

Effect of Hepatic Pathology on Liver Regeneration: The Main Metabolic Mechanisms Causing Impaired Hepatic Regeneration

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
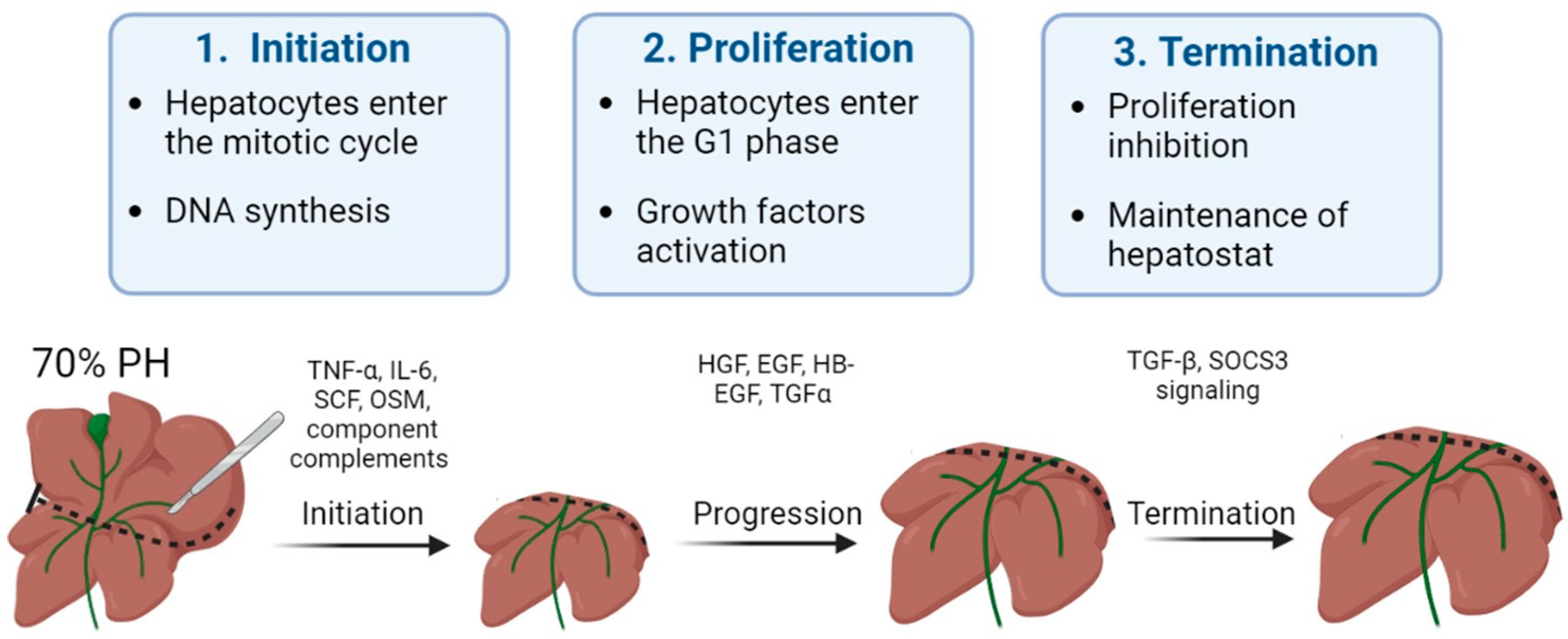
1. Mechanisms of Liver Regeneration
2. Liver Regeneration in Hepatic Pathology
2.1. Non-Alcoholic Fatty Liver Disease
2.2. Diabetes-Provided Fatty Liver Disease
2.3. Fibrosis and Cirrhosis
2.4. Alcoholic Liver Disease
3. Future Perspectives
3.1. Promising Approaches to Stimulate Liver Regeneration
3.2. Prospects for Predictive Liver Assessment
4. Conclusions and Outlooks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Michalopoulos, G.K. The Liver: Biology and Pathobiology, 6th ed.; Arias, I.M., Alter, H.J., Eds.; Wiley-Blackwell: Hoboken, NJ, USA, 2020; Volume 1, pp. 566–584. [Google Scholar]
- Michalopoulos, G.K. Hepatostat: Liver regeneration and normal liver tissue maintenance. Hepatology 2017, 65, 1384–1392. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Zhao, Y.L.; Zhang, X.Q.; Li, L.J. The vascular endothelial growth factor signaling pathway regulates liver sinusoidal endothelial cells during liver regeneration after partial hepatectomy. Expert Rev. Gastroenterol. Hepatol. 2021, 15, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Higgins, G.M. Experimental pathology of the liver. Restoration of the liver of the white rat following partial surgical removal. Arch. Pathol. Lab. Med. 1931, 12, 1186–1202. [Google Scholar]
- Bhushan, B.; Walesky, C.; Manley, M.; Gallagher, T.; Borude, P.; Edwards, G.; Monga, P.S.; Apte, U. Pro-regenerative signaling after acetaminophen-induced acute liver injury in mice identified using a novel incremental dose model. Am. J. Pathol. 2014, 184, 3013–3025. [Google Scholar] [CrossRef]
- Forbes, S.J.; Newsome, P.N. Liver regeneration—Mechanisms and models to clinical application. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 473–485. [Google Scholar] [CrossRef]
- Miyaoka, Y.; Ebato, K.; Kato, H.; Arakawa, S.; Shimizu, S.; Miyajima, A. Hypertrophy and unconventional cell division of hepatocytes underlie liver regeneration. Curr. Biol. 2012, 22, 1166–1175. [Google Scholar] [CrossRef]
- Bangru, S.; Arif, W.; Seimetz, J.; Bhate, A.; Chen, J.; Rashan, E.H.; Carstens, R.P.; Anakk, S.; Kalsotra, A. Alternative splicing rewires Hippo signaling pathway in hepatocytes to promote liver regeneration. Nat. Struct. Mol. Biol. 2018, 25, 928–939. [Google Scholar] [CrossRef]
- Abu Rmilah, A.; Zhou, W.; Nelson, E.; Lin, L.; Amiot, B.; Nyberg, S.L. Understanding the marvels behind liver regeneration. Wiley Interdiscip. Rev. Dev. Biol. 2019, 8, e340. [Google Scholar] [CrossRef]
- Rio Bartulos, C.; Senk, K.; Schumacher, M.; Plath, J.; Kaiser, N.; Bade, R.; Woetzel, J.; Wiggermann, P. Assessment of Liver Function With MRI: Where Do We Stand? Front. Med. 2022, 9, 729. [Google Scholar] [CrossRef]
- Gentric, G.; Desdouets, C. Polyploidization in liver tissue. Am. J. Pathol. 2014, 184, 322–331. [Google Scholar] [CrossRef]
- Wilkinson, P.D.; Alencastro, F.; Delgado, E.R.; Leek, M.P.; Weirich, M.P.; Otero, P.A.; Roy, N.; Brown, W.K.; Oertel, M.; Duncan, A.W. Polyploid hepatocytes facilitate adaptation and regeneration to chronic liver injury. Am. J. Pathol. 2019, 189, 1241–1255. [Google Scholar] [CrossRef] [PubMed]
- Pandit, S.K.; Westendorp, B.; de Bruin, A. Physiological significance of polyploidization in mammalian cells. Trends Cell Biol. 2013, 23, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Gentric, G.; Celton-Morizur, S.; Desdouets, C. Polyploidy and liver proliferation. Clin. Res. Hepatol. Gastroenterol. 2012, 36, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Donne, R.; Saroul-Aïnama, M.; Cordier, P.; Celton-Morizur, S.; Desdouets, C. Polyploidy in liver development, homeostasis and disease. Nat. Rev. Gastroenterol. Hepat. 2020, 17, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Delgado, E.R.; Stahl, E.C.; Roy, N.; Wilkinson, P.D.; Duncan, A.W. The Liver: Biology and Pathobiology, 6th ed.; John Wiley and Sons Ltd.: New York, NY, USA, 2020; pp. 603–613. [Google Scholar]
- Wang, B.; Zhao, L.; Fish, M.; Logan, C.Y.; Nusse, R. Self-renewing diploid Axin2+ cells fuel homeostatic renewal of the liver. Nature 2015, 524, 180–185. [Google Scholar] [CrossRef]
- Font-Burgada, J.; Shalapour, S.; Ramaswamy, S.; Hsueh, B.; Rossell, D.; Umemura, A.; Taniguchi, K.; Nakagawa, H.; Valasek, M.A.; Ye, L.; et al. Hybrid periportal hepatocytes regenerate the injured liver without giving rise to cancer. Cell 2015, 162, 766–779. [Google Scholar] [CrossRef]
- Gandillet, A.; Alexandre, E.; Holl, V.; Royer, C.; Bischoff, P.; Cinqualbre, J.; Wolf, P.; Jaeck, D.; Richert, L. Hepatocyte ploidy in normal young rat. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2003, 134, 665–673. [Google Scholar] [CrossRef]
- Asahina, K.; Teramoto, K.; Teraoka, H. Embryonic stem cells: Hepatic differentiation and regenerative medicine for the treatment of liver disease. Curr. Stem Cell Res. Ther. 2006, 1, 139–156. [Google Scholar] [CrossRef]
- Planas-Paz, L.; Orsini, V.; Boulter, L.; Calabrese, D.; Pikiolek, M.; Nigsch, F.; Xie, Y.; Roma, G.; Donovan, A.; Marti, P.; et al. The RSPO–LGR4/5–ZNRF3/RNF43 module controls liver zonation and size. Nat. Cell Biol. 2016, 18, 467–479. [Google Scholar] [CrossRef]
- Kreutz, C.; MacNelly, S.; Follo, M.; Wäldin, A.; Binninger-Lacour, P.; Timmer, J.; Bartolomé-Rodríguez, M.M. Hepatocyte ploidy is a diversity factor for liver homeostasis. Front. Physiol. 2017, 8, 862. [Google Scholar] [CrossRef]
- Michalopoulos, G.K.; Khan, Z. Liver stem cells: Experimental findings and implications for human liver disease. Gastroenterology 2015, 149, 876–882. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Itoh, T.; Tanimizu, N.; Miyajima, A. Liver stem/progenitor cells: Their characteristics and regulatory mechanisms. J. Biochem. 2011, 149, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.A.; Glorioso, J.M.; Nyberg, S.L. Liver regeneration. Transl. Res. 2014, 163, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Dusabineza, A.C.; Hul, N.K.V.; Abarca-Quinones, J.; Starkel, P.; Najimi, M.; Leclercq, I.A. Participation of liver progenitor cells in liver regeneration: Lack of evidence in the AAF/PH rat model. Lab. Investig. 2012, 92, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Stueck, A.E.; Wanless, I.R. Hepatocyte buds derived from progenitor cells repopulate regions of parenchymal extinction in human cirrhosis. Hepatology 2015, 61, 1696–1707. [Google Scholar] [CrossRef] [PubMed]
- Raven, A.; Lu, W.Y.; Man, T.Y.; Ferreira-Gonzalez, S.; O’Duibhir, E.; Dwyer, B.J.; Thomson, J.P.; Meehan, R.R.; Bogorad, R.; Koteliansky, V.; et al. Cholangiocytes act as facultative liver stem cells during impaired hepatocyte regeneration. Nature 2017, 547, 350–354. [Google Scholar] [CrossRef]
- Li, B.; Dorrell, C.; Canaday, P.S.; Pelz, C.; Haft, A.; Finegold, M.; Grompe, M. Adult mouse liver contains two distinct populations of cholangiocytes. Stem Cell Rep. 2017, 9, 478–489. [Google Scholar] [CrossRef]
- Michalopoulos, G.K.; Bhushan, B. Liver regeneration: Biological and pathological mechanisms and implications. Nat. Rev. Gastroenterol. Hepat. 2021, 18, 40–55. [Google Scholar] [CrossRef]
- Huch, M.; Gehart, H.; Van Boxtel, R.; Hamer, K.; Blokzijl, F.; Verstegen, M.M.; Ellis, E.; Wenum, M.; Fuchs, S.A.; de Ligt, J.; et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell 2015, 160, 299–312. [Google Scholar] [CrossRef]
- Lu, W.Y.; Bird, T.G.; Boulter, L.; Tsuchiya, A.; Cole, A.M.; Hay, T.; Guest, R.V.; Wojtacha, D.; Man, T.Y.; Mackinnon, A.; et al. Hepatic progenitor cells of biliary origin with liver repopulation capacity. Nat. Cell Biol. 2015, 17, 971–983. [Google Scholar] [CrossRef]
- Schaub, J.R.; Malato, Y.; Gormond, C.; Willenbring, H. Evidence against a stem cell origin of new hepatocytes in a common mouse model of chronic liver injury. Cell Rep. 2014, 8, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Yanger, K.; Knigin, D.; Zong, Y.; Maggs, L.; Gu, G.; Akiyama, H.; Pikarsky, E.; Stanger, B.Z. Adult hepatocytes are generated by self-duplication rather than stem cell differentiation. Cell Stem Cell 2014, 15, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Butcher, R.L. Factors affecting luteal regulation following parabiosis in the rat. Endocrinology 1966, 4, 57–460. [Google Scholar] [CrossRef] [PubMed]
- Fausto, N.; Campbell, J.S.; Riehle, K.J. Liver regeneration. Hepatology 2006, 43, S45–S53. [Google Scholar] [CrossRef] [PubMed]
- Böhm, F.; Köhler, U.A.; Speicher, T.; Werner, S. Regulation of liver regeneration by growth factors and cytokines. EMBO Mol. Med. 2010, 2, 294–305. [Google Scholar] [CrossRef]
- Fajardo-Puerta, A.B.; Prado, M.M.; Frampton, A.E.; Jiao, L.R. Gene of the month: HGF. J. Clin. Pathol. 2016, 69, 575–579. [Google Scholar] [CrossRef]
- Tao, Y.; Wang, M.; Chen, E.; Tang, H. Liver regeneration: Analysis of the main relevant signaling molecules. Mediat. Inflamm. 2017, 2017, 4256352. [Google Scholar] [CrossRef]
- Paranjpe, S.; Bowen, W.C.; Mars, W.M.; Orr, A.; Haynes, M.M.; DeFrances, M.C.; Liu, S.; Tseng, G.C.; Tsagianni, A.; Michalopoulos, G.K. Combined systemic elimination of MET and EGFR signaling completely abolishes liver regeneration and leads to liver decompensation. Hepatology 2016, 64, 1711. [Google Scholar] [CrossRef]
- Guglielmi, A.; Ruzzenente, A.; Conci, S.; Valdegamberi, A.; Iacono, C. How much remnant is enough in liver resection? Digest. Surg. 2012, 29, 6–17. [Google Scholar] [CrossRef]
- Golse, N.; Bucur, P.O.; Adam, R.; Castaing, D.; Sa Cunha, A.; Vibert, E. New paradigms in post-hepatectomy liver failure. J. Gastrointest. Surg. 2013, 17, 593–605. [Google Scholar] [CrossRef]
- Nilsson, H.; Karlgren, S.; Blomqvist, L.; Jonas, E. The inhomogeneous distribution of liver function: Possible impact on the prediction of post-operative remnant liver function. Hpb 2015, 17, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Truant, S.; Scatton, O.; Dokmak, S.; Regimbeau, J.M.; Lucidi, V.; Laurent, A.; Gauzolino, R.; Castro Benitez, C.; Pequignot, A.; Donckier, V. Associating liver partition and portal vein ligation for staged hepatectomy (ALPPS): Impact of the inter-stages course on morbi-mortality and implications for management. Eur. J. Surg. Oncol. 2015, 41, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Moris, D.; Vernadakis, S.; Papalampros, A.; Vailas, M.; Dimitrokallis, N.; Petrou, A.; Dimitroulis, D. Mechanistic insights of rapid liver regeneration after associating liver partition and portal vein ligation for stage hepatectomy. World J. Gastroenterol. 2016, 22, 7613. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Stewart, A.G.; Woodman, O.L.; Ritchie, R.H.; Qin, C.X. Non-alcoholic steatohepatitis: A review of its mechanism, models and medical treatments. Front. Pharmacol. 2020, 11, 603926. [Google Scholar] [CrossRef]
- Hamano, M.; Ezaki, H.; Kiso, S.; Furuta, K.; Egawa, M.; Kizu, T.; Chatani, N.; Kamada, Y.; Yoshida, Y.; Takehara, T. Lipid overloading during liver regeneration causes delayed hepatocyte DNA replication by increasing ER stress in mice with simple hepatic steatosis. J. Gastroenterol. 2014, 49, 305–316. [Google Scholar] [CrossRef]
- Allaire, M.; Gilgenkrantz, H. The aged liver: Beyond cellular senescence. Clin. Res. Hepatol. Gastroenterol. 2020, 44, 6–11. [Google Scholar] [CrossRef]
- Brunt, E.M. Nonalcoholic fatty liver disease and the ongoing role of liver biopsy evaluation. Hepatol. Commun. 2017, 1, 370–378. [Google Scholar] [CrossRef]
- Veteläinen, R.; van Vliet, A.; Gouma, D.J.; van Gulik, T.M. Steatosis as a risk factor in liver surgery. Ann. Surg. 2007, 245, 20. [Google Scholar] [CrossRef]
- De Meijer, V.E.; Kalish, B.T.; Puder, M.; IJzermans, J.N.M. Systematic review and meta-analysis of steatosis as a risk factor in major hepatic resection. J. Br. Surg. 2010, 97, 1331–1339. [Google Scholar] [CrossRef]
- Kele, P.G.; van der Jagt, E.J.; Gouw, A.S.; Lisman, T.; Porte, R.J.; de Boer, M.T. The impact of hepatic steatosis on liver regeneration after partial hepatectomy. Liver Int. 2013, 33, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Chu, M.J.; Hickey, A.J.; Phillips, A.R.; Bartlett, A.S. The impact of hepatic steatosis on hepatic ischemia-reperfusion injury in experimental studies: A systematic review. BioMed Res. Int. 2013, 2013, 192029. [Google Scholar] [CrossRef] [PubMed]
- Basaranoglu, M.; Neuschwander-Tetri, B.A. Nonalcoholic fatty liver disease: Clinical features and pathogenesis. Gastroenterol. Hepatol. 2006, 2, 282. [Google Scholar]
- Krawczyk, M.; Bonfrate, L.; Portincasa, P. Nonalcoholic fatty liver disease. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Michalopoulos, G.K. Liver regeneration after partial hepatectomy: Critical analysis of mechanistic dilemmas. Am. J. Pathol. 2010, 176, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Hijona, E.; Hijona, L.; Arenas, J.I.; Bujanda, L. Inflammatory mediators of hepatic steatosis. Mediat. Inflamm. 2010, 2010, 837419. [Google Scholar] [CrossRef]
- Cusi, K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: Pathophysiology and clinical implications. Gastroenterology 2012, 142, 711–725. [Google Scholar] [CrossRef]
- Hirsova, P.; Ibrahim, S.H.; Gores, G.J.; Malhi, H. Lipotoxic lethal and sublethal stress signaling in hepatocytes: Relevance to NASH pathogenesis. J. Lipid Res. 2016, 57, 1758–1770. [Google Scholar] [CrossRef]
- Mota, M.; Banini, B.A.; Cazanave, S.C.; Sanyal, A.J. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism 2016, 65, 1049–1061. [Google Scholar] [CrossRef]
- Wei, Y.; Rector, R.S.; Thyfault, J.P.; Ibdah, J.A. Nonalcoholic fatty liver disease and mitochondrial dysfunction. World J. Gastroenterol. 2008, 14, 193. [Google Scholar] [CrossRef] [PubMed]
- Van Zutphen, T.; Ciapaite, J.; Bloks, V.W.; Ackereley, C.; Gerding, A.; Jurdzinski, A.; Moraes, R.A.; Zhang, L.; Wolters, J.C.; Bischoff, R.; et al. Malnutrition-associated liver steatosis and ATP depletion is caused by peroxisomal and mitochondrial dysfunction. J. Hepatol. 2016, 65, 1198–1208. [Google Scholar] [CrossRef] [PubMed]
- Ibdah, J.A.; Bennett, M.J.; Rinaldo, P.; Zhao, Y.; Gibson, B.; Sims, H.F.; Strauss, A.W. A fetal fatty-acid oxidation disorder as a cause of liver disease in pregnant women. N. Engl. J. Med. 1999, 340, 1723–1731. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Ristow, M. Mitochondria and metabolic homeostasis. Antioxid. Redox Signal. 2013, 19, 240–242. [Google Scholar] [CrossRef]
- Koves, T.R.; Ussher, J.R.; Noland, R.C.; Slentz, D.; Mosedale, M.; Ilkayeva, O. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008, 7, 45–56. [Google Scholar] [CrossRef]
- Nassir, F.; Rector, R.S.; Hammoud, G.M.; Ibdah, J.A. Pathogenesis and prevention of hepatic steatosis. Gastroenterol. Hepatol. 2015, 11, 167. [Google Scholar]
- Marsman, H.A.; De Graaf, W.; Heger, M.; Van Golen, R.F.; Ten Kate, F.J.W.; Bennink, R.; Van Gulik, T.M. Hepatic regeneration and functional recovery following partial liver resection in an experimental model of hepatic steatosis treated with omega-3 fatty acids. J. Br. Surg. 2013, 100, 674–683. [Google Scholar] [CrossRef]
- Gargouri, M.; Magné, C.; El Feki, A. Hyperglycemia, oxidative stress, liver damage and dysfunction in alloxan-induced diabetic rat are prevented by Spirulina supplementation. Nutr. Res. 2016, 36, 1255–1268. [Google Scholar] [CrossRef]
- Gilgenkrantz, H.; de l’Hortet, A.C. Understanding liver regeneration: From mechanisms to regenerative medicine. Am. J. Pathol. 2018, 188, 1316–1327. [Google Scholar] [CrossRef]
- Sarin, S.K.; Choudhury, A. Acute-on-chronic liver failure: Terminology, mechanisms and management. Nat. Rev. Gastroenterol. Hepat. 2016, 13, 131–149. [Google Scholar] [CrossRef]
- Lucchesi, A.N.; Freitas, N.T.D.; Cassettari, L.L.; Marques, S.F.G.; Spadella, C.T. Diabetes mellitus triggers oxidative stress in the liver of alloxan-treated rats: A mechanism for diabetic chronic liver disease. Acta Cir. Bras. 2013, 28, 502–508. [Google Scholar] [CrossRef]
- Francés, D.E.; Ronco, M.T.; Ingaramo, P.I.; Monti, J.A.; Pisani, G.B.; Parody, J.; Pellegrino, J.; Carrillo, M.C.; Martín-Sanz, P.; Carnovale, C.E. Role of reactive oxygen species in the early stages of liver regeneration in streptozotocin-induced diabetic rats. Free Radic. Res. 2011, 45, 1143–1153. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Sun, T.; Han, Y.; Lin, L.; Liu, C.; Liu, J.; Yan, G.; Human, R.T. umbilical cord mesenchymal stem cells implantation accelerates cutaneous wound healing in diabetic rats via the Wnt signaling pathway. Eur. J. Med. Res. 2019, 24, 10. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.L.; He, Y.; Ji, L.L.; Wang, K.Y.; Wang, Y.L.; Chen, D.F.; Geng, Y.; OuYang, P.; Lai, W.M. Hepatoprotective potential of isoquercitrin against type 2 diabetes-induced hepatic injury in rats. Oncotarget 2017, 8, 101545. [Google Scholar] [CrossRef] [PubMed]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Karsdal, M.A.; Manon-Jensen, T.; Genovese, F.; Kristensen, J.H.; Nielsen, M.J.; Sand, J.M.; Hansen, N.U.B.; Bay-Jensen, A.C.; Bager, C.L.; Krag, A.; et al. Novel insights into the function and dynamics of extracellular matrix in liver fibrosis. Am. J. Physiol.-Gastrointest. Liver Physiol. 2015, 308, G807–G830. [Google Scholar]
- Liang, S.; Kisseleva, T.; Brenner, D.A. The role of NADPH oxidases (NOXs) in liver fibrosis and the activation of myofibroblasts. Front. Physiol. 2016, 7, 17. [Google Scholar] [CrossRef]
- Crespo Yanguas, S.; Cogliati, B.; Willebrords, J.; Maes, M.; Colle, I.; Van den Bossche, B.; Souza de Oliveira, C.P.M.; Andraus, W.; Alves, V.A.; Leclercq, I.; et al. Experimental models of liver fibrosis. Arch. Toxicol. 2016, 90, 1025–1048. [Google Scholar] [CrossRef]
- Tanaka, M.; Miyajima, A. Liver regeneration and fibrosis after inflammation. Inflamm. Regen. 2016, 36, 19. [Google Scholar] [CrossRef]
- Dewhurst, M.R.; Ow, J.R.; Zafer, G.; van Hul, N.K.; Wollmann, H.; Bisteau, X.; Brough, D.; Choi, H.; Kaldis, P. Loss of hepatocyte cell division leads to liver inflammation and fibrosis. PLoS Genet. 2020, 16, e1009084. [Google Scholar] [CrossRef]
- Dahlke, M.H.; Popp, F.C.; Bahlmann, F.H.; Aselmann, H.; Jäger, M.D.; Neipp, M.; Piso, P.; Klempnauer, J.; Schlitt, H.J. Liver regeneration in a retrorsine/CCl4–induced acute liver failure model: Do bone marrow-derived cells contribute? J. Hepatol. 2003, 39, 365–373. [Google Scholar] [CrossRef]
- Hafez, M.M.; Al-Shabanah, O.A.; Al-Harbi, N.O.; Al-Harbi, M.M.; Al-Rejaie, S.S.; Alsurayea, S.M.; Sayed-Ahmed, M.M. Association between paraoxonases gene expression and oxidative stress in hepatotoxicity induced by CCl4. Oxidative Med. Cell. Longev. 2014, 2014, 893212. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Pacher, P.; De Lisle, R.C.; Huang, H.; Ding, W.X. A mechanistic review of cell death in alcohol-induced liver injury. Alcohol. Clin. Exp. Res. 2016, 40, 1215–1223. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Yi, X.; Chen, W.; Yuan, Y.; Zhang, X.; Li, J.; Tong, M.; Liu, G.; You, S.; Kong, X. Augmenter of liver regeneration (ALR) gene therapy attenuates CCl4-induced liver injury and fibrosis in rats. Biochem. Biophys. Res. Commun. 2011, 415, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Ulger, O.; Kubat, G.B.; Cicek, Z.; Celik, E.; Atalay, O.; Suvay, S.; Ozler, M. The effects of mitochondrial transplantation in acetaminophen-induced liver toxicity in rats. Life Sci. 2021, 279, 119669. [Google Scholar] [CrossRef] [PubMed]
- Bhushan, B.; Apte, U. Liver regeneration after acetaminophen hepatotoxicity: Mechanisms and therapeutic opportunities. Am. J. Pathol. 2019, 189, 719–729. [Google Scholar] [CrossRef]
- Bhushan, B.; Chavan, H.; Borude, P.; Xie, Y.; Du, K.; McGill, M.R.; Lebofsky, M.; Jaeschke, H.; Krishnamurthy, P.; Apte, U. Dual role of epidermal growth factor receptor in liver injury and regeneration after acetaminophen overdose in mice. Toxicol. Sci. 2017, 155, 363–378. [Google Scholar] [CrossRef]
- Seitz, H.K.; Bataller, R.; Cortez-Pinto, H.; Gao, B.; Gual, A.; Lackner, C.; Mathurin, P.; Mueller, S.; Szabo, G.; Tsukamoto, H. Alcoholic liver disease. Nat. Rev. Dis. Prim. 2018, 4, 16. [Google Scholar] [CrossRef]
- Barr, T.; Helms, C.; Grant, K.; Messaoudi, I. Opposing effects of alcohol on the immune system. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2016, 65, 242–251. [Google Scholar] [CrossRef]
- Dippold, R.P.; Vadigepalli, R.; Gonye, G.E.; Patra, B.; Hoek, J.B. Chronic Ethanol Feeding Alters miRNA Expression Dynamics During Liver Regeneration. Alcohol. Clin. Exp. Res. 2013, 37, E59–E69. [Google Scholar] [CrossRef]
- Louvet, A. Mathurin, Alcoholic liver disease: Mechanisms of injury and targeted treatment. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 231–242. [Google Scholar] [CrossRef]
- Ohashi, K.; Pimienta, M.; Seki, E. Alcoholic liver disease: A current molecular and clinical perspective. Liver Res. 2018, 2, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.R.; Wang, C.; Patel, R.; Trujillo, A.; Patel, N.A.; Prather, J.; Gould, L.J.; Wu, M.H. Human adipose-derived stem cell conditioned media and exosomes containing MALAT1 promote human dermal fibroblast migration and ischemic wound healing. Adv. Wound Care 2018, 7, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, F.G.; Carvalho, M.M.; Panchalingam, K.M.; Rodrigues, A.J.; Mendes-Pinheiro, B.; Anjo, S.; Manadas, B.; Behie, L.A.; Sousa, N.; Salgado, A.J. Impact of the secretome of human mesenchymal stem cells on brain structure and animal behavior in a rat model of Parkinson’s disease. Stem Cells Transl. Med. 2017, 6, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Togel, F.; Weiss, K.; Yang, Y.; Hu, Z.; Zhang, P.; Westenfelder, C. Vasculotropic, paracrine actions of infused mesenchymal stem cells are important to the recovery from acute kidney injury. Am. J. Physiol.-Ren. Physiol. 2007, 292, F1626–F1635. [Google Scholar] [CrossRef]
- Inukai, T.; Katagiri, W.; Yoshimi, R.; Osugi, M.; Kawai, T.; Hibi, H.; Ueda, M. Novel application of stem cell-derived factors for periodontal regeneration. Biochem. Biophys. Res. Commun. 2013, 430, 763–768. [Google Scholar] [CrossRef]
- Heo, S.C.; Jeon, E.S.; Lee, I.H.; Kim, H.S.; Kim, M.B.; Kim, J.H. Tumor necrosis factor-α-activated human adipose tissue–derived mesenchymal stem cells accelerate cutaneous wound healing through paracrine mechanisms. J. Investig. Dermatol. 2011, 131, 1559–1567. [Google Scholar] [CrossRef]
- Hu, C.; Zhao, L.; Wu, Z.; Li, L. Transplantation of mesenchymal stem cells and their derivatives effectively promotes liver regeneration to attenuate acetaminophen-induced liver injury. Stem Cell Res. 2020, 11, 88. [Google Scholar] [CrossRef]
- Nazarie, S.R.; Gharbia, S.; Hermenean, A.; Dinescu, S.; Costache, M. Regenerative potential of mesenchymal stem cells’(MSCs) secretome for liver fibrosis therapies. Int. J. Mol. Sci. 2021, 22, 13292. [Google Scholar] [CrossRef]
- Driscoll, J.; Patel, T. The mesenchymal stem cell secretome as an acellular regenerative therapy for liver disease. J. Gastroenterol. 2019, 54, 763–773. [Google Scholar] [CrossRef]
- Gómez-Aguado, I.; Rodríguez-Castejón, J.; Vicente-Pascual, M.; Rodríguez-Gascón, A.; Solinís, M.Á.; del Pozo-Rodríguez, A. Nanomedicines to deliver mRNA: State of the art and future perspectives. Nanomaterials 2020, 10, 364. [Google Scholar] [CrossRef]
- Kowalski, P.S.; Rudra, A.; Miao, L.; Anderson, D.G. Delivering the messenger: Advances in technologies for therapeutic mRNA delivery. Mol. Ther. 2019, 27, 710–728. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Sharma, A.D.; Roll, G.R.; Ng, R.; Lee, A.Y.; Blelloch, R.H.; Frandsen, N.M.; Willenbring, H. MicroRNAs control hepatocyte proliferation during liver regeneration. Hepatology 2010, 51, 1735–1743. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Shi, Y.; Zhou, X.; Wang, X.; Wang, J.; Lan, Y.; Wang, M.; Zheng, L.; Li, H.; Wu, Q. A set of microRNAs mediate direct conversion of human umbilical cord lining-derived mesenchymal stem cells into hepatocytes. Cell Death Dis. 2013, 4, e918. [Google Scholar] [CrossRef] [PubMed]
- Yi, P.S.; Zhang, M.; Xu, M.Q. Role of microRNA in liver regeneration. Hepatobiliary Pancreat. Dis. Int. 2016, 15, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.L.; Paoletti, C.; Campisi, M.; Osaki, T.; Adriani, G.; Kamm, R.D.; Mattu, C.; Chiono, V. MicroRNA delivery through nanoparticles. J. Control. Release 2019, 313, 80–95. [Google Scholar] [CrossRef] [PubMed]
- Hajj, K.A.; Whitehead, K.A. Tools for translation: Non-viral materials for therapeutic mRNA delivery. Nat. Rev. Mat. 2017, 2, 17056. [Google Scholar] [CrossRef]
- Yang, N. An overview of viral and nonviral delivery systems for microRNA. Int. J. Ppharm. Investig. 2015, 5, 179. [Google Scholar] [CrossRef]
- Monteiro, N.; Martins, A.; Reis, R.L.; Neves, N.M. Liposomes in tissue engineering and regenerative medicine. J. R. Soc. Interface 2014, 11, 20140459. [Google Scholar] [CrossRef]
- Gan, L.; Wang, J.; Zhao, Y.; Chen, D.; Zhu, C.; Liu, J.; Gan, Y. Hyaluronan-modified core-shell liponanoparticles targeting CD44-positive retinal pigment epithelium cells via intravitreal injection. Biomaterials 2013, 34, 5978–5987. [Google Scholar] [CrossRef]
- Apaolaza, P.S.; del Pozo-Rodríguez, A.; Solinís, M.A.; Rodríguez, J.M.; Friedrich, U.; Torrecilla, J.; Weber, B.H.F.; Rodríguez-Gascón, A. Structural recovery of the retina in a retinoschisin-deficient mouse after gene replacement therapy by solid lipid nanoparticles. Biomaterials 2016, 90, 40–49. [Google Scholar] [CrossRef]
- Stewart, M.P.; Sharei, A.; Ding, X.; Sahay, G.; Langer, R.; Jensen, K.F. In vitro and ex vivo strategies for intracellular delivery. Nature 2016, 538, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.A.; Selby, L.I.; Johnston, A.P.; Such, G.K. The endosomal escape of nanoparticles: Toward more efficient cellular delivery. Bioconjugate Chem. 2018, 30, 263–272. [Google Scholar] [CrossRef] [PubMed]
- McNaughton, D.A.; Abu-Yousef, M.M. Doppler US of the liver made simple. Radiographics 2011, 31, 161–188. [Google Scholar] [CrossRef] [PubMed]
- Will, O.M.; Damm, T.; Campbell, G.M.; von Schönfells, W.; Açil, Y.; Will, M.; Chalaris-Rissmann, A.; Ayna, M.; Drucker, C.; Gluer, C.C. Longitudinal micro-computed tomography monitoring of progressive liver regeneration in a mouse model of partial hepatectomy. Lab. Anim. 2017, 51, 422–426. [Google Scholar] [CrossRef]
- Assy, N.; Minuk, G.Y. Liver regeneration: Methods for monitoring and their applications. J. Hepatol. 1997, 26, 945–952. [Google Scholar] [CrossRef]
- Di Martino, M.; Koryukova, K.; Bezzi, M.; Catalano, C. Imaging features of non-alcoholic fatty liver disease in children and adolescents. Children 2017, 4, 73. [Google Scholar] [CrossRef]
- Hoekstra, L.T.; de Graaf, W.; Nibourg, G.A.; Heger, M.; Bennink, R.J.; Stieger, B.; van Gulik, T.M. Physiological and biochemical basis of clinical liver function tests: A review. Ann. Surg. 2013, 257, 27–36. [Google Scholar] [CrossRef]
- Stockmann, M.; Lock, J.F.; Malinowski, M.; Niehues, S.M.; Seehofer, D.; Neuhaus, P. The LiMAx test: A new liver function test for predicting postoperative outcome in liver surgery. Hpb 2010, 12, 139–146. [Google Scholar] [CrossRef]
- Thomas, M.N.; Weninger, E.; Angele, M.; Bösch, F.; Pratschke, S.; Andrassy, J.; Rentsch, M.; Stangl, M.; Hartwig, W.; Werner, J. Intraoperative simulation of remnant liver function during anatomic liver resection with indocyanine green clearance (LiMON) measurements. Hpb 2015, 17, 471–476. [Google Scholar] [CrossRef]
- Wei, W.; Dirsch, O.; Mclean, A.L.; Zafarnia, S.; Schwier, M.; Dahmen, U. Rodent models and imaging techniques to study liver regeneration. Eur. Surg. Res. 2015, 54, 97–113. [Google Scholar] [CrossRef]
- Helmke, S.; Colmenero, J.; Everson, G.T. Non-invasive assessment of liver function. Curr. Opin. Gastroenterol. 2015, 31, 199. [Google Scholar] [CrossRef] [PubMed]
- Iimuro, Y. ICG clearance test and 99mTc-GSA SPECT/CT fusion images. Visc. Med. 2017, 33, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Sumiyoshi, T.; Shima, Y.; Okabayashi, T.; Noda, Y.; Hata, Y.; Murata, Y.; Kozuki, A.; Tokumaru, T.; Nakamura, T.; Uka, K. Functional discrepancy between two liver lobes after hemilobe biliary drainage in patients with jaundice and bile duct cancer: An appraisal using 99mTc-GSA SPECT/CT fusion imaging. Radiology 2014, 273, 444–451. [Google Scholar] [CrossRef]
- Skala, M.C.; Riching, K.M.; Gendron-Fitzpatrick, A.; Eickhoff, J.; Eliceiri, K.W.; White, J.G.; Ramanujam, N. In vivo multiphoton microscopy of NADH and FAD redox states, fluorescence lifetimes, and cellular morphology in precancerous epithelia. Proc. Natl. Acad. Sci. USA 2007, 104, 19494–19499. [Google Scholar] [CrossRef] [PubMed]
- Cicchi, R.; Vogler, N.; Kapsokalyvas, D.; Dietzek, B.; Popp, J.; Pavone, F.S. From molecular structure to tissue architecture: Collagen organization probed by SHG microscopy. J. Biophotonics 2013, 6, 129–142. [Google Scholar] [CrossRef]
- Wang, H.; Liang, X.; Mohammed, Y.H.; Thomas, J.A.; Bridle, K.R.; Thorling, C.A.; Grice, J.E.; Xu, Z.P.; Liu, X.; Crawford, D.H.G.; et al. Real-time histology in liver disease using multiphoton microscopy with fluorescence lifetime imaging. Biomed. Opt. Express 2015, 6, 780. [Google Scholar] [CrossRef]
- Wilson, D.F. Oxidative phosphorylation: Regulation and role in cellular and tissue metabolism. J. Physiol. 2017, 595, 7023–7038. [Google Scholar] [CrossRef]
- Chan, T.S.; Cassim, S.; Raymond, V.A.; Gottschalk, S.; Merlen, G.; Zwingmann, C.; Lapierre, P.; Darby, P.; Mazer, C.D.; Bilodeau, M. Upregulation of Krebs cycle and anaerobic glycolysis activity early after onset of liver ischemia. PLoS ONE 2018, 13, e0199177. [Google Scholar] [CrossRef]
- Datta, R.; Gillette, A.; Stefely, M.; Skala, M.C. Recent innovations in fluorescence lifetime imaging microscopy for biology and medicine. J. Biomed. Opt. 2021, 26, 070603. [Google Scholar] [CrossRef]
- Shirshin, E.A.; Shirmanova, M.V.; Gayer, A.V.; Lukina, M.M.; Nikonova, E.E.; Yakimov, B.; Budylin, G.S.; Dudenkova, V.V.; Ignatova, N.I.; Komarov, D.V.; et al. Label-free sensing of cells with fluorescence lifetime imaging: The quest for metabolic heterogeneity. Proc. Natl. Acad. Sci. USA 2022, 119, e2118241119. [Google Scholar] [CrossRef]
- Verma, B.K.; Subramaniam, P.; Vadigepalli, R. Model-based virtual patient analysis of human liver regeneration predicts critical perioperative factors controlling the dynamic mode of response to resection. BMC Syst. Biol. 2019, 13, 9. [Google Scholar] [CrossRef] [PubMed]
- Verma, B.K.; Subramaniam, P.; Vadigepalli, R. Modeling the dynamics of human liver failure post liver resection. Processes 2018, 6, 115. [Google Scholar] [CrossRef] [PubMed]
- Verma, B.K.; Subramaniam, P.; Vadigepalli, R. Characterizing different class of patients based on their liver regeneration capacity post hepatectomy and the prediction of safe future liver for improved recovery. In Proceedings of the International Conference on Bioinformatics and Systems Biology, Allahabad, India, 26–28 October 2018; pp. 152–156. [Google Scholar]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodimova, S.; Mozherov, A.; Elagin, V.; Karabut, M.; Shchechkin, I.; Kozlov, D.; Krylov, D.; Gavrina, A.; Bobrov, N.; Zagainov, V.; et al. Effect of Hepatic Pathology on Liver Regeneration: The Main Metabolic Mechanisms Causing Impaired Hepatic Regeneration. Int. J. Mol. Sci. 2023, 24, 9112. https://doi.org/10.3390/ijms24119112
Rodimova S, Mozherov A, Elagin V, Karabut M, Shchechkin I, Kozlov D, Krylov D, Gavrina A, Bobrov N, Zagainov V, et al. Effect of Hepatic Pathology on Liver Regeneration: The Main Metabolic Mechanisms Causing Impaired Hepatic Regeneration. International Journal of Molecular Sciences. 2023; 24(11):9112. https://doi.org/10.3390/ijms24119112
Chicago/Turabian StyleRodimova, Svetlana, Artem Mozherov, Vadim Elagin, Maria Karabut, Ilya Shchechkin, Dmitry Kozlov, Dmitry Krylov, Alena Gavrina, Nikolai Bobrov, Vladimir Zagainov, and et al. 2023. "Effect of Hepatic Pathology on Liver Regeneration: The Main Metabolic Mechanisms Causing Impaired Hepatic Regeneration" International Journal of Molecular Sciences 24, no. 11: 9112. https://doi.org/10.3390/ijms24119112
APA StyleRodimova, S., Mozherov, A., Elagin, V., Karabut, M., Shchechkin, I., Kozlov, D., Krylov, D., Gavrina, A., Bobrov, N., Zagainov, V., Zagaynova, E., & Kuznetsova, D. (2023). Effect of Hepatic Pathology on Liver Regeneration: The Main Metabolic Mechanisms Causing Impaired Hepatic Regeneration. International Journal of Molecular Sciences, 24(11), 9112. https://doi.org/10.3390/ijms24119112