

CRISPR-Mediated In Situ Introduction or Integration of F9-Padua in Human iPSCs for Gene Therapy of Hemophilia B
 ,
,  , and
, and 
Abstract
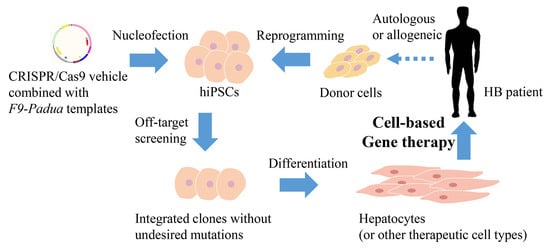
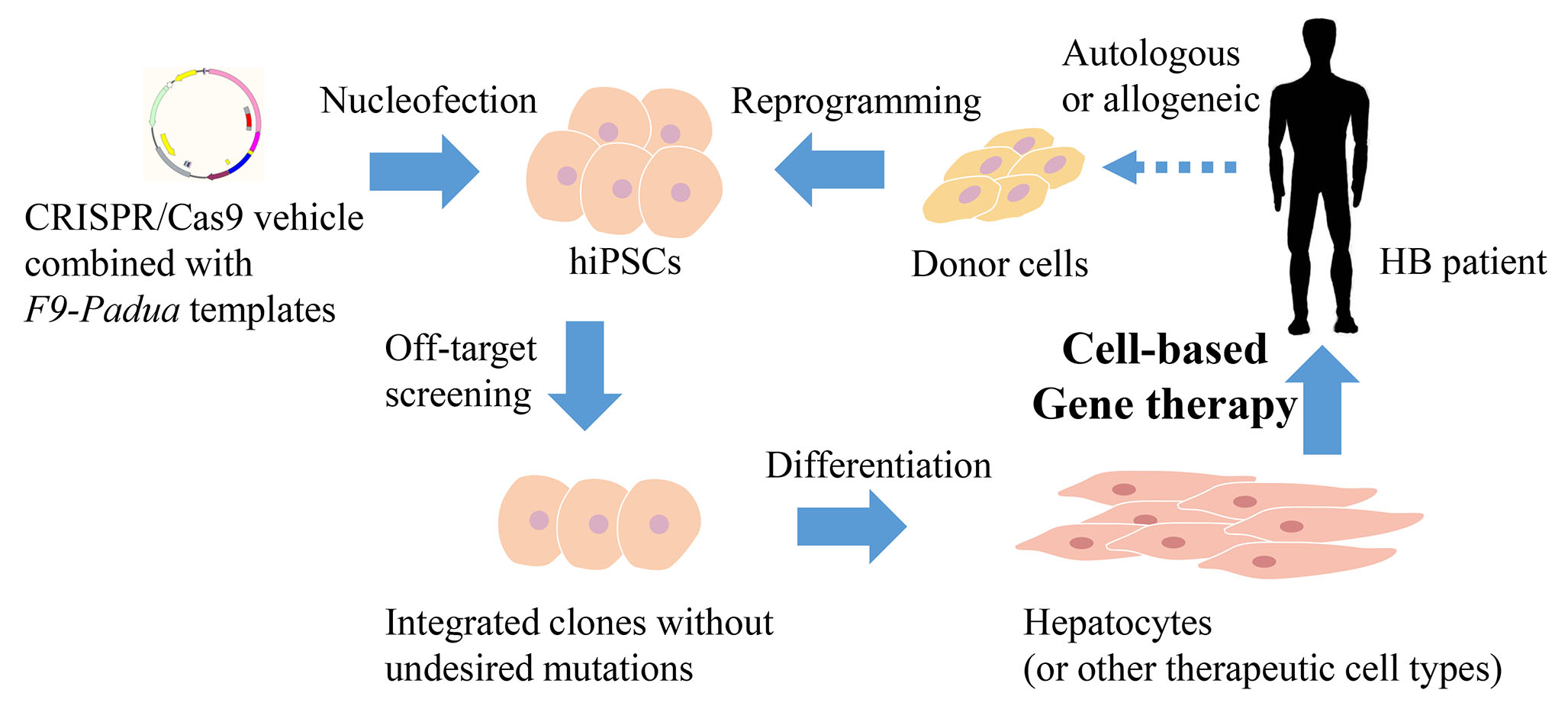{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Introduction of the F9-Padua Mutation into hiPSCs
2.2. Generation and Characterization of HB-hiPSCs
2.3. Integration of Modified Full-Length F9 cDNA with the F9-Padua Mutation in HB-hiPSCs
2.4. Derivation of Hepatocytes from Edited Clones
2.5. Validation of the Hyperactivity of FIX-Padua Proteins Produced by Hepatocytes from the F9-Padua hiPSCs Model and Integrated HB-hiPSCs
3. Discussion
4. Materials and Methods
4.1. Plasmid Construction
4.2. Generation of HB-hiPSCs from Renal Tubular Epithelial Cells
4.3. Alkaline Phosphatase (ALP) Staining
4.4. Karyotyping
4.5. Teratoma Formation Experiments
4.6. Cell Culture
4.7. HEK293T Transfection
4.8. T7E1 Cleavage Assay
4.9. hiPSCs Nucleofection and Genotyping
4.10. Off-Target Analysis
4.11. Immunofluorescence Analysis
4.12. Hepatocyte Differentiation
4.13. qRT-PCR Analysis
4.14. ICG Uptake Assay
4.15. PAS Staining
4.16. FIX Coagulation Activity Test
4.17. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shapiro, A.D.; Hardesty, B.M.; Peyvandi, F.; Iorio, A. Prevalence of selected bleeding and thrombotic events in persons with hemophilia versus the general population: A scoping review. Res. Pract. Thromb. Haemost. 2023, 7, 100007. [Google Scholar] [CrossRef] [PubMed]
- Nienhuis, A.W.; Nathwani, A.C.; Davidoff, A.M. Gene Therapy for Hemophilia. Mol. Ther. 2017, 25, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- Baas, L.; van der Graaf, R.; van Hoorn, E.S.; Bredenoord, A.L.; Meijer, K. The ethics of gene therapy for hemophilia: A narrative review. J. Thromb. Haemost. 2023, 21, 413–420. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, J.; Hughes, D.; Camp, C.; Burke, T.; Carroll, L.; Diego, D.G. The cost of severe haemophilia in Europe: The CHESS study. Orphanet J. Rare Dis. 2017, 12, 106. [Google Scholar] [CrossRef] [PubMed]
- Manco-Johnson, M.J.; Abshire, T.C.; Shapiro, A.D.; Riske, B.; Hacker, M.R.; Kilcoyne, R.; Ingram, J.D.; Manco-Johnson, M.L.; Funk, S.; Jacobson, L.; et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N. Engl. J. Med. 2007, 357, 535–544. [Google Scholar] [CrossRef]
- Lyu, C.; Shen, J.; Wang, R.; Gu, H.; Zhang, J.; Xue, F.; Liu, X.; Liu, W.; Fu, R.; Zhang, L.; et al. Targeted genome engineering in human induced pluripotent stem cells from patients with hemophilia B using the CRISPR-Cas9 system. Stem Cell Res. Ther. 2018, 9, 92. [Google Scholar] [CrossRef]
- Simioni, P.; Tormene, D.; Tognin, G.; Gavasso, S.; Bulato, C.; Iacobelli, N.P.; Finn, J.D.; Spiezia, L.; Radu, C.; Arruda, V.R. X-linked thrombophilia with a mutant factor IX (factor IX Padua). N. Engl. J. Med. 2009, 361, 1671–1675. [Google Scholar] [CrossRef]
- Crudele, J.M.; Finn, J.D.; Siner, J.I.; Martin, N.B.; Niemeyer, G.P.; Zhou, S.; Mingozzi, F.; Lothrop, C.D., Jr.; Arruda, V.R. AAV liver expression of FIX-Padua prevents and eradicates FIX inhibitor without increasing thrombogenicity in hemophilia B dogs and mice. Blood 2015, 125, 1553–1561. [Google Scholar] [CrossRef]
- French, R.A.; Samelson-Jones, B.J.; Niemeyer, G.P.; Lothrop, C.D., Jr.; Merricks, E.P.; Nichols, T.C.; Arruda, V.R. Complete correction of hemophilia B phenotype by FIX-Padua skeletal muscle gene therapy in an inhibitor-prone dog model. Blood Adv. 2018, 2, 505–508. [Google Scholar] [CrossRef]
- Monahan, P.E.; Sun, J.; Gui, T.; Hu, G.; Hannah, W.B.; Wichlan, D.G.; Wu, Z.; Grieger, J.C.; Li, C.; Suwanmanee, T.; et al. Employing a gain-of-function factor IX variant R338L to advance the efficacy and safety of hemophilia B human gene therapy: Preclinical evaluation supporting an ongoing adeno-associated virus clinical trial. Hum. Gene Ther. 2015, 26, 69–81. [Google Scholar] [CrossRef]
- Cantore, A.; Ranzani, M.; Bartholomae, C.C.; Volpin, M.; Valle, P.D.; Sanvito, F.; Sergi, L.S.; Gallina, P.; Benedicenti, F.; Bellinger, D.; et al. Liver-directed lentiviral gene therapy in a dog model of hemophilia B. Sci. Transl. Med. 2015, 7, 277ra228. [Google Scholar] [CrossRef]
- Samelson-Jones, B.J.; George, L.A. Adeno-Associated Virus Gene Therapy for Hemophilia. Annu. Rev. Med. 2023, 74, 231–247. [Google Scholar] [CrossRef]
- Arruda, V.R.; Doshi, B.S.; Samelson-Jones, B.J. Novel approaches to hemophilia therapy: Successes and challenges. Blood 2017, 130, 2251–2256. [Google Scholar] [CrossRef]
- Samelson-Jones, B.J.; Finn, J.D.; George, L.A.; Camire, R.M.; Arruda, V.R. Hyperactivity of factor IX Padua (R338L) depends on factor VIIIa cofactor activity. JCI Insight 2019, 5, e128683. [Google Scholar] [CrossRef]
- Heo, Y.A. Etranacogene Dezaparvovec: First Approval. Drugs 2023, 83, 347–352. [Google Scholar] [CrossRef]
- Doshi, B.S.; Arruda, V.R. Gene therapy for hemophilia: What does the future hold? Ther. Adv. Hematol. 2018, 9, 273–293. [Google Scholar] [CrossRef]
- Nathwani, A.C.; Rosales, C.; McIntosh, J.; Rastegarlari, G.; Nathwani, D.; Raj, D.; Nawathe, S.; Waddington, S.N.; Bronson, R.; Jackson, S.; et al. Long-term safety and efficacy following systemic administration of a self-complementary AAV vector encoding human FIX pseudotyped with serotype 5 and 8 capsid proteins. Mol. Ther. 2011, 19, 876–885. [Google Scholar] [CrossRef]
- Nathwani, A.C. Gene therapy for hemophilia. Hematol. Am. Soc. Hematol. Educ. Progr. 2022, 2022, 569–578. [Google Scholar] [CrossRef]
- Ganesan, L.P.; Mohanty, S.; Kim, J.; Clark, K.R.; Robinson, J.M.; Anderson, C.L. Rapid and efficient clearance of blood-borne virus by liver sinusoidal endothelium. PLoS Pathog. 2011, 7, e1002281. [Google Scholar] [CrossRef]
- Dirisala, A.; Uchida, S.; Toh, K.; Li, J.; Osawa, S.; Tockary, T.A.; Liu, X.; Abbasi, S.; Hayashi, K.; Mochida, Y.; et al. Transient stealth coating of liver sinusoidal wall by anchoring two-armed PEG for retargeting nanomedicines. Sci. Adv. 2020, 6, eabb8133. [Google Scholar] [CrossRef]
- Shieh, P.B.; Bönnemann, C.G.; Müller-Felber, W.; Blaschek, A.; Dowling, J.J.; Kuntz, N.L.; Seferian, A.M. Re: “Moving Forward After Two Deaths in a Gene Therapy Trial of Myotubular Myopathy” by Wilson and Flotte. Hum. Gene Ther. 2020, 31, 787. [Google Scholar] [CrossRef] [PubMed]
- Hinderer, C.; Katz, N.; Buza, E.L.; Dyer, C.; Goode, T.; Bell, P.; Richman, L.K.; Wilson, J.M. Severe Toxicity in Nonhuman Primates and Piglets Following High-Dose Intravenous Administration of an Adeno-Associated Virus Vector Expressing Human SMN. Hum. Gene Ther. 2018, 29, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Knott, G.J.; Doudna, J.A. CRISPR-Cas guides the future of genetic engineering. Science 2018, 361, 866–869. [Google Scholar] [CrossRef] [PubMed]
- Pulecio, J.; Verma, N.; Mejía-Ramírez, E.; Huangfu, D.; Raya, A. CRISPR/Cas9-Based Engineering of the Epigenome. Cell Stem Cell 2017, 21, 431–447. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, J.; Duan, N.; Liu, W.; Zhang, Y.; Zhou, M.; Hu, Z.; Feng, M.; Liu, X.; Wu, L.; et al. Paired CRISPR/Cas9 Nickases Mediate Efficient Site-Specific Integration of F9 into rDNA Locus of Mouse ESCs. Int. J. Mol. Sci. 2018, 19, 3035. [Google Scholar] [CrossRef]
- Guan, Y.; Ma, Y.; Li, Q.; Sun, Z.; Ma, L.; Wu, L.; Wang, L.; Zeng, L.; Shao, Y.; Chen, Y.; et al. CRISPR/Cas9-mediated somatic correction of a novel coagulator factor IX gene mutation ameliorates hemophilia in mouse. EMBO Mol. Med. 2016, 8, 477–488. [Google Scholar] [CrossRef]
- Guo, C.; Ma, X.; Gao, F.; Guo, Y. Off-target effects in CRISPR/Cas9 gene editing. Front. Bioeng. Biotechnol. 2023, 11, 1143157. [Google Scholar] [CrossRef]
- Rallapalli, P.M.; Kemball-Cook, G.; Tuddenham, E.G.; Gomez, K.; Perkins, S.J. An interactive mutation database for human coagulation factor IX provides novel insights into the phenotypes and genetics of hemophilia B. J. Thromb. Haemost. 2013, 11, 1329–1340. [Google Scholar] [CrossRef]
- Mazetto Bde, M.; Orsi, F.L.; Siqueira, L.H.; de Mello, T.B.; de Paula, E.V.; Annichino-Bizzacchi, J.M. Prevalence of Factor IX-R338L (Factor IX Padua) in a cohort of patients with venous thromboembolism and mild elevation of factor IX levels. Thromb. Res. 2010, 126, e165. [Google Scholar] [CrossRef]
- Koenderman, J.S.; Bertina, R.M.; Reitsma, P.H.; de Visser, M.C. Factor IX-R338L (Factor IX Padua) screening in a Dutch population of sibpairs with early onset venous thromboembolism. Thromb. Res. 2011, 128, 603. [Google Scholar] [CrossRef]
- Vielhaber, E.; Jacobson, D.P.; Ketterling, R.P.; Liu, J.Z.; Sommer, S.S. A mutation in the 3′ untranslated region of the factor IX gene in four families with hemophilia B. Hum. Mol. Genet. 1993, 2, 1309–1310. [Google Scholar] [CrossRef]
- Anokye-Danso, F.; Trivedi, C.M.; Juhr, D.; Gupta, M.; Cui, Z.; Tian, Y.; Zhang, Y.; Yang, W.; Gruber, P.J.; Epstein, J.A.; et al. Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell 2011, 8, 376–388. [Google Scholar] [CrossRef]
- Zhao, Y.; Cui, Y.; Luan, J.; Wang, J.; Shi, L.; Han, Z.; Han, J. Development and identification of a induced pluripotent stem cells line (SMBCi013-A) derived from urine cells of a patient with Wilson’s disease. Stem Cell Res 2021, 59, 102650. [Google Scholar] [CrossRef]
- Tatsumi, K.; Ohashi, K.; Mukobata, S.; Kubo, A.; Koyama, F.; Nakajima, Y.; Shima, M.; Okano, T. Hepatocyte Is a Sole Cell Type Responsible for the Production of Coagulation Factor IX In Vivo. Cell Med. 2012, 3, 25–31. [Google Scholar] [CrossRef]
- Toba, Y.; Kiso, A.; Nakamae, S.; Sakurai, F.; Takayama, K.; Mizuguchi, H. FGF signal is not required for hepatoblast differentiation of human iPS cells. Sci. Rep. 2019, 9, 3713. [Google Scholar] [CrossRef]
- Boti, M.A.; Athanasopoulou, K.; Adamopoulos, P.G.; Sideris, D.C.; Scorilas, A. Recent Advances in Genome-Engineering Strategies. Genes 2023, 14, 129. [Google Scholar] [CrossRef]
- Zhang, H.X.; Zhang, Y.; Yin, H. Genome Editing with mRNA Encoding ZFN, TALEN, and Cas9. Mol. Ther. 2019, 27, 735–746. [Google Scholar] [CrossRef]
- Veres, A.; Gosis, B.S.; Ding, Q.; Collins, R.; Ragavendran, A.; Brand, H.; Erdin, S.; Cowan, C.A.; Talkowski, M.E.; Musunuru, K. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell 2014, 15, 27–30. [Google Scholar] [CrossRef]
- Park, C.Y.; Kim, D.H.; Son, J.S.; Sung, J.J.; Lee, J.; Bae, S.; Kim, J.H.; Kim, D.W.; Kim, J.S. Functional Correction of Large Factor VIII Gene Chromosomal Inversions in Hemophilia A Patient-Derived iPSCs Using CRISPR-Cas9. Cell Stem Cell 2015, 17, 213–220. [Google Scholar] [CrossRef]
- Ramaswamy, S.; Tonnu, N.; Menon, T.; Lewis, B.M.; Green, K.T.; Wampler, D.; Monahan, P.E.; Verma, I.M. Autologous and Heterologous Cell Therapy for Hemophilia B toward Functional Restoration of Factor IX. Cell Rep. 2018, 23, 1565–1580. [Google Scholar] [CrossRef]
- Pasi, K.J.; Fischer, K.; Ragni, M.; Kulkarni, R.; Ozelo, M.C.; Mahlangu, J.; Shapiro, A.; P’Ng, S.; Chambost, H.; Nolan, B.; et al. Long-term safety and sustained efficacy for up to 5 years of treatment with recombinant factor IX Fc fusion protein in subjects with haemophilia B: Results from the B-YOND extension study. Haemophilia 2020, 26, e262–e271. [Google Scholar] [CrossRef] [PubMed]
- Nathwani, A.C.; Tuddenham, E.G.; Rangarajan, S.; Rosales, C.; McIntosh, J.; Linch, D.C.; Chowdary, P.; Riddell, A.; Pie, A.J.; Harrington, C.; et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N. Engl. J. Med. 2011, 365, 2357–2365. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhong, X.; Li, Q.; Su, J.; Liu, Y.; Mo, L.; Deng, H.; Yang, Y. CRISPR-Cas9-Mediated In Vivo Gene Integration at the Albumin Locus Recovers Hemostasis in Neonatal and Adult Hemophilia B Mice. Mol. Ther. Methods Clin. Dev. 2020, 18, 520–531. [Google Scholar] [CrossRef]
- Knight, K.A.; Coyle, C.W.; Radford, C.E.; Parker, E.T.; Fedanov, A.; Shields, J.M.; Szlam, F.; Purchel, A.; Chen, M.; Denning, G.; et al. Identification of coagulation factor IX variants with enhanced activity through ancestral sequence reconstruction. Blood Adv. 2021, 5, 3333–3343. [Google Scholar] [CrossRef] [PubMed]
- Samelson-Jones, B.J.; Finn, J.D.; Raffini, L.J.; Merricks, E.P.; Camire, R.M.; Nichols, T.C.; Arruda, V.R. Evolutionary insights into coagulation factor IX Padua and other high-specific-activity variants. Blood Adv. 2021, 5, 1324–1332. [Google Scholar] [CrossRef]
- Lin, C.N.; Kao, C.Y.; Miao, C.H.; Hamaguchi, N.; Wu, H.L.; Shi, G.Y.; Liu, Y.L.; High, K.A.; Lin, S.W. Generation of a novel factor IX with augmented clotting activities in vitro and in vivo. J. Thromb. Haemost. 2010, 8, 1773–1783. [Google Scholar] [CrossRef]
- Kao, C.Y.; Yang, S.J.; Tao, M.H.; Jeng, Y.M.; Yu, I.S.; Lin, S.W. Incorporation of the factor IX Padua mutation into FIX-Triple improves clotting activity in vitro and in vivo. Thromb. Haemost. 2013, 110, 244–256. [Google Scholar] [CrossRef]
- Le Quellec, S.; Dane, A.P.; Barbon, E.; Bordet, J.C.; Mingozzi, F.; Dargaud, Y.; Marais, T.; Biferi, M.G.; Négrier, C.; Nathawani, A.C.; et al. Recombinant Adeno-Associated Viral Vectors Expressing Human Coagulation FIX-E456H Variant in Hemophilia B Mice. Thromb. Haemost. 2019, 119, 1956–1967. [Google Scholar] [CrossRef]
- Pan, C.; Kumar, C.; Bohl, S.; Klingmueller, U.; Mann, M. Comparative proteomic phenotyping of cell lines and primary cells to assess preservation of cell type-specific functions. Mol. Cell Proteomics 2009, 8, 443–450. [Google Scholar] [CrossRef]
- Alge, C.S.; Hauck, S.M.; Priglinger, S.G.; Kampik, A.; Ueffing, M. Differential protein profiling of primary versus immortalized human RPE cells identifies expression patterns associated with cytoskeletal remodeling and cell survival. J. Proteome Res. 2006, 5, 862–878. [Google Scholar] [CrossRef]
- Foley, J.H.; Shehu, E.; Riddell, A.; Gray, E.; Goodale, A.; Yu, I.M.; Verhoef, D.; Little, J.; Shattock, D.; Kitchen, S.; et al. Differences in wild-type- and R338L-tenase complex formation are at the root of R338L-factor IX assay discrepancies. Blood Adv. 2023, 7, 458–467. [Google Scholar] [CrossRef]
- Mannucci, P.M. Hemophilia treatment innovation: 50 years of progress and more to come. J. Thromb. Haemost. 2023, 21, 403–412. [Google Scholar] [CrossRef]
- Gollomp, K.L.; Doshi, B.S.; Arruda, V.R. Gene therapy for hemophilia: Progress to date and challenges moving forward. Transfus. Apher. Sci. 2019, 58, 602–612. [Google Scholar] [CrossRef]
- Cavazza, A.; Moiani, A.; Mavilio, F. Mechanisms of retroviral integration and mutagenesis. Hum. Gene Ther. 2013, 24, 119–131. [Google Scholar] [CrossRef]
- Wong, S.P.; Argyros, O.; Harbottle, R.P. Sustained expression from DNA vectors. Adv. Genet. 2015, 89, 113–152. [Google Scholar] [CrossRef]
- Hasbrouck, N.C.; High, K.A. AAV-mediated gene transfer for the treatment of hemophilia B: Problems and prospects. Gene. Ther. 2008, 15, 870–875. [Google Scholar] [CrossRef]
- Ernst, M.P.T.; Broeders, M.; Herrero-Hernandez, P.; Oussoren, E.; van der Ploeg, A.T.; Pijnappel, W. Ready for Repair? Gene Editing Enters the Clinic for the Treatment of Human Disease. Mol. Ther. Methods Clin. Dev. 2020, 18, 532–557. [Google Scholar] [CrossRef]
- Wu, Y.M.; Huang, Y.J.; Chen, P.; Hsu, Y.C.; Lin, S.W.; Lai, H.S.; Lee, H.S. Hepatocyte-Like Cells Derived From Mouse Induced Pluripotent Stem Cells Produce Functional Coagulation Factor IX in a Hemophilia B Mouse Model. Cell Transplant. 2016, 25, 1237–1246. [Google Scholar] [CrossRef]
- Okamoto, R.; Takayama, K.; Akita, N.; Nagamoto, Y.; Hosokawa, D.; Iizuka, S.; Sakurai, F.; Suemizu, H.; Ohashi, K.; Mizuguchi, H. Human iPS Cell-based Liver-like Tissue Engineering at Extrahepatic Sites in Mice as a New Cell Therapy for Hemophilia B. Cell Transplant. 2018, 27, 299–309. [Google Scholar] [CrossRef]
- Köchl, S.; Niederstätter, H.; Parson, W. DNA extraction and quantitation of forensic samples using the phenol-chloroform method and real-time PCR. Methods Mol. Biol. 2005, 297, 13–30. [Google Scholar] [CrossRef]
- Toni, L.S.; Garcia, A.M.; Jeffrey, D.A.; Jiang, X.; Stauffer, B.L.; Miyamoto, S.D.; Sucharov, C.C. Optimization of phenol-chloroform RNA extraction. MethodsX 2018, 5, 599–608. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, Q.; Hu, Z.; Zhao, J.; Zhou, T.; Tang, S.; Wang, P.; Xiao, R.; Chen, Y.; Wu, L.; Zhou, M.; et al. CRISPR-Mediated In Situ Introduction or Integration of F9-Padua in Human iPSCs for Gene Therapy of Hemophilia B. Int. J. Mol. Sci. 2023, 24, 9013. https://doi.org/10.3390/ijms24109013
Tang Q, Hu Z, Zhao J, Zhou T, Tang S, Wang P, Xiao R, Chen Y, Wu L, Zhou M, et al. CRISPR-Mediated In Situ Introduction or Integration of F9-Padua in Human iPSCs for Gene Therapy of Hemophilia B. International Journal of Molecular Sciences. 2023; 24(10):9013. https://doi.org/10.3390/ijms24109013
Chicago/Turabian StyleTang, Qiyu, Zhiqing Hu, Junya Zhao, Tao Zhou, Shuqing Tang, Peiyun Wang, Rou Xiao, Yan Chen, Lingqian Wu, Miaojin Zhou, and et al. 2023. "CRISPR-Mediated In Situ Introduction or Integration of F9-Padua in Human iPSCs for Gene Therapy of Hemophilia B" International Journal of Molecular Sciences 24, no. 10: 9013. https://doi.org/10.3390/ijms24109013
APA StyleTang, Q., Hu, Z., Zhao, J., Zhou, T., Tang, S., Wang, P., Xiao, R., Chen, Y., Wu, L., Zhou, M., & Liang, D. (2023). CRISPR-Mediated In Situ Introduction or Integration of F9-Padua in Human iPSCs for Gene Therapy of Hemophilia B. International Journal of Molecular Sciences, 24(10), 9013. https://doi.org/10.3390/ijms24109013