
Glioblastoma Metabolism: Insights and Therapeutic Strategies
 , ,
, ,  and
and
Abstract
1. Introduction
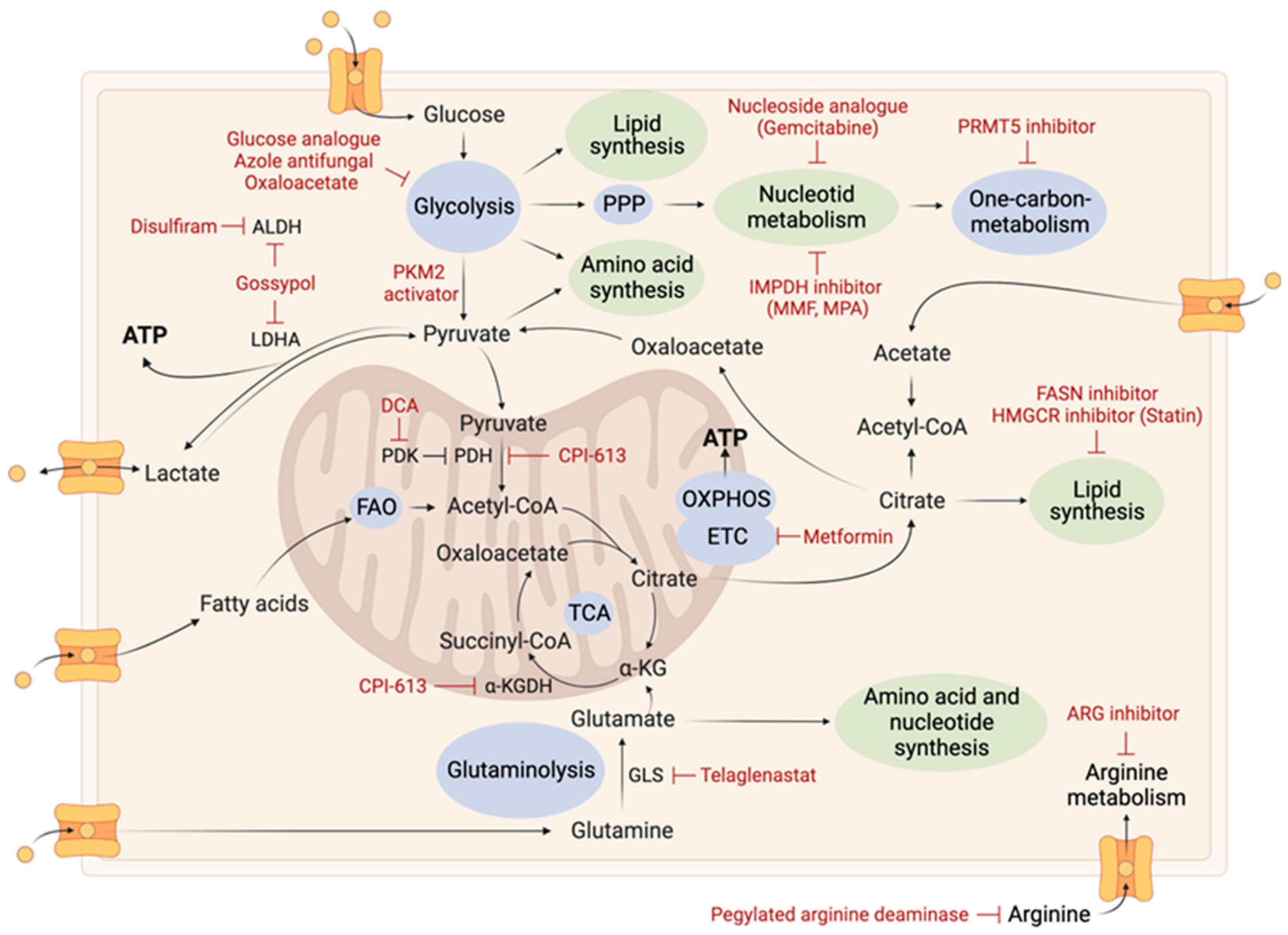
2. Tumoral Metabolic Reprogramming

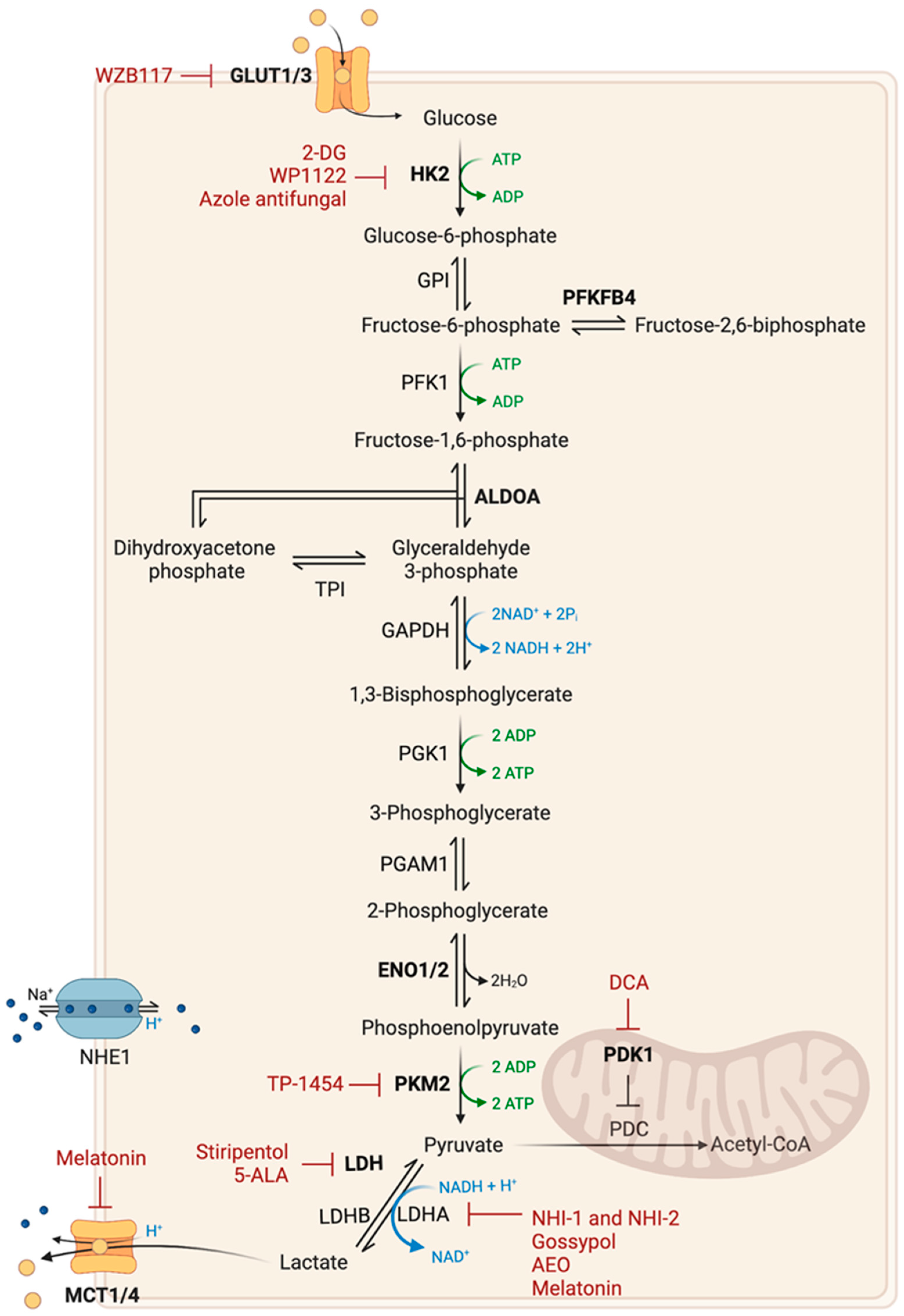
2.1. Glucose Metabolism
2.2. Amino Acid Metabolism
2.2.1. Glutamine Metabolism
2.2.2. Arginine Metabolism
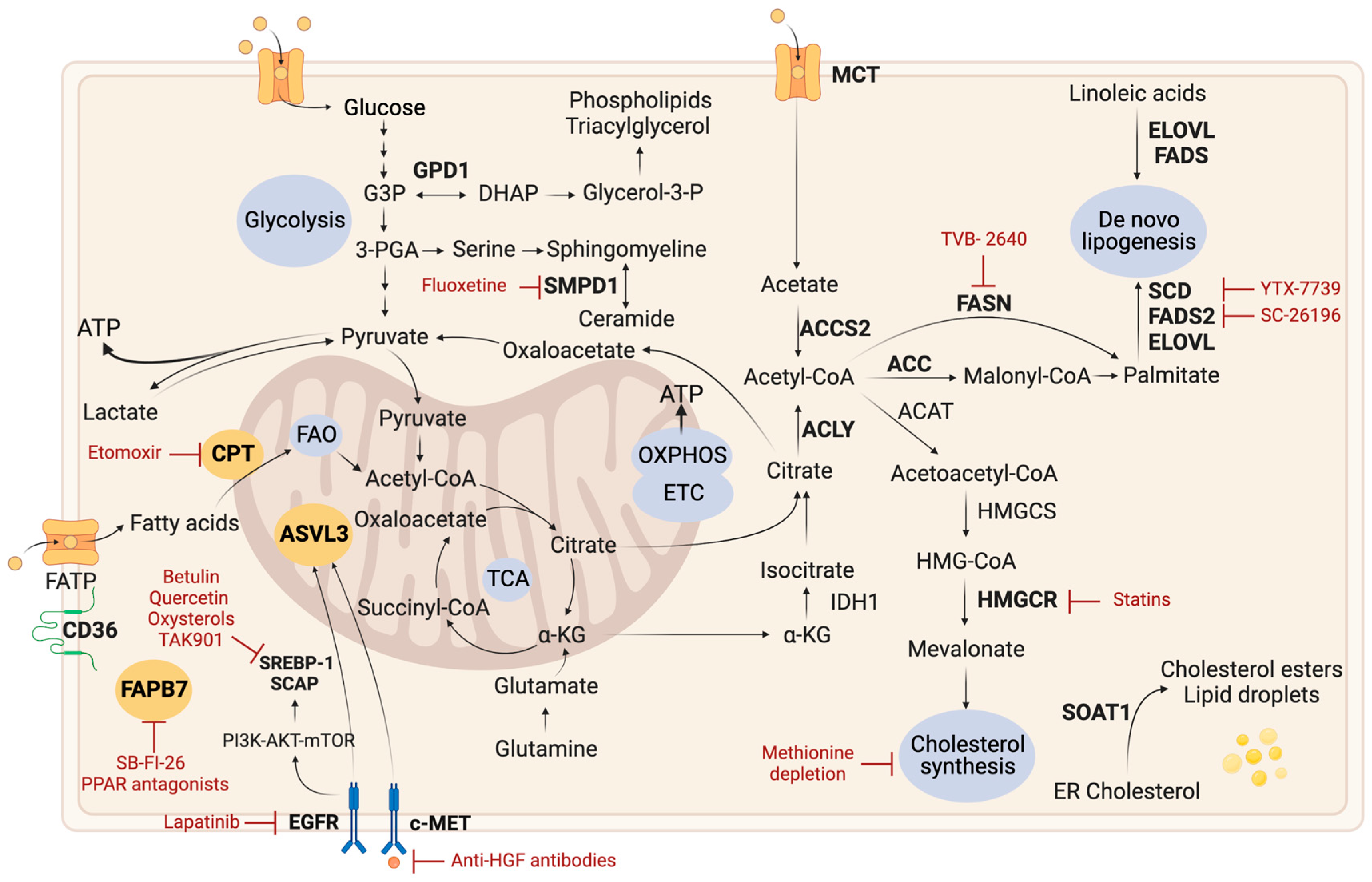
2.3. Lipid Metabolism
2.3.1. Lipid Droplets
2.3.2. De Novo Lipogenesis
2.3.3. Fatty Acid Oxidation
2.3.4. Cholesterol Metabolism
2.3.5. Other Lipid-Related Pathways
2.4. Nucleotide Metabolism
2.5. Monocarbon Metabolism
2.6. Nicotinamide Metabolism
2.7. TCA Cycle
2.8. Electron Transport Chain and Oxidative Phosphorylation
2.9. Transporters and Ion Channels
3. Complexity, Heterogeneity, and Plasticity of Tumor Metabolism
3.1. Metabolic Heterogeneity and Plasticity
3.2. Metabolic Interactions in the Tumor Microenvironment
4. Combination of Therapies
4.1. Metabolic-Related Therapy
4.2. Metabolic Therapies and Radiotherapy
4.3. Metabolic Therapies and Chemotherapy
4.4. Metabolic and Targeted Therapies
4.5. Metabolic Therapies and Immunotherapy
4.6. Diet Interventions
5. Discussion and Perspectives
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Jiang, Y.; Uhrbom, L. On the Origin of Glioma. Ups. J. Med. Sci. 2012, 117, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Bauchet, L.; Davis, F.G.; Deltour, I.; Fisher, J.L.; Langer, C.E.; Pekmezci, M.; Schwartzbaum, J.A.; Turner, M.C.; Walsh, K.M.; et al. The Epidemiology of Glioma in Adults: A State of the Science Review. Neuro Oncol. 2014, 16, 896–913. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; DeCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef] [PubMed]
- Cruz Da Silva, E.; Mercier, M.-C.; Etienne-Selloum, N.; Dontenwill, M.; Choulier, L. A Systematic Review of Glioblastoma-Targeted Therapies in Phases II, III, IV Clinical Trials. Cancers 2021, 13, 1795. [Google Scholar] [CrossRef] [PubMed]
- Garnier, D.; Renoult, O.; Alves-Guerra, M.-C.C.; Paris, F.; Pecqueur, C. Glioblastoma Stem-Like Cells, Metabolic Strategy to Kill a Challenging Target. Front. Oncol. 2019, 9, 118. [Google Scholar] [CrossRef]
- Zhou, W.; Wahl, D.R. Metabolic Abnormalities in Glioblastoma and Metabolic Strategies to Overcome Treatment Resistance. Cancers 2019, 11, 1231. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Libby, C.J.; Tran, A.N.; Scott, S.E.; Griguer, C.; Hjelmeland, A.B. The Pro-Tumorigenic Effects of Metabolic Alterations in Glioblastoma Including Brain Tumor Initiating Cells. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 175–188. [Google Scholar]
- Deshmukh, R.; Allega, M.F.; Tardito, S. A Map of the Altered Glioma Metabolism. Trends Mol. Med. 2021, 27, 1045–1059. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic Reprogramming and Cancer Progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef]
- Yang, K.; Rich, J.N. A Delicate Initiation: Lipolysis of Lipid Droplets Fuels Glioblastoma. Mol. Cell 2021, 81, 2686–2687. [Google Scholar] [CrossRef]
- Mashimo, T.; Pichumani, K.; Vemireddy, V.; Hatanpaa, K.J.; Singh, D.K.; Sirasanagandla, S.; Nannepaga, S.; Piccirillo, S.G.; Kovacs, Z.; Foong, C.; et al. Acetate Is a Bioenergetic Substrate for Human Glioblastoma and Brain Metastases. Cell 2014, 159, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; et al. Akt Stimulates Aerobic Glycolysis in Cancer Cells. Cancer Res. 2004, 64, 3892–3899. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic Glycolysis: Meeting the Metabolic Requirements of Cell Proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef]
- Maher, E.A.; Marin-Valencia, I.; Bachoo, R.M.; Mashimo, T.; Raisanen, J.; Hatanpaa, K.J.; Jindal, A.; Jeffrey, F.M.; Choi, C.; Madden, C.; et al. Metabolism of [U-13C]Glucose in Human Brain Tumors in Vivo. NMR Biomed. 2012, 25, 1234–1244. [Google Scholar] [CrossRef]
- Wise, D.R.; Thompson, C.B. Glutamine Addiction: A New Therapeutic Target in Cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef]
- Agnihotri, S.; Zadeh, G. Metabolic Reprogramming in Glioblastoma: The Influence of Cancer Metabolism on Epigenetics and Unanswered Questions. Neuro Oncol. 2016, 18, 160–172. [Google Scholar] [CrossRef]
- El Khayari, A.; Bouchmaa, N.; Taib, B.; Wei, Z.; Zeng, A.; El Fatimy, R. Metabolic Rewiring in Glioblastoma Cancer: EGFR, IDH and Beyond. Front. Oncol. 2022, 12, 901951. [Google Scholar] [CrossRef]
- Park, J.H.; Pyun, W.Y.; Park, H.W. Cancer Metabolism: Phenotype, Signaling and Therapeutic Targets. Cells 2020, 9, 2308. [Google Scholar] [CrossRef]
- Masui, K.; Tanaka, K.; Akhavan, D.; Babic, I.; Gini, B.; Matsutani, T.; Iwanami, A.; Liu, F.; Villa, G.R.; Gu, Y.; et al. MTOR Complex 2 Controls Glycolytic Metabolism in Glioblastoma through FoxO Acetylation and Upregulation of C-Myc. Cell Metab. 2013, 18, 726–739. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-Inducible Factors Regulate Tumorigenic Capacity of Glioma Stem Cells. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef]
- Heddleston, J.M.; Li, Z.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. The Hypoxic Microenvironment Maintains Glioblastoma Stem Cells and Promotes Reprogramming towards a Cancer Stem Cell Phenotype. Cell Cycle 2009, 8, 3274–3284. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The Biology of Cancer: Metabolic Reprogramming Fuels Cell Growth and Proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.; Xu, L.; Chen, Y.; Gohongi, T.; Seed, B.; Jain, R.K. Hypoxia and Acidosis Independently Up-Regulate Vascular Endothelial Growth Factor Transcription in Brain Tumors in Vivo. Cancer Res. 2001, 61, 6020–6024. [Google Scholar] [PubMed]
- de la Cruz-López, K.G.; Castro-Muñoz, L.J.; Reyes-Hernández, D.O.; García-Carrancá, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef]
- Moreno-Sánchez, R.; Rodríguez-Enríquez, S.; Marín-Hernández, A.; Saavedra, E. Energy Metabolism in Tumor Cells. FEBS J. 2007, 274, 1393–1418. [Google Scholar] [CrossRef] [PubMed]
- Dwarakanath, B.; Singh, D.; Banerji, A.; Sarin, R.; Venkataramana, N.; Jalali, R.; Vishwanath, P.; Mohanti, B.; Tripathi, R.; Kalia, V.; et al. Clinical Studies for Improving Radiotherapy with 2-Deoxy-D-Glucose: Present Status and Future Prospects. J. Cancer Res. Ther. 2009, 5, 21. [Google Scholar] [CrossRef]
- Singh, D.; Banerji, A.K.; Dwarakanath, B.S.; Tripathi, R.P.; Gupta, J.P.; Mathew, T.L.; Ravindranath, T.; Jain, V. Optimizing Cancer Radiotherapy with 2-Deoxy-D-Glucose: Dose Escalation Studies in Patients with Glioblastoma Multiforme. Strahlenther. Onkol. 2005, 181, 507–514. [Google Scholar] [CrossRef]
- Priebe, W.; Zielinski, R.; Fokt, I.; Felix, E.; Radjendirane, V.; Arumugam, J.; Tai Khuong, M.; Krasinski, M.; Skora, S. EXTH-07. design and evaluation of wp1122, an inhibitor of glycolysis with increased cns uptake. Neuro Oncol. 2018, 20, vi86. [Google Scholar] [CrossRef][Green Version]
- Labak, C.M.; Wang, P.Y.; Arora, R.; Guda, M.R.; Asuthkar, S.; Tsung, A.J.; Velpula, K.K. Glucose Transport: Meeting the Metabolic Demands of Cancer, and Applications in Glioblastoma Treatment. Am. J. Cancer Res. 2016, 6, 1599–1608. [Google Scholar]
- Christensen, D.R.; Calder, P.C.; Houghton, F.D. GLUT3 and PKM2 Regulate OCT4 Expression and Support the Hypoxic Culture of Human Embryonic Stem Cells. Sci. Rep. 2015, 5, 17500. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Wu, Q.; Hitomi, M.; Rahim, N.; Kim, Y.; Sloan, A.E.; Weil, R.J.; Nakano, I.; Sarkaria, J.N.; Stringer, B.W.; et al. Brain Tumor Initiating Cells Adapt to Restricted Nutrition through Preferential Glucose Uptake. Nat. Neurosci. 2013, 16, 1373–1382. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, K.; Okada, M.; Suzuki, S.; Seino, M.; Seino, S.; Takeda, H.; Kitanaka, C. Targeting the Facilitative Glucose Transporter GLUT1 Inhibits the Self-Renewal and Tumor-Initiating Capacity of Cancer Stem Cells. Oncotarget 2015, 6, 651–661. [Google Scholar] [CrossRef]
- Cosset, É.; Ilmjärv, S.; Dutoit, V.; Elliott, K.; von Schalscha, T.; Camargo, M.F.; Reiss, A.; Moroishi, T.; Seguin, L.; Gomez, G.; et al. Glut3 Addiction Is a Druggable Vulnerability for a Molecularly Defined Subpopulation of Glioblastoma. Cancer Cell 2017, 32, 856–868.e5. [Google Scholar] [CrossRef]
- Kuang, R.; Jahangiri, A.; Mascharak, S.; Nguyen, A.; Chandra, A.; Flanigan, P.M.; Yagnik, G.; Wagner, J.R.; De Lay, M.; Carrera, D.; et al. GLUT3 Upregulation Promotes Metabolic Reprogramming Associated with Antiangiogenic Therapy Resistance. JCI Insight 2017, 2, e88815. [Google Scholar] [CrossRef]
- Kwak, S.; Park, S.-H.; Kim, S.-H.; Sung, G.-J.; Song, J.-H.; Jeong, J.-H.; Kim, H.; Ha, C.H.; Kim, S.W.; Choi, K.-C. MiR-3189-Targeted GLUT3 Repression by HDAC2 Knockdown Inhibits Glioblastoma Tumorigenesis through Regulating Glucose Metabolism and Proliferation. J. Exp. Clin. Cancer Res. 2022, 41, 87. [Google Scholar] [CrossRef]
- Goidts, V.; Bageritz, J.; Puccio, L.; Nakata, S.; Zapatka, M.; Barbus, S.; Toedt, G.; Campos, B.; Korshunov, A.; Momma, S.; et al. RNAi Screening in Glioma Stem-like Cells Identifies PFKFB4 as a Key Molecule Important for Cancer Cell Survival. Oncogene 2012, 31, 3235–3243. [Google Scholar] [CrossRef]
- Sanzey, M.; Abdul Rahim, S.A.; Oudin, A.; Dirkse, A.; Kaoma, T.; Vallar, L.; Herold-Mende, C.; Bjerkvig, R.; Golebiewska, A.; Niclou, S.P. Comprehensive Analysis of Glycolytic Enzymes as Therapeutic Targets in the Treatment of Glioblastoma. PLoS ONE 2015, 10, e0123544. [Google Scholar] [CrossRef]
- Wolf, A.; Agnihotri, S.; Micallef, J.; Mukherjee, J.; Sabha, N.; Cairns, R.; Hawkins, C.; Guha, A. Hexokinase 2 Is a Key Mediator of Aerobic Glycolysis and Promotes Tumor Growth in Human Glioblastoma Multiforme. J. Exp. Med. 2011, 208, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Agnihotri, S.; Mansouri, S.; Burrell, K.; Li, M.; Mamatjan, Y.; Liu, J.; Nejad, R.; Kumar, S.; Jalali, S.; Singh, S.K.; et al. Ketoconazole and Posaconazole Selectively Target HK2-Expressing Glioblastoma Cells. Clin. Cancer Res. 2019, 25, 844–855. [Google Scholar] [CrossRef]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 Splice Isoform of Pyruvate Kinase Is Important for Cancer Metabolism and Tumour Growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Xia, Y.; Hawke, D.; Li, X.; Liang, J.; Xing, D.; Aldape, K.; Hunter, T.; Alfred Yung, W.K.; Lu, Z. PKM2 Phosphorylates Histone H3 and Promotes Gene Transcription and Tumorigenesis. Cell 2012, 150, 685–696. [Google Scholar] [CrossRef]
- Liang, J.; Cao, R.; Wang, X.; Zhang, Y.; Wang, P.; Gao, H.; Li, C.; Yang, F.; Zeng, R.; Wei, P.; et al. Mitochondrial PKM2 Regulates Oxidative Stress-Induced Apoptosis by Stabilizing Bcl2. Cell Res. 2017, 27, 329–351. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Yang, J.; Gong, H.; Lin, Y.; Liu, J. Trametinib Inhibits the Growth and Aerobic Glycolysis of Glioma Cells by Targeting the PKM2/c-Myc Axis. Front. Pharmacol. 2021, 12, 760055. [Google Scholar] [CrossRef]
- Muller, F.L.; Colla, S.; Aquilanti, E.; Manzo, V.E.; Genovese, G.; Lee, J.; Eisenson, D.; Narurkar, R.; Deng, P.; Nezi, L.; et al. Passenger Deletions Generate Therapeutic Vulnerabilities in Cancer. Nature 2012, 488, 337–342. [Google Scholar] [CrossRef]
- Larrieu, C.M.; Storevik, S.; Guyon, J.; Pagano Zottola, A.C.; Bouchez, C.L.; Derieppe, M.-A.; Tan, T.Z.; Miletic, H.; Lorens, J.; Tronstad, K.J.; et al. Refining the Role of Pyruvate Dehydrogenase Kinases in Glioblastoma Development. Cancers 2022, 14, 3769. [Google Scholar] [CrossRef]
- Shen, H.; Hau, E.; Joshi, S.; Dilda, P.J.; McDonald, K.L. Sensitization of Glioblastoma Cells to Irradiation by Modulating the Glucose Metabolism. Mol. Cancer Ther. 2015, 14, 1794–1804. [Google Scholar] [CrossRef]
- Valvona, C.J.; Fillmore, H.L.; Nunn, P.B.; Pilkington, G.J. The Regulation and Function of Lactate Dehydrogenase A: Therapeutic Potential in Brain Tumor. Brain Pathol. 2016, 26, 3–17. [Google Scholar] [CrossRef]
- Kim, J.; Han, J.; Jang, Y.; Kim, S.J.; Lee, M.J.; Ryu, M.J.; Kweon, G.R.; Heo, J.Y. High-Capacity Glycolytic and Mitochondrial Oxidative Metabolisms Mediate the Growth Ability of Glioblastoma. Int. J. Oncol. 2015, 47, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Daniele, S.; Giacomelli, C.; Zappelli, E.; Granchi, C.; Trincavelli, M.L.; Minutolo, F.; Martini, C. Lactate Dehydrogenase-A Inhibition Induces Human Glioblastoma Multiforme Stem Cell Differentiation and Death. Sci. Rep. 2015, 5, 15556. [Google Scholar] [CrossRef]
- Guyon, J.; Fernandez-Moncada, I.; Larrieu, C.; Bouchez, C.; Chouleur, T.; Espedal, H.; Røsland, G.; Daher, B.; Barre, A.; Dartigues, B. Specific Expression of Lactate Dehydrogenases in Glioblastoma Controls Intercellular Lactate Transfer to Promote Tumor Growth and Invasion. Res. Sq. 2021. [Google Scholar] [CrossRef]
- Guyon, J.; Fernandez-Moncada, I.; Larrieu, C.M.; Bouchez, C.L.; Pagano Zottola, A.C.; Galvis, J.; Chouleur, T.; Burban, A.; Joseph, K.; Ravi, V.M.; et al. Lactate Dehydrogenases Promote Glioblastoma Growth and Invasion via a Metabolic Symbiosis. EMBO Mol. Med. 2022, 14, e15343. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Lee, B.I.; Jeon, J.H.; Kim, D.K.; Kang, S.G.; Shim, J.K.; Kim, S.Y.; Kang, S.W.; Jang, H. Gossypol Suppresses Growth of Temozolomide-Resistant Glioblastoma Tumor Spheres. Biomolecules 2019, 9, 595. [Google Scholar] [CrossRef]
- Fiveash, J.B.; Chowdhary, S.A.; Peereboom, D.; Mikkelsen, T.; Nabors, L.B.; Lesser, G.J.; Rosenfeld, M.R.; Ye, X.; Grossman, S.A. NABTT-0702: A Phase II Study of R-(-)-Gossypol (AT-101) in Recurrent Glioblastoma Multiforme (GBM). J. Clin. Oncol. 2009, 27, 2010. [Google Scholar] [CrossRef]
- Grigalavicius, M.; Ezzatpanah, S.; Papakyriakou, A.; Raabe, T.T.H.; Yannakopoulou, K.; Theodossiou, T.A. 5-ALA Is a Potent Lactate Dehydrogenase Inhibitor but Not a Substrate: Implications for Cell Glycolysis and New Avenues in 5-ALA-Mediated Anticancer Action. Cancers 2022, 14, 4003. [Google Scholar] [CrossRef]
- Fernandez-Gil, B.I.; Otamendi-Lopez, A.; Bechtle, A.; Vazquez-Ramos, C.A.; Qosja, N.; Suarez-Meade, P.; Sarabia-Estrada, R.; Jentoft, M.E.; Guerrero-Cázares, H.; Escames, G.; et al. Melatonin Treatment Triggers Metabolic and Intracellular PH Imbalance in Glioblastoma. Cells 2022, 11, 3467. [Google Scholar] [CrossRef]
- Lu, G.; Wang, X.; Li, F.; Wang, S.; Zhao, J.; Wang, J.; Liu, J.; Lyu, C.; Ye, P.; Tan, H.; et al. Engineered Biomimetic Nanoparticles Achieve Targeted Delivery and Efficient Metabolism-Based Synergistic Therapy against Glioblastoma. Nat. Commun. 2022, 13, 4214. [Google Scholar] [CrossRef] [PubMed]
- Ijare, O.; Conway, D.; Cash, A.; Baskin, D.; Pichumani, K. CBMT-49. Oxaloacetate Alters Glucose Metabolism in Glioblastoma: 13C Isotopomer Study. Neuro Oncol. 2019, 21, vi43–vi44. [Google Scholar] [CrossRef]
- Ruban, A.; Berkutzki, T.; Cooper, I.; Mohar, B.; Teichberg, V.I. Blood Glutamate Scavengers Prolong the Survival of Rats and Mice with Brain-Implanted Gliomas. Investig. New Drugs 2012, 30, 2226–2235. [Google Scholar] [CrossRef]
- Natarajan, S.K.; Venneti, S. Glutamine Metabolism in Brain Tumors. Cancers 2019, 11, 1628. [Google Scholar] [CrossRef] [PubMed]
- Kucharzewska, P.; Christianson, H.C.; Belting, M. Global Profiling of Metabolic Adaptation to Hypoxic Stress in Human Glioblastoma Cells. PLoS ONE 2015, 10, e0116740. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond Aerobic Glycolysis: Transformed Cells Can Engage in Glutamine Metabolism That Exceeds the Requirement for Protein and Nucleotide Synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef]
- Rosati, A.; Poliani, P.L.; Todeschini, A.; Cominelli, M.; Medicina, D.; Cenzato, M.; Simoncini, E.L.; Magrini, S.M.; Buglione, M.; Grisanti, S.; et al. Glutamine Synthetase Expression as a Valuable Marker of Epilepsy and Longer Survival in Newly Diagnosed Glioblastoma Multiforme. Neuro Oncol. 2013, 15, 618–625. [Google Scholar] [CrossRef]
- Tardito, S.; Oudin, A.; Ahmed, S.U.; Fack, F.; Keunen, O.; Zheng, L.; Miletic, H.; Sakariassen, P.Ø.; Weinstock, A.; Wagner, A.; et al. Glutamine Synthetase Activity Fuels Nucleotide Biosynthesis and Supports Growth of Glutamine-Restricted Glioblastoma. Nat. Cell Biol. 2015, 17, 1556–1568. [Google Scholar] [CrossRef]
- Obara-Michlewska, M.; Szeliga, M. Targeting Glutamine Addiction in Gliomas. Cancers 2020, 12, 310. [Google Scholar] [CrossRef]
- Cheng, T.; Sudderth, J.; Yang, C.; Mullen, A.R.; Jin, E.S.; Mates, J.M.; DeBerardinis, R.J. Pyruvate Carboxylase Is Required for Glutamine-Independent Growth of Tumor Cells. Proc. Natl. Acad. Sci. USA 2011, 108, 8674–8679. [Google Scholar] [CrossRef]
- Oizel, K.; Chauvin, C.; Oliver, L.; Gratas, C.; Geraldo, F.; Jarry, U.; Scotet, E.; Rabe, M.; Alves-Guerra, M.C.; Teusan, R.; et al. Efficient Mitochondrial Glutamine Targeting Prevails over Glioblastoma Metabolic Plasticity. Clin. Cancer Res. 2017, 23, 6292–6305. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; Deberardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.-Y.X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc Regulates a Transcriptional Program That Stimulates Mitochondrial Glutaminolysis and Leads to Glutamine Addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Sasayama, T.; Irino, Y.; Takata, K.; Nagashima, H.; Satoh, N.; Kyotani, K.; Mizowaki, T.; Imahori, T.; Ejima, Y.; et al. Compensatory Glutamine Metabolism Promotes Glioblastoma Resistance to MTOR Inhibitor Treatment. J. Clin. Investig. 2015, 125, 1591–1602. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, G.; Mao, Q.; Li, S.; Xiong, W.; Lin, Y.; Ge, J. Glutamate Dehydrogenase (GDH) Regulates Bioenergetics and Redox Homeostasis in Human Glioma. Oncotarget 2016, 5, 1–12. [Google Scholar] [CrossRef]
- Franceschi, S.; Corsinovi, D.; Lessi, F.; Tantillo, E.; Aretini, P.; Menicagli, M.; Scopelliti, C.; Civita, P.; Pasqualetti, F.; Naccarato, A.G.; et al. Mitochondrial Enzyme GLUD2 Plays a Critical Role in Glioblastoma Progression. EBioMedicine 2018, 37, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Przystal, J.M.; Hajji, N.; Khozoie, C.; Renziehausen, A.; Zeng, Q.; Abaitua, F.; Hajitou, A.; Suwan, K.; Want, E.; Bomalaski, J.; et al. Efficacy of Arginine Depletion by ADI-PEG20 in an Intracranial Model of GBM. Cell Death Dis. 2018, 9, 1192. [Google Scholar] [CrossRef]
- Hall, P.E.; Lewis, R.; Syed, N.; Shaffer, R.; Evanson, J.; Ellis, S.; Williams, M.; Feng, X.; Johnston, A.; Thomson, J.A.; et al. A Phase I Study of Pegylated Arginine Deiminase (Pegargiminase), Cisplatin, and Pemetrexed in Argininosuccinate Synthetase 1-Deficient Recurrent High-Grade Glioma. Clin. Cancer Res. 2019, 25, 2708–2716. [Google Scholar] [CrossRef]
- Hou, X.; Chen, S.; Zhang, P.; Guo, D.; Wang, B. Targeted Arginine Metabolism Therapy: A Dilemma in Glioma Treatment. Front. Oncol. 2022, 12, 3484. [Google Scholar] [CrossRef]
- Broadfield, L.A.; Pane, A.A.; Talebi, A.; Swinnen, J.V.; Fendt, S.-M. Lipid Metabolism in Cancer: New Perspectives and Emerging Mechanisms. Dev. Cell 2021, 56, 1363–1393. [Google Scholar] [CrossRef]
- Hoang-Minh, L.B.; Siebzehnrubl, F.A.; Yang, C.; Suzuki-Hatano, S.; Dajac, K.; Loche, T.; Andrews, N.; Schmoll Massari, M.; Patel, J.; Amin, K.; et al. Infiltrative and Drug-resistant Slow-cycling Cells Support Metabolic Heterogeneity in Glioblastoma. EMBO J. 2018, 37, e98772. [Google Scholar] [CrossRef]
- Guo, D. Lipid Droplets, Potential Biomarker and Metabolic Target in Glioblastoma. Intern. Med. Rev. 2017, 3. [Google Scholar] [CrossRef]
- Liu, R.; Lee, J.-H.; Li, J.; Yu, R.; Tan, L.; Xia, Y.; Zheng, Y.; Bian, X.-L.; Lorenzi, P.L.; Chen, Q.; et al. Choline Kinase Alpha 2 Acts as a Protein Kinase to Promote Lipolysis of Lipid Droplets. Mol. Cell 2021, 81, 2722–2735.e9. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Bell, E.H.; Chakravarti, A. Lipid Metabolism Emerges as a Promising Target for Malignant Glioma Therapy. CNS Oncol. 2013, 2, 289–299. [Google Scholar] [CrossRef]
- Jones, J.E.C.; Esler, W.P.; Patel, R.; Lanba, A.; Vera, N.B.; Pfefferkorn, J.A.; Vernochet, C. Inhibition of Acetyl-CoA Carboxylase 1 (ACC1) and 2 (ACC2) Reduces Proliferation and De Novo Lipogenesis of EGFRvIII Human Glioblastoma Cells. PLoS ONE 2017, 12, e0169566. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Kridel, S.; Thorburn, A.; Kooshki, M.; Little, J.; Hebbar, S.; Robbins, M. Fatty Acid Synthase: A Novel Target for Antiglioma Therapy. Br. J. Cancer 2006, 95, 869–878. [Google Scholar] [CrossRef]
- Grube, S.; Dünisch, P.; Freitag, D.; Klausnitzer, M.; Sakr, Y.; Walter, J.; Kalff, R.; Ewald, C. Overexpression of Fatty Acid Synthase in Human Gliomas Correlates with the WHO Tumor Grade and Inhibition with Orlistat Reduces Cell Viability and Triggers Apoptosis. J. Neurooncol. 2014, 118, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Yasumoto, Y.; Miyazaki, H.; Vaidyan, L.K.; Kagawa, Y.; Ebrahimi, M.; Yamamoto, Y.; Ogata, M.; Katsuyama, Y.; Sadahiro, H.; Suzuki, M.; et al. Inhibition of Fatty Acid Synthase Decreases Expression of Stemness Markers in Glioma Stem Cells. PLoS ONE 2016, 11, e0147717. [Google Scholar] [CrossRef]
- Gimple, R.C.; Kidwell, R.L.; Kim, L.J.Y.; Sun, T.; Gromovsky, A.D.; Wu, Q.; Wolf, M.; Lv, D.; Bhargava, S.; Jiang, L.; et al. Glioma Stem Cell–Specific Superenhancer Promotes Polyunsaturated Fatty-Acid Synthesis to Support EGFR Signaling. Cancer Discov. 2019, 9, 1248–1267. [Google Scholar] [CrossRef]
- Eyme, K.M.; Sammarco, A.; Jha, R.; Mnatsakanyan, H.; Pechdimaljian, C.; Carvalho, L.; Neustadt, R.; Moses, C.; Alnasser, A.; Tardiff, D.F.; et al. Targeting de Novo Lipid Synthesis Induces Lipotoxicity and Impairs DNA Damage Repair in Glioblastoma Mouse Models. Sci. Transl. Med. 2023, 15, eabq6288. [Google Scholar] [CrossRef]
- Parik, S.; Fernández-García, J.; Lodi, F.; De Vlaminck, K.; Derweduwe, M.; De Vleeschouwer, S.; Sciot, R.; Geens, W.; Weng, L.; Bosisio, F.M.; et al. GBM Tumors Are Heterogeneous in Their Fatty Acid Metabolism and Modulating Fatty Acid Metabolism Sensitizes Cancer Cells Derived from Recurring GBM Tumors to Temozolomide. Front. Oncol. 2022, 12, 4708. [Google Scholar] [CrossRef]
- Morihiro, Y.; Yasumoto, Y.; Vaidyan, L.K.; Sadahiro, H.; Uchida, T.; Inamura, A.; Sharifi, K.; Ideguchi, M.; Nomura, S.; Tokuda, N.; et al. Fatty Acid Binding Protein 7 as a Marker of Glioma Stem Cells. Pathol. Int. 2013, 63, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Patel, S.; Affeck, V.S.; Wilson, I.; Turnbull, D.M.; Joshi, A.R.; Maxwell, R.; Stoll, E.A.; Affleck, V.S.; Wilson, I.; et al. Fatty Acid Oxidation Is Required for the Respiration and Proliferation of Malignant Glioma Cells. Neuro Oncol. 2017, 19, 43–54. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, A.; Pellegatta, S.; Rossi, M.; Tunici, P.; Magnoni, L.; Speranza, M.C.; Malusa, F.; Miragliotta, V.; Mori, E.; Finocchiaro, G.; et al. A Radial Glia Gene Marker, Fatty Acid Binding Protein 7 (FABP7), Is Involved in Proliferation and Invasion of Glioblastoma Cells. PLoS ONE 2012, 7, e52113. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Xie, B.; Xiao, W.; Fan, M.; Xu, S.; Duan, Y.; Hamsafar, Y.; Evans, A.C.; Huang, J.; Zhou, W.; et al. Fatty Acid Oxidation Fuels Glioblastoma Radioresistance with CD47-Mediated Immune Evasion. Nat. Commun. 2022, 13, 1511. [Google Scholar] [CrossRef]
- Ahmad, F.; Sun, Q.; Patel, D.; Stommel, J.M. Cholesterol Metabolism: A Potential Therapeutic Target in Glioblastoma. Cancers 2019, 11, 146. [Google Scholar] [CrossRef]
- Villa, G.R.; Hulce, J.J.; Zanca, C.; Bi, J.; Ikegami, S.; Cahill, G.L.; Gu, Y.; Lum, K.M.; Masui, K.; Yang, H.; et al. An LXR-Cholesterol Axis Creates a Metabolic Co-Dependency for Brain Cancers. Cancer Cell 2016, 30, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Huang, Z.; Wu, Q.; Prager, B.C.; Mack, S.C.; Yang, K.; Kim, L.J.Y.; Gimple, R.C.; Shi, Y.; Lai, S.; et al. MYC-Regulated Mevalonate Metabolism Maintains Brain Tumor–Initiating Cells. Cancer Res. 2017, 77, 4947–4960. [Google Scholar] [CrossRef] [PubMed]
- Altwairgi, A.K.; Alghareeb, W.A.; AlNajjar, F.H.; Alhussain, H.; Alsaeed, E.; Balbaid, A.A.O.; Aldanan, S.; Orz, Y.; Alsharm, A.A. Atorvastatin in Combination with Radiotherapy and Temozolomide for Glioblastoma: A Prospective Phase II Study. Investig. New Drugs 2021, 39, 226–231. [Google Scholar] [CrossRef]
- Lo, H.-W. Targeting Ras-RAF-ERK and Its Interactive Pathways as a Novel Therapy for Malignant Gliomas. Curr. Cancer Drug Targets 2010, 10, 840–848. [Google Scholar] [CrossRef]
- Ma, Y.; Gong, Y.; Cheng, Z.; Loganathan, S.; Kao, C.; Sarkaria, J.N.; Abel, T.W.; Wang, J. Critical Functions of RhoB in Support of Glioblastoma Tumorigenesis. Neuro Oncol. 2015, 17, 516–525. [Google Scholar] [CrossRef]
- Al-Koussa, H.; El Atat, O.; Jaafar, L.; Tashjian, H.; El-Sibai, M. The Role of Rho GTPases in Motility and Invasion of Glioblastoma Cells. Anal. Cell. Pathol. 2020, 2020, 9274016. [Google Scholar] [CrossRef]
- Yanae, M.; Tsubaki, M.; Satou, T.; Itoh, T.; Imano, M.; Yamazoe, Y.; Nishida, S. Statin-Induced Apoptosis via the Suppression of ERK1/2 and Akt Activation by Inhibition of the Geranylgeranyl-Pyrophosphate Biosynthesis in Glioblastoma. J. Exp. Clin. Cancer Res. 2011, 30, 74. [Google Scholar] [CrossRef] [PubMed]
- Yokogami, K.; Kikuchi, T.; Watanabe, T.; Nakatake, Y.; Yamashita, S.; Mizuguchi, A.; Takeshima, H. Methionine Regulates Self-Renewal, Pluripotency, and Cell Death of GIC through Cholesterol—RRNA Axis. BMC Cancer 2022, 22, 1351. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Xia, S.; Lal, B.; Shi, X.; Yang, K.S.; Watkins, P.A.; Laterra, J. Lipid Metabolism Enzyme ACSVL3 Supports Glioblastoma Stem Cell Maintenance and Tumorigenicity. BMC Cancer 2014, 14, 401. [Google Scholar] [CrossRef] [PubMed]
- Pei, Z.; Sun, P.; Huang, P.; Lal, B.; Laterra, J.; Watkins, P.A. Acyl-CoA Synthetase VL3 Knockdown Inhibits Human Glioma Cell Proliferation and Tumorigenicity. Cancer Res. 2009, 69, 9175–9182. [Google Scholar] [CrossRef]
- Bi, J.; Khan, A.; Tang, J.; Armando, A.M.; Wu, S.; Zhang, W.; Gimple, R.C.; Reed, A.; Jing, H.; Koga, T.; et al. Targeting Glioblastoma Signaling and Metabolism with a Re-Purposed Brain-Penetrant Drug. Cell Rep. 2021, 37, 109957. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Prins, R.M.; Dang, J.; Kuga, D.; Iwanami, A.; Soto, H.; Lin, K.Y.; Huang, T.T.; Akhavan, D.; Hock, M.B.; et al. EGFR Signaling Through an Akt-SREBP-1–Dependent, Rapamycin-Resistant Pathway Sensitizes Glioblastomas to Antilipogenic Therapy. Sci. Signal. 2009, 2, ra82. [Google Scholar] [CrossRef]
- Cheng, C.; Ru, P.; Geng, F.; Liu, J.; Yoo, J.Y.; Wu, X.; Cheng, X.; Euthine, V.; Hu, P.; Guo, J.Y.; et al. Glucose-Mediated N-Glycosylation of SCAP Is Essential for SREBP-1 Activation and Tumor Growth. Cancer Cell 2015, 28, 569–581. [Google Scholar] [CrossRef]
- Sassi, K.; Nury, T.; Samadi, M.; Fennira, F.B.-A.; Vejux, A.; Lizard, G. Cholesterol Derivatives as Promising Anticancer Agents in Glioblastoma Metabolic Therapy. In Gliomas; Exon Publications: Brisbane, Australia, 2021; pp. 97–120. [Google Scholar]
- Zhan, X.; Qiu, R.; He, Y.; Zhao, Z.; Huang, M.; Liu, Q.; Zhi, F.; Long, W. The Aurora Kinase Inhibitor TAK901 Inhibits Glioblastoma Growth by Blocking SREBP1-Mediated Lipid Metabolism. Cancers 2022, 14, 5805. [Google Scholar] [CrossRef]
- Geng, F.; Cheng, X.; Wu, X.; Yoo, J.Y.; Cheng, C.; Guo, J.Y.; Mo, X.; Ru, P.; Hurwitz, B.; Kim, S.-H.; et al. Inhibition of SOAT1 Suppresses Glioblastoma Growth via Blocking SREBP-1–Mediated Lipogenesis. Clin. Cancer Res. 2016, 22, 5337–5348. [Google Scholar] [CrossRef]
- Rusu, P.; Shao, C.; Neuerburg, A.; Acikgöz, A.A.; Wu, Y.; Zou, P.; Phapale, P.; Shankar, T.S.; Döring, K.; Dettling, S.; et al. GPD1 Specifically Marks Dormant Glioma Stem Cells with a Distinct Metabolic Profile. Cell Stem Cell 2019, 25, 241–257.e8. [Google Scholar] [CrossRef] [PubMed]
- Hale, J.S.; Otvos, B.; Sinyuk, M.; Alvarado, A.G.; Hitomi, M.; Stoltz, K.; Wu, Q.; Flavahan, W.; Levison, B.; Johansen, M.L.; et al. Cancer Stem Cell-Specific Scavenger Receptor CD36 Drives Glioblastoma Progression. Stem Cells 2014, 32, 1746–1758. [Google Scholar] [CrossRef]
- Guo, D.; Hildebrandt, I.J.; Prins, R.M.; Soto, H.; Mazzotta, M.M.; Dang, J.; Czernin, J.; Shyy, J.Y.-J.; Watson, A.D.; Phelps, M.; et al. The AMPK Agonist AICAR Inhibits the Growth of EGFRvIII-Expressing Glioblastomas by Inhibiting Lipogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 12932–12937. [Google Scholar] [CrossRef] [PubMed]
- Laks, D.R.; Ta, L.; Crisman, T.J.; Gao, F.; Coppola, G.; Radu, C.G.; Nathanson, D.A.; Kornblum, H.I. Inhibition of Nucleotide Synthesis Targets Brain Tumor Stem Cells in a Subset of Glioblastoma. Mol. Cancer Ther. 2016, 15, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Echizenya, S.; Ishii, Y.; Kitazawa, S.; Tanaka, T.; Matsuda, S.; Watanabe, E.; Umekawa, M.; Terasaka, S.; Houkin, K.; Hatta, T.; et al. Discovery of a New Pyrimidine Synthesis Inhibitor Eradicating Glioblastoma-Initiating Cells. Neuro Oncol. 2020, 22, 229–239. [Google Scholar] [CrossRef]
- Lafita-Navarro, M.C.; Venkateswaran, N.; Kilgore, J.A.; Kanji, S.; Han, J.; Barnes, S.; Williams, N.S.; Buszczak, M.; Burma, S.; Conacci-Sorrell, M. Inhibition of the de Novo Pyrimidine Biosynthesis Pathway Limits Ribosomal RNA Transcription Causing Nucleolar Stress in Glioblastoma Cells. PLOS Genet. 2020, 16, e1009117. [Google Scholar] [CrossRef] [PubMed]
- Spina, R.; Mills, I.; Ahmad, F.; Chen, C.; Ames, H.M.; Winkles, J.A.; Woodworth, G.F.; Bar, E.E. DHODH Inhibition Impedes Glioma Stem Cell Proliferation, Induces DNA Damage, and Prolongs Survival in Orthotopic Glioblastoma Xenografts. Oncogene 2022, 41, 5361–5372. [Google Scholar] [CrossRef]
- Wang, X.; Yang, K.; Wu, Q.; Kim, L.J.Y.Y.; Morton, A.R.; Gimple, R.C.; Prager, B.C.; Shi, Y.; Zhou, W.; Bhargava, S.; et al. Targeting Pyrimidine Synthesis Accentuates Molecular Therapy Response in Glioblastoma Stem Cells. Sci. Transl. Med. 2019, 11, eaau4972. [Google Scholar] [CrossRef]
- Nikaki, A.; Angelidis, G.; Efthimiadou, R.; Tsougos, I.; Valotassiou, V.; Fountas, K.; Prasopoulos, V.; Georgoulias, P. 18F-Fluorothymidine PET Imaging in Gliomas: An Update. Ann. Nucl. Med. 2017, 31, 495–505. [Google Scholar] [CrossRef]
- Wang, X.; Yang, K.; Xie, Q.; Wu, Q.; Mack, S.C.; Shi, Y.; Kim, L.J.Y.; Prager, B.C.; Flavahan, W.A.; Liu, X.; et al. Purine Synthesis Promotes Maintenance of Brain Tumor Initiating Cells in Glioma. Nat. Neurosci. 2017, 20, 661–673. [Google Scholar] [CrossRef]
- Zhou, W.; Yao, Y.; Scott, A.J.; Wilder-Romans, K.; Dresser, J.J.; Werner, C.K.; Sun, H.; Pratt, D.; Sajjakulnukit, P.; Zhao, S.G.; et al. Purine Metabolism Regulates DNA Repair and Therapy Resistance in Glioblastoma. Nat. Commun. 2020, 11, 3811. [Google Scholar] [CrossRef]
- Shireman, J.M.; Atashi, F.; Lee, G.; Ali, E.S.; Saathoff, M.R.; Park, C.H.; Savchuk, S.; Baisiwala, S.; Miska, J.; Lesniak, M.S.; et al. De Novo Purine Biosynthesis Is a Major Driver of Chemoresistance in Glioblastoma. Brain 2021, 144, 1230–1246. [Google Scholar] [CrossRef] [PubMed]
- Metro, G.; Fabi, A.; Mirri, M.A.; Vidiri, A.; Pace, A.; Carosi, M.; Russillo, M.; Maschio, M.; Giannarelli, D.; Pellegrini, D.; et al. Phase II Study of Fixed Dose Rate Gemcitabine as Radiosensitizer for Newly Diagnosed Glioblastoma Multiforme. Cancer Chemother. Pharmacol. 2010, 65, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Bastiancich, C.; Bastiat, G.; Lagarce, F. Gemcitabine and Glioblastoma: Challenges and Current Perspectives. Drug Discov. Today 2018, 23, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Banasavadi-Siddegowda, Y.K.; Welker, A.M.; An, M.; Yang, X.; Zhou, W.; Shi, G.; Imitola, J.; Li, C.; Hsu, S.; Wang, J.; et al. PRMT5 as a Druggable Target for Glioblastoma Therapy. Neuro Oncol. 2018, 20, 753–763. [Google Scholar] [CrossRef]
- Kim, D.; Fiske, B.P.; Birsoy, K.; Freinkman, E.; Kami, K.; Possemato, R.L.; Chudnovsky, Y.; Pacold, M.E.; Chen, W.W.; Cantor, J.R.; et al. SHMT2 Drives Glioma Cell Survival in Ischaemia but Imposes a Dependence on Glycine Clearance. Nature 2015, 520, 363–367. [Google Scholar] [CrossRef]
- Kryukov, G.V.; Wilson, F.H.; Ruth, J.R.; Paulk, J.; Tsherniak, A.; Marlow, S.E.; Vazquez, F.; Weir, B.A.; Fitzgerald, M.E.; Tanaka, M.; et al. MTAP Deletion Confers Enhanced Dependency on the PRMT5 Arginine Methyltransferase in Cancer Cells. Science 2016, 351, 1214–1218. [Google Scholar] [CrossRef]
- Sachamitr, P.; Ho, J.C.; Ciamponi, F.E.; Ba-Alawi, W.; Coutinho, F.J.; Guilhamon, P.; Kushida, M.M.; Cavalli, F.M.G.; Lee, L.; Rastegar, N.; et al. PRMT5 Inhibition Disrupts Splicing and Stemness in Glioblastoma. Nat. Commun. 2021, 12, 979. [Google Scholar] [CrossRef]
- Gao, X.; Sanderson, S.M.; Dai, Z.; Reid, M.A.; Cooper, D.E.; Lu, M.; Richie, J.P.; Ciccarella, A.; Calcagnotto, A.; Mikhael, P.G.; et al. Dietary Methionine Influences Therapy in Mouse Cancer Models and Alters Human Metabolism. Nature 2019, 572, 397–401. [Google Scholar] [CrossRef]
- Okada, M.; Suzuki, S.; Togashi, K.; Sugai, A.; Yamamoto, M.; Kitanaka, C. Targeting Folate Metabolism Is Selectively Cytotoxic to Glioma Stem Cells and Effectively Cooperates with Differentiation Therapy to Eliminate Tumor-Initiating Cells in Glioma Xenografts. Int. J. Mol. Sci. 2021, 22, 11633. [Google Scholar] [CrossRef]
- Tan, B.; Young, D.A.; Lu, Z.-H.; Wang, T.; Meier, T.I.; Shepard, R.L.; Roth, K.; Zhai, Y.; Huss, K.; Kuo, M.-S.; et al. Pharmacological Inhibition of Nicotinamide Phosphoribosyltransferase (NAMPT), an Enzyme Essential for NAD+ Biosynthesis, in Human Cancer Cells. J. Biol. Chem. 2013, 288, 3500–3511. [Google Scholar] [CrossRef]
- Tateishi, K.; Iafrate, A.J.; Ho, Q.; Curry, W.T.; Batchelor, T.T.; Flaherty, K.T.; Onozato, M.L.; Lelic, N.; Sundaram, S.; Cahill, D.P.; et al. Myc-Driven Glycolysis Is a Therapeutic Target in Glioblastoma. Clin. Cancer Res. 2016, 22, 4452–4465. [Google Scholar] [CrossRef]
- Jung, J.; Kim, L.J.Y.; Wang, X.; Wu, Q.; Sanvoranart, T.; Hubert, C.G.; Prager, B.C.; Wallace, L.C.; Jin, X.; Mack, S.C.; et al. Nicotinamide Metabolism Regulates Glioblastoma Stem Cell Maintenance. JCI Insight 2017, 2, e90019. [Google Scholar] [CrossRef] [PubMed]
- Stuart, S.D.; Schauble, A.; Gupta, S.; Kennedy, A.D.; Keppler, B.R.; Bingham, P.M.; Zachar, Z. A Strategically Designed Small Molecule Attacks Alpha-Ketoglutarate Dehydrogenase in Tumor Cells through a Redox Process. Cancer Metab. 2014, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Zachar, Z.; Marecek, J.; Maturo, C.; Gupta, S.; Stuart, S.D.; Howell, K.; Schauble, A.; Lem, J.; Piramzadian, A.; Karnik, S.; et al. Non-Redox-Active Lipoate Derivates Disrupt Cancer Cell Mitochondrial Metabolism and Are Potent Anticancer Agents in Vivo. J. Mol. Med. 2011, 89, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Yu, J.; Tu, L.; Huang, N.; Li, H.; Luo, Y. Isocitrate Dehydrogenase Mutations in Glioma: From Basic Discovery to Therapeutics Development. Front. Oncol. 2019, 9, 506. [Google Scholar] [CrossRef]
- Calvert, A.E.; Chalastanis, A.; Wu, Y.; Hurley, L.A.; Kouri, F.M.; Bi, Y.; Kachman, M.; May, J.L.; Bartom, E.; Hua, Y.; et al. Cancer-Associated IDH1 Promotes Growth and Resistance to Targeted Therapies in the Absence of Mutation. Cell Rep. 2017, 19, 1858–1873. [Google Scholar] [CrossRef]
- Wahl, D.R.; Dresser, J.; Wilder-Romans, K.; Parsels, J.D.; Zhao, S.G.; Davis, M.; Zhao, L.; Kachman, M.; Wernisch, S.; Burant, C.F.; et al. Glioblastoma Therapy Can Be Augmented by Targeting IDH1-Mediated NADPH Biosynthesis. Cancer Res. 2017, 77, 960–970. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, R.E.; Keatley, K.; Littlewood, D.T.J.; Meunier, B.; Holt, W.V.; An, Q.; Higgins, S.C.; Polyzoidis, S.; Stephenson, K.F.; Ashkan, K.; et al. Identification and Functional Prediction of Mitochondrial Complex III and IV Mutations Associated with Glioblastoma. Neuro Oncol. 2015, 17, 942–952. [Google Scholar] [CrossRef]
- Griguer, C.E.; Cantor, A.B.; Fathallah-Shaykh, H.M.; Gillespie, G.Y.; Gordon, A.S.; Markert, J.M.; Radovanovic, I.; Clement-Schatlo, V.; Shannon, C.N.; Oliva, C.R. Prognostic Relevance of Cytochrome c Oxidase in Primary Glioblastoma Multiforme. PLoS ONE 2013, 8, e61035. [Google Scholar] [CrossRef] [PubMed]
- Oliva, C.R.; Markert, T.; Gillespie, G.Y.; Griguer, C.E. Nuclear-Encoded Cytochrome c Oxidase Subunit 4 Regulates BMI1 Expression and Determines Proliferative Capacity of High-Grade Gliomas. Oncotarget 2015, 6, 4330–4344. [Google Scholar] [CrossRef]
- Griguer, C.E.; Oliva, C.R.; Gobin, E.; Marcorelles, P.; Benos, D.J.; Lancaster, J.R.; Gillespie, G.Y. CD133 Is a Marker of Bioenergetic Stress in Human Glioma. PLoS ONE 2008, 3, e3655. [Google Scholar] [CrossRef]
- Janiszewska, M.; Suvà, M.L.; Riggi, N.; Houtkooper, R.H.; Auwerx, J.; Clément-Schatlo, V.; Radovanovic, I.; Rheinbay, E.; Provero, P.; Stamenkovic, I.; et al. Imp2 Controls Oxidative Phosphorylation and Is Crucial for Preserving Glioblastoma Cancer Stem Cells. Genes Dev. 2012, 26, 1926–1944. [Google Scholar] [CrossRef] [PubMed]
- Chai, Y.; Wang, C.; Liu, W.; Fan, Y.; Zhang, Y. MPC1 Deletion Is Associated with Poor Prognosis and Temozolomide Resistance in Glioblastoma. J. Neurooncol. 2019, 144, 293–301. [Google Scholar] [CrossRef]
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. Metformin Decreases the Dose of Chemotherapy for Prolonging Tumor Remission in Mouse Xenografts Involving Multiple Cancer Cell Types. Cancer Res. 2011, 71, 3196–3201. [Google Scholar] [CrossRef] [PubMed]
- Griss, T.; Vincent, E.E.; Egnatchik, R.; Chen, J.; Ma, E.H.; Faubert, B.; Viollet, B.; DeBerardinis, R.J.; Jones, R.G. Metformin Antagonizes Cancer Cell Proliferation by Suppressing Mitochondrial-Dependent Biosynthesis. PLoS Biol. 2015, 13, e1002309. [Google Scholar] [CrossRef] [PubMed]
- Mazurek, M.; Litak, J.; Kamieniak, P.; Kulesza, B.; Jonak, K.; Baj, J.; Grochowski, C. Metformin as Potential Therapy for High-Grade Glioma. Cancers 2020, 12, 210. [Google Scholar] [CrossRef]
- Yang, S.H.; Li, S.; Lu, G.; Xue, H.; Kim, D.H.; Zhu, J.-J.; Liu, Y. Metformin Treatment Reduces Temozolomide Resistance of Glioblastoma Cells. Oncotarget 2016, 7, 78787–78803. [Google Scholar] [CrossRef]
- Feng, S.-W.; Chang, P.-C.; Chen, H.-Y.; Hueng, D.-Y.; Li, Y.-F.; Huang, S.-M. Exploring the Mechanism of Adjuvant Treatment of Glioblastoma Using Temozolomide and Metformin. Int. J. Mol. Sci. 2022, 23, 8171. [Google Scholar] [CrossRef]
- Würth, R.; Pattarozzi, A.; Gatti, M.; Bajetto, A.; Corsaro, A.; Parodi, A.; Sirito, R.; Massollo, M.; Marini, C.; Zona, G.; et al. Metformin Selectively Affects Human Glioblastoma Tumor-Initiating Cell Viability. Cell Cycle 2013, 12, 145–156. [Google Scholar] [CrossRef]
- Ohno, M.; Kitanaka, C.; Miyakita, Y.; Tanaka, S.; Sonoda, Y.; Mishima, K.; Ishikawa, E.; Takahashi, M.; Yanagisawa, S.; Ohashi, K.; et al. Metformin with Temozolomide for Newly Diagnosed Glioblastoma: Results of Phase I Study and a Brief Review of Relevant Studies. Cancers 2022, 14, 4222. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Ko, B.; Hensley, C.T.; Jiang, L.; Wasti, A.T.; Kim, J.; Sudderth, J.; Calvaruso, M.A.; Lumata, L.; Mitsche, M.; et al. Glutamine Oxidation Maintains the TCA Cycle and Cell Survival during Impaired Mitochondrial Pyruvate Transport. Mol. Cell 2014, 56, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Arif, T.; Krelin, Y.; Nakdimon, I.; Benharroch, D.; Paul, A.; Dadon-Klein, D.; Shoshan-Barmatz, V. VDAC1 Is a Molecular Target in Glioblastoma, with Its Depletion Leading to Reprogrammed Metabolism and Reversed Oncogenic Properties. Neuro Oncol. 2017, 19, 951–964. [Google Scholar] [CrossRef] [PubMed]
- Mohan, A.A.; Tomaszewski, W.H.; Haskell-Mendoza, A.P.; Hotchkiss, K.M.; Singh, K.; Reedy, J.L.; Fecci, P.E.; Sampson, J.H.; Khasraw, M. Targeting Immunometabolism in Glioblastoma. Front. Oncol. 2021, 11, 696402. [Google Scholar] [CrossRef] [PubMed]
- Garofano, L.; Migliozzi, S.; Oh, Y.T.; D’Angelo, F.; Najac, R.D.; Ko, A.; Frangaj, B.; Caruso, F.P.; Yu, K.; Yuan, J.; et al. Pathway-Based Classification of Glioblastoma Uncovers a Mitochondrial Subtype with Therapeutic Vulnerabilities. Nat. Cancer 2021, 2, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-B.; Karpova, A.; Gritsenko, M.A.; Kyle, J.E.; Cao, S.; Li, Y.; Rykunov, D.; Colaprico, A.; Rothstein, J.H.; Hong, R.; et al. Proteogenomic and Metabolomic Characterization of Human Glioblastoma. Cancer Cell 2021, 39, 509–528.e20. [Google Scholar] [CrossRef] [PubMed]
- Shakya, S.; Gromovsky, A.D.; Hale, J.S.; Knudsen, A.M.; Prager, B.; Wallace, L.C.; Penalva, L.O.F.; Brown, H.A.; Kristensen, B.W.; Rich, J.N.; et al. Altered Lipid Metabolism Marks Glioblastoma Stem and Non-Stem Cells in Separate Tumor Niches. Acta Neuropathol. Commun. 2021, 9, 101. [Google Scholar] [CrossRef]
- Cabodevilla, A.G.; Sánchez-Caballero, L.; Nintou, E.; Boiadjieva, V.G.; Picatoste, F.; Gubern, A.; Claro, E. Cell Survival during Complete Nutrient Deprivation Depends on Lipid Droplet-Fueled β-Oxidation of Fatty Acids. J. Biol. Chem. 2013, 288, 27777–27788. [Google Scholar] [CrossRef]
- Seliger, C.; Meyer, A.-L.; Leidgens, V.; Rauer, L.; Moeckel, S.; Jachnik, B.; Proske, J.; Dettmer, K.; Rothhammer-Hampl, T.; Kaulen, L.D.; et al. Metabolic Heterogeneity of Brain Tumor Cells of Proneural and Mesenchymal Origin. Int. J. Mol. Sci. 2022, 23, 11629. [Google Scholar] [CrossRef]
- Liparulo, I.; Bergamini, C.; Bortolus, M.; Calonghi, N.; Gasparre, G.; Kurelac, I.; Masin, L.; Rizzardi, N.; Rugolo, M.; Wang, W.; et al. Coenzyme Q Biosynthesis Inhibition Induces HIF-1α Stabilization and Metabolic Switch toward Glycolysis. FEBS J. 2021, 288, 1956–1974. [Google Scholar] [CrossRef]
- Sonveaux, P.; Copetti, T.; De Saedeleer, C.J.; Végran, F.; Verrax, J.; Kennedy, K.M.; Moon, E.J.; Dhup, S.; Danhier, P.; Frérart, F.; et al. Targeting the Lactate Transporter MCT1 in Endothelial Cells Inhibits Lactate-Induced HIF-1 Activation and Tumor Angiogenesis. PLoS ONE 2012, 7, e33418. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, G.; Chu, H.; Wang, X.; Xiong, L.; Cai, G.; Liu, R.; Gao, H.; Tao, B.; Li, W.; et al. Macrophage-Associated PGK1 Phosphorylation Promotes Aerobic Glycolysis and Tumorigenesis. Mol. Cell 2018, 71, 201–215.e7. [Google Scholar] [CrossRef]
- Di Ianni, N.; Musio, S.; Pellegatta, S. Altered Metabolism in Glioblastoma: Myeloid-Derived Suppressor Cell (MDSC) Fitness and Tumor-Infiltrating Lymphocyte (TIL) Dysfunction. Int. J. Mol. Sci. 2021, 22, 4460. [Google Scholar] [CrossRef]
- Kim, E.H.; Lee, J.-H.; Oh, Y.; Koh, I.; Shim, J.-K.; Park, J.; Choi, J.; Yun, M.; Jeon, J.Y.; Huh, Y.M.; et al. Inhibition of Glioblastoma Tumorspheres by Combined Treatment with 2-Deoxyglucose and Metformin. Neuro Oncol. 2017, 19, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhao, J.; Cui, X.; Zhan, Q.; Yi, K.; Wang, Q.; Xiao, M.; Tan, Y.; Hong, B.; Fang, C.; et al. TCA-Phospholipid-Glycolysis Targeted Triple Therapy Effectively Suppresses ATP Production and Tumor Growth in Glioblastoma. Theranostics 2022, 12, 7032–7050. [Google Scholar] [CrossRef] [PubMed]
- McKelvey, K.J.; Wilson, E.B.; Short, S.; Melcher, A.A.; Biggs, M.; Diakos, C.I.; Howell, V.M. Glycolysis and Fatty Acid Oxidation Inhibition Improves Survival in Glioblastoma. Front. Oncol. 2021, 11, 633210. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Park, S.J.; Park, J.; Cho, H.J.; Shim, J.-K.; Seon, J.; Choi, R.J.; Yoon, S.-J.; Moon, J.H.; Kim, E.H.; et al. Dual Inhibition of CPT1A and G6PD Suppresses Glioblastoma Tumorspheres. J. Neurooncol. 2022, 160, 677–689. [Google Scholar] [CrossRef]
- Zhan, Q.; Yi, K.; Cui, X.; Li, X.; Yang, S.; Wang, Q.; Fang, C.; Tan, Y.; Li, L.; Xu, C.; et al. Blood Exosomes-Based Targeted Delivery of CPLA2 SiRNA and Metformin to Modulate Glioblastoma Energy Metabolism for Tailoring Personalized Therapy. Neuro Oncol. 2022, 24, 1871–1883. [Google Scholar] [CrossRef]
- Zhang, B.; Peng, H.; Zhou, M.; Bao, L.; Wang, C.; Cai, F.; Zhang, H.; Wang, J.E.; Niu, Y.; Chen, Y.; et al. Targeting BCAT1 Combined with α-Ketoglutarate Triggers Metabolic Synthetic Lethality in Glioblastoma. Cancer Res. 2022, 82, 2388–2402. [Google Scholar] [CrossRef]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Matsutani, T.; Masui, K.; Poulou, M.; Popescu, R.; Della Donna, L.; Evers, P.; Dekmezian, C.; et al. Metabolic State of Glioma Stem Cells and Nontumorigenic Cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16062–16067. [Google Scholar] [CrossRef]
- Vartanian, A.; Agnihotri, S.; Wilson, M.R.; Burrell, K.E.; Tonge, P.D.; Alamsahebpour, A.; Jalali, S.; Taccone, M.S.; Mansouri, S.; Golbourn, B.; et al. Targeting Hexokinase 2 Enhances Response to Radio-Chemotherapy in Glioblastoma. Oncotarget 2016, 7, 69518–69535. [Google Scholar] [CrossRef]
- Sampath, D.; Zabka, T.S.; Misner, D.L.; O’Brien, T.; Dragovich, P.S. Inhibition of Nicotinamide Phosphoribosyltransferase (NAMPT) as a Therapeutic Strategy in Cancer. Pharmacol. Ther. 2015, 151, 16–31. [Google Scholar] [CrossRef] [PubMed]
- McBrayer, S.K.; Mayers, J.R.; DiNatale, G.J.; Shi, D.D.; Khanal, J.; Chakraborty, A.A.; Sarosiek, K.A.; Briggs, K.J.; Robbins, A.K.; Sewastianik, T.; et al. Transaminase Inhibition by 2-Hydroxyglutarate Impairs Glutamate Biosynthesis and Redox Homeostasis in Glioma. Cell 2018, 175, 101–116.e25. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.; Carson, G.; McConnell, M.; Herst, P. High Dose Ascorbate Causes Both Genotoxic and Metabolic Stress in Glioma Cells. Antioxidants 2017, 6, 58. [Google Scholar] [CrossRef]
- Hajji, N.; Garcia-Revilla, J.; Soto, M.S.; Perryman, R.; Symington, J.; Quarles, C.C.; Healey, D.R.; Guo, Y.; Orta-Vázquez, M.L.; Mateos-Cordero, S.; et al. Arginine Deprivation Alters Microglial Polarity and Synergizes with Radiation to Eradicate Non-Arginine-Auxotrophic Glioblastoma Tumors. J. Clin. Investig. 2022, 132, e142137. [Google Scholar] [CrossRef]
- Oliva, C.R.; Nozell, S.E.; Diers, A.; McClugage, S.G.; Sarkaria, J.N.; Markert, J.M.; Darley-Usmar, V.M.; Bailey, S.M.; Gillespie, G.Y.; Landar, A.; et al. Acquisition of Temozolomide Chemoresistance in Gliomas Leads to Remodeling of Mitochondrial Electron Transport Chain. J. Biol. Chem. 2010, 285, 39759–39767. [Google Scholar] [CrossRef]
- Caragher, S.; Miska, J.; Shireman, J.; Park, C.H.; Muroski, M.; Lesniak, M.S.; Ahmed, A.U. Temozolomide Treatment Increases Fatty Acid Uptake in Glioblastoma Stem Cells. Cancers 2020, 12, 3126. [Google Scholar] [CrossRef]
- Wang, N.; Huang, R.; Yang, K.; He, Y.; Gao, Y.; Dong, D. Interfering with Mitochondrial Dynamics Sensitizes Glioblastoma Multiforme to Temozolomide Chemotherapy. J. Cell. Mol. Med. 2022, 26, 893–912. [Google Scholar] [CrossRef] [PubMed]
- Wicks, R.T.; Azadi, J.; Mangraviti, A.; Zhang, I.; Hwang, L.; Joshi, A.; Bow, H.; Hutt-Cabezas, M.; Martin, K.L.; Rudek, M.A.; et al. Local Delivery of Cancer-Cell Glycolytic Inhibitors in High-Grade Glioma. Neuro Oncol. 2015, 17, 70–80. [Google Scholar] [CrossRef]
- Yuan, S.; Wang, F.; Chen, G.; Zhang, H.; Feng, L.; Wang, L.; Colman, H.; Keating, M.J.; Li, X.; Xu, R.-H.; et al. Effective Elimination of Cancer Stem Cells By a Novel Drug Combination Strategy. Stem Cells 2013, 31, 23–34. [Google Scholar] [CrossRef]
- Sun, X.; Fan, T.; Sun, G.; Zhou, Y.; Huang, Y.; Zhang, N.; Zhao, L.; Zhong, R.; Peng, Y. 2-Deoxy-D-Glucose Increases the Sensitivity of Glioblastoma Cells to BCNU through the Regulation of Glycolysis, ROS and ERS Pathways: In Vitro and In Vivo Validation. Biochem. Pharmacol. 2022, 199, 115029. [Google Scholar] [CrossRef]
- Yu, Z.; Zhao, G.; Xie, G.; Zhao, L.; Chen, Y.; Yu, H.; Zhang, Z.; Li, C.; Li, Y. Metformin and Temozolomide Act Synergistically to Inhibit Growth of Glioma Cells and Glioma Stem Cells in Vitro and in Vivo. Oncotarget 2015, 6, 32930–32943. [Google Scholar] [CrossRef]
- Park, H.H.; Park, J.; Cho, H.J.; Shim, J.-K.; Moon, J.H.; Kim, E.H.; Chang, J.H.; Kim, S.Y.; Kang, S.-G. Combinatorial Therapeutic Effect of Inhibitors of Aldehyde Dehydrogenase and Mitochondrial Complex I, and the Chemotherapeutic Drug, Temozolomide against Glioblastoma Tumorspheres. Molecules 2021, 26, 282. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.-K.; Choi, S.; Yoon, S.-J.; Choi, R.J.; Park, J.; Lee, E.H.; Cho, H.J.; Lee, S.; Teo, W.-Y.; Moon, J.H.; et al. Etomoxir, a Carnitine Palmitoyltransferase 1 Inhibitor, Combined with Temozolomide Reduces Stemness and Invasiveness in Patient-Derived Glioblastoma Tumorspheres. Cancer Cell Int. 2022, 22, 309. [Google Scholar] [CrossRef]
- Mazor, G.; Levin, L.; Picard, D.; Ahmadov, U.; Carén, H.; Borkhardt, A.; Reifenberger, G.; Leprivier, G.; Remke, M.; Rotblat, B. The LncRNA TP73-AS1 Is Linked to Aggressiveness in Glioblastoma and Promotes Temozolomide Resistance in Glioblastoma Cancer Stem Cells. Cell Death Dis. 2019, 10, 246. [Google Scholar] [CrossRef] [PubMed]
- Fack, F.; Espedal, H.; Keunen, O.; Golebiewska, A.; Obad, N.; Harter, P.N.; Mittelbronn, M.; Bähr, O.; Weyerbrock, A.; Stuhr, L.; et al. Bevacizumab Treatment Induces Metabolic Adaptation toward Anaerobic Metabolism in Glioblastomas. Acta Neuropathol. 2015, 129, 115–131. [Google Scholar] [CrossRef]
- Keunen, O.; Johansson, M.; Oudin, A.; Sanzey, M.; Rahim, S.A.A.; Fack, F.; Thorsen, F.; Taxt, T.; Bartos, M.; Jirik, R.; et al. Anti-VEGF Treatment Reduces Blood Supply and Increases Tumor Cell Invasion in Glioblastoma. Proc. Natl. Acad. Sci. USA 2011, 108, 3749–3754. [Google Scholar] [CrossRef]
- Divé, I.; Klann, K.; Michaelis, J.B.; Heinzen, D.; Steinbach, J.P.; Münch, C.; Ronellenfitsch, M.W. Inhibition of MTOR Signaling Protects Human Glioma Cells from Hypoxia-Induced Cell Death in an Autophagy-Independent Manner. Cell Death Discov. 2022, 8, 409. [Google Scholar] [CrossRef]
- Romero-Garcia, S.; Moreno-Altamirano, M.M.B.; Prado-Garcia, H.; Sánchez-García, F.J. Lactate Contribution to the Tumor Microenvironment: Mechanisms, Effects on Immune Cells and Therapeutic Relevance. Front. Immunol. 2016, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Hao, M.; Hou, S.; Li, W.; Li, K.; Xue, L.; Hu, Q.; Zhu, L.; Chen, Y.; Sun, H.; Ju, C.; et al. Combination of Metabolic Intervention and T Cell Therapy Enhances Solid Tumor Immunotherapy. Sci. Transl. Med. 2020, 12, eaaz6667. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, D.A.; Chang, A.L.; Dey, M.; Balyasnikova, I.V.; Kim, C.K.; Tobias, A.; Cheng, Y.; Kim, J.W.; Qiao, J.; Zhang, L.; et al. Durable Therapeutic Efficacy Utilizing Combinatorial Blockade against IDO, CTLA-4, and PD-L1 in Mice with Brain Tumors. Clin. Cancer Res. 2014, 20, 5290–5301. [Google Scholar] [CrossRef]
- Medikonda, R.; Choi, J.; Pant, A.; Saleh, L.; Routkevitch, D.; Tong, L.; Belcaid, Z.; Kim, Y.H.; Jackson, C.M.; Jackson, C.; et al. Synergy between Glutamate Modulation and Anti–Programmed Cell Death Protein 1 Immunotherapy for Glioblastoma. J. Neurosurg. 2022, 136, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Safdie, F.; Brandhorst, S.; Wei, M.; Wang, W.; Lee, C.; Hwang, S.; Conti, P.S.; Chen, T.C.; Longo, V.D. Fasting Enhances the Response of Glioma to Chemo- and Radiotherapy. PLoS ONE 2012, 7, e44603. [Google Scholar] [CrossRef] [PubMed]
- Kanarek, N.; Petrova, B.; Sabatini, D.M. Dietary Modifications for Enhanced Cancer Therapy. Nature 2020, 579, 507–517. [Google Scholar] [CrossRef]
- Voss, M.; Wagner, M.; von Mettenheim, N.; Harter, P.N.; Wenger, K.J.; Franz, K.; Bojunga, J.; Vetter, M.; Gerlach, R.; Glatzel, M.; et al. ERGO2: A Prospective, Randomized Trial of Calorie-Restricted Ketogenic Diet and Fasting in Addition to Reirradiation for Malignant Glioma. Int. J. Radiat. Oncol. 2020, 108, 987–995. [Google Scholar] [CrossRef]
- Astuti, D.; Latif, F.; Dallol, A.; Dahia, P.L.M.; Douglas, F.; George, E.; Sköldberg, F.; Husebye, E.S.; Eng, C.; Maher, E.R. Gene Mutations in the Succinate Dehydrogenase Subunit SDHB Cause Susceptibility to Familial Pheochromocytoma and to Familial Paraganglioma. Am. J. Hum. Genet. 2001, 69, 49–54. [Google Scholar] [CrossRef]
- Nusblat, L.M.; Tanna, S.; Roth, C.M. Gene Silencing of HIF-2α Disrupts Glioblastoma Stem Cell Phenotype. Cancer Drug Resist. 2020, 3, 199. [Google Scholar] [CrossRef]
- Simpson, J.E.; Gammoh, N. The Impact of Autophagy during the Development and Survival of Glioblastoma. Open Biol. 2020, 10, 200184. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapy | Mechanism | Trial Phase | Study Participants/Therapeutic Strategy | References |
|---|---|---|---|---|
| 2-Deoxy-D-Glucose | Glucose analogue | I/II | Newly diagnosed GB patients treated with 2-DG in combination with radiotherapy | [30,31] |
| WP1122 | Glucose analogue | I | Healthy volunteers | NCT05195723 |
| Ketoconazole Posaconazole | HK2 inhibitor | I I | GB patients scheduled for radiotherapy | NCT04869449 NCT04825275 |
| TP-1454 | PKM2 activator | I | Patients with progressive solid tumors, treated with TP-1454 alone or in combination with immunotherapy | NCT04328740 |
| Dichloroacetate | PDK1 inhibitor | I II | Recurrent GB Newly diagnosed and recurrent GB | NCT01111097 NCT00540176 |
| Gossypol | Bcl-2 protein family and dehydrogenases inhibitor | I | Newly diagnosed GB treated in combination with TMZ with or without radiotherapy | NCT00390403 |
| II | Progressive or recurrent GB | NCT00540722 | ||
| Anhydrous Enol-Oxaloacetate (AEO) | Oxaloacetate pro-drug | II | Newly diagnosed GB treated in combination with standard therapy | NCT04450160 |
| Telaglenastat | GLS inhibitor | I/II | Advanced/metastatic solid tumors patients in combination with talazoparib (PARPs inhibitor) | NCT03875313 |
| ADI-PEG20 | Pegylated arginine deiminase | I | Recurrent high-grade gliomas in combination with chemotherapy | [76,77] |
| INCB001158 | Arginase inhibitor | I/II | Advanced solid tumors in combination with chemotherapy | NCT03314935 |
| TVB-2640 | FASN inhibitor | II | Recurrent GB in combination with bevacizumab | NCT03032484 |
| Atorvastatin | HMG-CoA reductase inhibitor | II | Newly diagnosed GB patients treated in combination with radiotherapy and temozolomide | NCT02029573 |
| Gemcitabine | Nucleoside analogue | II | High-grade glioma patients treated in combination with radiotherapy | [123] |
| GSK3326595 | PRMT5 inhibitor | I | GB | NCT02783300 |
| CPI-613 | TCA-targeted therapy | II | Solid tumor | NCT01832857 |
| Metformin | AMPK activator, complex I respiratory chain inhibitor | II II | Recurrent or refractory GB treated with low TMZ plus metformin Newly diagnosed IDH wild-type GB patients with the OXPHOS+ signature in combination with standard therapy | NCT03243851 NCT04945148 |
| Ascorbic Acid | Cofactor, antioxidant | II | Newly diagnosed GB treated in combination with the standard therapy | NCT02344355 |
| Disulfiram | ALDH inhibitor | I/II | Patients with presumed GB treated with disulfiram and copper before surgery and during adjuvant chemoradiotherapy | NCT02715609 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernhard, C.; Reita, D.; Martin, S.; Entz-Werle, N.; Dontenwill, M. Glioblastoma Metabolism: Insights and Therapeutic Strategies. Int. J. Mol. Sci. 2023, 24, 9137. https://doi.org/10.3390/ijms24119137
Bernhard C, Reita D, Martin S, Entz-Werle N, Dontenwill M. Glioblastoma Metabolism: Insights and Therapeutic Strategies. International Journal of Molecular Sciences. 2023; 24(11):9137. https://doi.org/10.3390/ijms24119137
Chicago/Turabian StyleBernhard, Chloé, Damien Reita, Sophie Martin, Natacha Entz-Werle, and Monique Dontenwill. 2023. "Glioblastoma Metabolism: Insights and Therapeutic Strategies" International Journal of Molecular Sciences 24, no. 11: 9137. https://doi.org/10.3390/ijms24119137
APA StyleBernhard, C., Reita, D., Martin, S., Entz-Werle, N., & Dontenwill, M. (2023). Glioblastoma Metabolism: Insights and Therapeutic Strategies. International Journal of Molecular Sciences, 24(11), 9137. https://doi.org/10.3390/ijms24119137