamazaki, T.; Kishimoto, K.; Miura, S.; Ezaki, O. Dietary β-conglycinin prevents fatty liver induced by a high-fat diet by a decrease in peroxisome proliferator-activated receptor γ2 protein. J. Nutr. Biochem. 2012, 23, 123–132. [Google Scholar] [CrossRef]
- Su, Y.; Liu, X.; Lian, J.; Deng, C. Epigenetic histone modulations of PPARγ and related pathways contribute to olanzapine-induced metabolic disorders. Pharmacol. Res. 2020, 155, 104703. [Google Scholar] [CrossRef]
- Bonnefont, J.P. Carnitine palmitoyltransferases 1 and 2: Biochemical, molecular and medical aspects. Mol. Aspects Med. 2004, 25, 495–520. [Google Scholar] [CrossRef]
- Schlaepfer, I.R.; Joshi, M. CPT1A-mediated Fat Oxidation, Mechanisms, and Therapeutic Potential. Endocrinology 2020, 161, bqz046. [Google Scholar] [CrossRef]
- Ren, Q.; Guo, M.; Yang, F.; Han, T.; Du, W.; Zhao, F.; Li, J.; Li, W.; Feng, Y.; Wang, S.; et al. Association of CPT1A gene polymorphism with the risk of gestational diabetes mellitus: A case-control study. J. Assist. Reprod. Genet. 2021, 38, 1861–1869. [Google Scholar] [CrossRef]
- Seok, S.; Kim, Y.-C.; Byun, S.; Choi, S.; Xiao, Z.; Iwamori, N.; Zhang, Y.; Wang, C.; Ma, J.; Ge, K.; et al. Fasting-induced JMJD3 histone demethylase epigenetically activates mitochondrial fatty acid β-oxidation. J. Clin. Investig. 2018, 128, 3144–3159. [Google Scholar] [CrossRef]
- Yang, Z.; Cappello, T.; Wang, L. Emerging role of microRNAs in lipid metabolism. Acta Pharm. Sin. B 2015, 5, 145–150. [Google Scholar] [CrossRef]
- Morgan, M.A.J.; Shilatifard, A. Reevaluating the roles of histone-modifying enzymes and their associated chromatin modifications in transcriptional regulation. Nat. Genet. 2020, 52, 1271–1281. [Google Scholar] [CrossRef]
- Demetriadou, C.; Koufaris, C.; Kirmizis, A. Histone N-alpha terminal modifications: Genome regulation at the tip of the tail. Epigenetics Chromatin 2020, 13, 29. [Google Scholar] [CrossRef] [PubMed]
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone lysine methylation dynamics: Establishment, regulation, and biological impact. Mol. Cell. 2012, 48, 491–507. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Jung, D.Y.; Nagappan, A.; Jung, M.H. Histone H3K9 demethylase JMJD2B induces hepatic steatosis through upregulation of PPARγ2. Sci. Rep. 2018, 8, 13734. [Google Scholar] [CrossRef] [PubMed]
- Moody, L.; Xu, G.B.; Chen, H.; Pan, Y.-X. Epigenetic regulation of carnitine palmitoyltransferase 1 (Cpt1a) by high fat diet. Biochim. Biophys. Acta (BBA)—Gene Regul. Mech. 2019, 1862, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Strakovsky, R.S.; Wang, H.; Engeseth, N.J.; Flaws, J.A.; Helferich, W.G.; Pan, Y.-X.; Lezmi, S. Developmental bisphenol A (BPA) exposure leads to sex-specific modification of hepatic gene expression and epigenome at birth that may exacerbate high-fat diet-induced hepatic steatosis. Toxicol. Appl. Pharmacol. 2015, 284, 101–112. [Google Scholar] [CrossRef]
- Husmann, D.; Gozani, O. Histone lysine methyltransferases in biology and disease. Nat. Struct. Mol. Biol. 2019, 26, 880–889. [Google Scholar] [CrossRef]
- Kumar, A.; Shalimar; Walia, G.K.; Gupta, V.; Sachdeva, M.P. Genetics of nonalcoholic fatty liver disease in Asian populations. J. Genet. 2019, 98, 29. [Google Scholar] [CrossRef]
- Wheeler, M.C.; Gekakis, N. Hsp90 modulates PPARγ activity in a mouse model of nonalcoholic fatty liver disease. J. Lipid. Res. 2014, 55, 1702–1710. [Google Scholar] [CrossRef]
- Staeker, J.; Leucht, S.; Steimer, W. Peroxisome proliferator-activated receptor gamma (PPARG) Pro12Ala: Lack of association with weight gain in psychiatric inpatients treated with olanzapine or clozapine. Mol. Diagn. Ther. 2012, 16, 93–98. [Google Scholar] [CrossRef]
- Brandl, E.J.; Tiwari, A.K.; Zai, C.C.; Chowdhury, N.I.; Lieberman, J.A.; Meltzer, H.Y.; Kennedy, J.L.; Müller, D.J. No evidence for a role of the peroxisome proliferator-activated receptor gamma (PPARG) and adiponectin (ADIPOQ) genes in antipsychotic-induced weight gain. Psychiatry Res. 2014, 219, 255–260. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, X.; Feng, X.; Liu, X.; Deng, C.; Hu, C.-H. Berberine Alleviates Olanzapine-Induced Adipogenesis via the AMPKα-SREBP Pathway in 3T3-L1 Cells. Int. J. Mol. Sci. 2016, 17, 1865. [Google Scholar] [CrossRef]
- Lee, J.E.; Schmidt, H.; Lai, B.; Ge, K. Transcriptional and Epigenomic Regulation of Adipogenesis. Mol. Cell. Biol. 2019, 39, e00601-18. [Google Scholar] [CrossRef]
- Siersbaek, R.; Nielsen, R.; Mandrup, S. PPARgamma in adipocyte differentiation and metabolism—Novel insights from genome-wide studies. FEBS Lett. 2010, 584, 3242–3249. [Google Scholar] [CrossRef]
- Kim, M.K.; Kim, S.H.; Yu, H.S.; Park, H.G.; Kang, U.G.; Ahn, Y.M.; Kim, Y.S. The effect of clozapine on the AMPK-ACC-CPT1 pathway in the rat frontal cortex. Int. J. Neuropsychopharmacol. 2012, 15, 907–917. [Google Scholar] [CrossRef]
- Lee, K.H.; Ju, U.-I.; Song, J.-Y.; Chun, Y.-S. The histone demethylase PHF2 promotes fat cell differentiation as an epigenetic activator of both C/EBPα and C/EBPδ. Mol. Cells. 2014, 37, 734–741. [Google Scholar] [CrossRef]
- Lan, F.; Zaratiegui, M.; Villén, J.; Vaughn, M.W.; Verdel, A.; Huarte, M.; Shi, Y.; Gygi, S.P.; Moazed, D.; Martienssen, R.A.; et al. S. pombe LSD1 homologs regulate heterochromatin propagation and euchromatic gene transcription. Mol. Cell. 2007, 26, 89–101. [Google Scholar] [CrossRef]
- Wang, L.; Jin, Q.; Lee, J.-E.; Su, I.-H.; Ge, K. Histone H3K27 methyltransferase Ezh2 represses Wnt genes to facilitate adipogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 7317–7322. [Google Scholar] [CrossRef]
- Weston-Green, K.; Huang, X.F.; Deng, C. Sensitivity of the female rat to olanzapine-induced weight gain--far from the clinic? Schizophr. Res. 2010, 116, 299–300. [Google Scholar] [CrossRef]
- Castellani, L.N.; Costa-Dookhan, K.A.; McIntyre, W.B.; Wright, D.C.; Flowers, S.A.; Hahn, M.K.; Ward, K.M. Preclinical and Clinical Sex Differences in Antipsychotic-Induced Metabolic Disturbances: A Narrative Review of Adiposity and Glucose Metabolism. J. Psychiatr. Brain. Sci. 2019, 4, e190013. [Google Scholar]
- Weston-Green, K.; Huang, X.F.; Deng, C. Olanzapine treatment and metabolic dysfunction: A dose response study in female Sprague Dawley rats. Behav. Brain. Res. 2011, 217, 337–346. [Google Scholar] [CrossRef]
- FDA. Estimating the Safe Starting Dose in Clinical Trials for Therapeutics in Adult Healthy Volunteers; U.S. FDA Center for Drug Evaluation and Research: Silver Spring, MD, USA, 2005.
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose translation from animal to human studies revisited. Faseb J. 2008, 22, 659–661. [Google Scholar] [CrossRef] [PubMed]
- Lecoutre, S.; Pourpe, C.; Butruille, L.; Marousez, L.; Laborie, C.; Guinez, C.; Lesage, J.; Vieau, D.; Eeckhoute, J.; Gabory, A.; et al. Reduced PPARγ2 expression in adipose tissue of male rat offspring from obese dams is associated with epigenetic modifications. Faseb J. 2018, 32, 2768–2778. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, Y.; Deng, C.; Liu, X.; Lian, J. Epigenetic Histone Methylation of PPARγ and CPT1A Signaling Contributes to Betahistine Preventing Olanzapine-Induced Dyslipidemia. Int. J. Mol. Sci. 2023, 24, 9143. https://doi.org/10.3390/ijms24119143
Su Y, Deng C, Liu X, Lian J. Epigenetic Histone Methylation of PPARγ and CPT1A Signaling Contributes to Betahistine Preventing Olanzapine-Induced Dyslipidemia. International Journal of Molecular Sciences. 2023; 24(11):9143. https://doi.org/10.3390/ijms24119143
Chicago/Turabian StyleSu, Yueqing, Chao Deng, Xuemei Liu, and Jiamei Lian. 2023. "Epigenetic Histone Methylation of PPARγ and CPT1A Signaling Contributes to Betahistine Preventing Olanzapine-Induced Dyslipidemia" International Journal of Molecular Sciences 24, no. 11: 9143. https://doi.org/10.3390/ijms24119143
APA StyleSu, Y., Deng, C., Liu, X., & Lian, J. (2023). Epigenetic Histone Methylation of PPARγ and CPT1A Signaling Contributes to Betahistine Preventing Olanzapine-Induced Dyslipidemia. International Journal of Molecular Sciences, 24(11), 9143. https://doi.org/10.3390/ijms24119143