

Neurotensin and Alcohol Use Disorders: Towards a Pharmacological Treatment




Abstract
1. Introduction
2. Neurotensin and NT Receptors
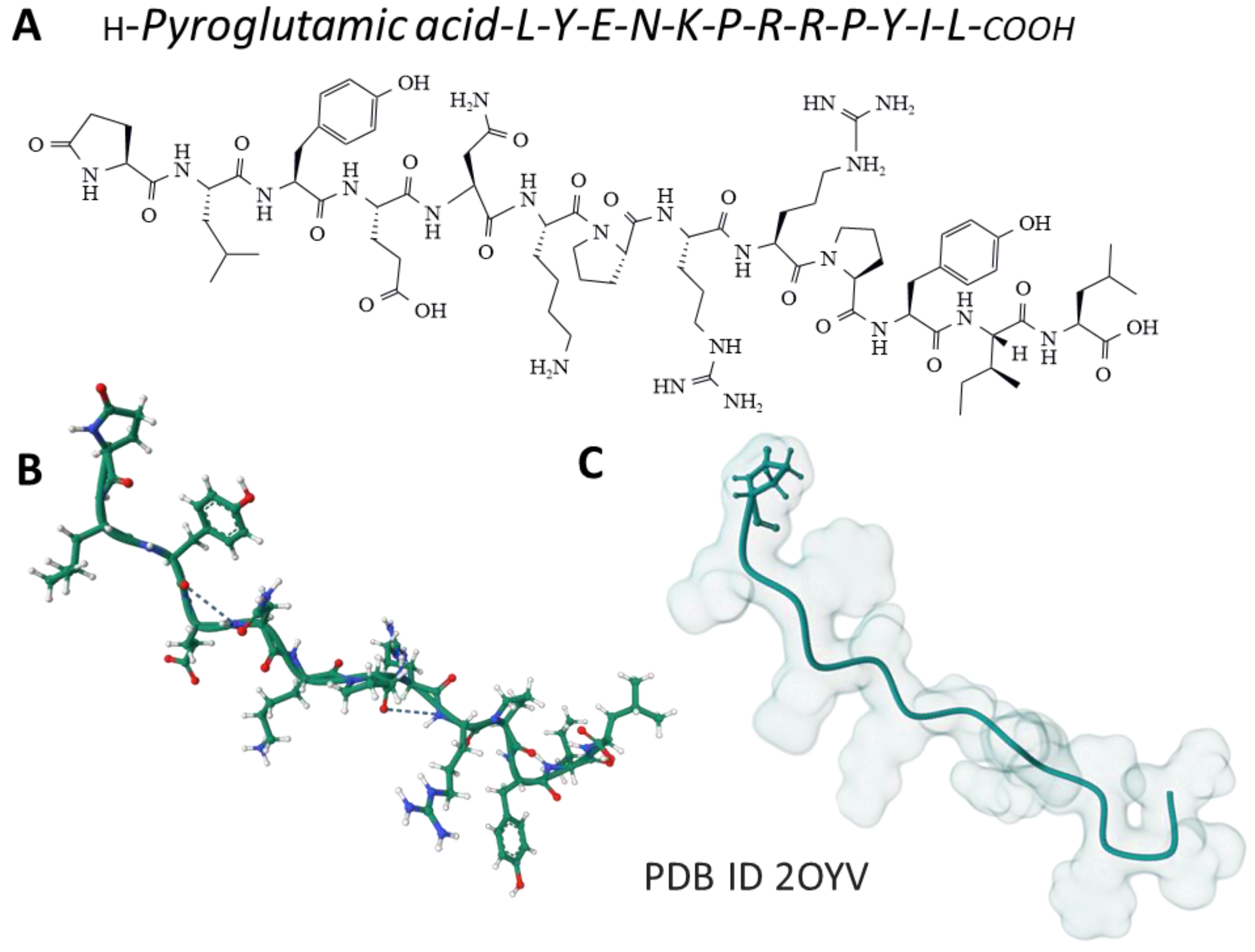
2.1. Neurotensin
2.2. Brain Distribution of Neurotensin
2.3. Neurotensin Receptors
2.3.1. NTR1
2.3.2. NTR2
2.3.3. NTR3/Sortilin
2.4. Neurotensin Signaling Pathways
3. Neurotensin, Reward, and Alcohol
3.1. Brain Reward Circuits
3.2. NT, Reward, and Stress
3.3. NT and Alcohol
4. NT Receptors Ligands as Possible Pharmacological Treatments of AUD
5. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Patussi, V.; Fanucchi, T.; Marcomini, F.; Balbinot, P.; Testino, G.; Caputo, F. Alcohol use disorders: Illness or other? Panminerva Med. 2019, 61, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Schuckit, M.A. Alcohol-use disorders. Lancet 2009, 373, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Heilig, M.; MacKillop, J.; Martinez, D.; Rehm, J.; Leggio, L.; Vanderschuren, L.J.M.J. Addiction as a brain disease revised: Why it still matters, and the need for consilience. Neuropsychopharmacology 2021, 46, 1715–1723. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.F.; Heilig, M.; Perez, A.; Probst, C.; Rehm, J. Alcohol use disorders. Lancet 2019, 394, 781–792. [Google Scholar] [CrossRef]
- World Health Organization. Global Status Report on Alcohol and Health 2018; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- Status Report on Alcohol Consumption, Harm, and Policy Responses in 30 European Countries. 2019. Available online: https://www.euro.who.int/__data/assets/pdf_file/0019/411418/Alcohol-consumption-harm-policy-responses-30-European-countries-2019.pdf (accessed on 30 January 2023).
- Technical Report on Alcohol 20–21. Consumption and Consequences. Available online: https://pnsd.sanidad.gob.es/profesionales/publicaciones/catalogo/catalogoPNSD/publicaciones/pdf/2022_Technical_Report_Alcohol_2021.pdf (accessed on 30 January 2023).
- John, U.; Rumpf, H.; Hanke, M.; Meyer, C. Severity of alcohol dependence and mortality after 20 years in an adult general population sample. Int. J. Methods Psychiatr. Res. 2022, 31, e1915. [Google Scholar] [CrossRef]
- Feltenstein, M.W.; See, R.E.; Fuchs, R.A. Neural substrates and circuits of drug addiction. Cold Spring Harb. Perspect. Med. 2021, 11, a039628. [Google Scholar] [CrossRef]
- Uhl, G.R.; Koob, G.F.; Cable, J. The neurobiology of addiction. Ann. N. Y. Acad. Sci. 2019, 1451, 5–28. [Google Scholar] [CrossRef]
- Horseman, C.; Meyer, A. Neurobiology of Addiction. Clin. Obstet. Gynecol. 2019, 62, 118–127. [Google Scholar] [CrossRef]
- Koob, G.F.; Volkow, N.D. Neurobiology of addiction: A neurocircuitry analysis. Lancet Psychiatry 2016, 3, 760–773. [Google Scholar] [CrossRef]
- Domi, E.; Domi, A.; Adermark, L.; Heilig, M.; Augier, E. Neurobiology of alcohol-seeking behavior. J. Neurochem. 2021, 157, 1585–1614. [Google Scholar] [CrossRef]
- Volkow, N.D.; Michaelides, M.; Baler, R. The neuroscience of drug reward and addiction. Physiol. Rev. 2019, 99, 2115–2140. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Singla, R.; Maheshwari, O.; Fontaine, C.J.; Gil-Mohapel, J. Alcohol use disorder: Neurobiology and therapeutics. Biomedicines 2022, 10, 1192. [Google Scholar] [CrossRef] [PubMed]
- Thorsell, A.; Mathé, A.A. Neuropeptide Y in alcohol addiction and affective disorders. Front. Endocrinol. 2017, 8, 178. [Google Scholar] [CrossRef] [PubMed]
- Cannella, N.; Borruto, A.M.; Petrella, M.; Micioni Di Bonaventura, M.V.; Soverchia, L.; Cifani, C.; De Carlo, S.; Domi, E.; Ubaldi, M. A Role for neuropeptide S in alcohol and cocaine seeking. Pharmaceuticals 2022, 15, 800. [Google Scholar] [CrossRef] [PubMed]
- Matzeu, A.; Martin-Fardon, R. Understanding the role of orexin neuropeptides in drug addiction: Preclinical studies and translational value. Front. Behav. Neurosci. 2021, 15, 787595. [Google Scholar] [CrossRef]
- Rodriguez, F.D.; Coveñas, R. Targeting NPY, CRF/UCNs, and NPS Neuropeptide systems to treat alcohol use disorder (AUD). Curr. Med. Chem. 2017, 24, 2528–2558. [Google Scholar] [CrossRef]
- Rodriguez, F.D.; Coveñas, R. Targeting opioid and neurokinin-1 receptors to treat alcoholism. Curr. Med. Chem. 2011, 18, 4321–4334. [Google Scholar] [CrossRef]
- Hartmann, M.C.; Pleil, K.E. Circuit and neuropeptide mechanisms of the paraventricular thalamus across stages of alcohol and drug use. Neuropharmacology 2021, 198, 108748. [Google Scholar] [CrossRef]
- The American Society of Addiction Medicine. Available online: https://www.asam.org/docs/default-source/quality-science/asam’s-2019-definition-of-addiction-(1).pdf?sfvrsn=b8b64fc2_2 (accessed on 26 January 2023).
- Mekonen, T.; Chan, G.C.K.; Connor, J.; Hall, W.; Hides, L.; Leung, J. Treatment rates for alcohol use disorders: A systematic review and meta-analysis. Addiction 2021, 116, 2617. [Google Scholar] [CrossRef]
- Gual, A.; Drummond, C. Killing me softly: Alcohol addiction today. Eur. Neuropsychopharmacol. 2022, 57, 30–32. [Google Scholar] [CrossRef]
- Connor, J.P.; Haber, P.S.; Hall, W.D. Alcohol use disorders. Lancet 2016, 387, 988. [Google Scholar] [CrossRef] [PubMed]
- Mihalak, K.B.; Carroll, F.I.; Luetje, C.W. Varenicline is a partial agonist at alpha4beta2 and a full agonist at alpha7 neuronal nicotinic receptors. Mol. Pharmacol. 2006, 70, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Witkiewitz, K.; Litten, R.Z.; Leggio, L. Advances in the science and treatment of alcohol use disorder. Sci. Adv. 2019, 5, eaax4043. [Google Scholar] [CrossRef]
- Ezquer, F.; Quintanilla, M.E.; Morales, P.; Santapau, D.; Munita, J.M.; Moya-Flores, F.; Ezquer, M.; Herrera-Marschitz, M.; Israel, Y. A dual treatment blocks alcohol binge-drinking relapse: Microbiota as a new player. Drug Alcohol Depend. 2022, 236, 109466. [Google Scholar] [CrossRef]
- Burnette, E.M.; Nieto, S.J.; Grodin, E.N.; Meredith, L.R.; Hurley, B.; Miotto, K.; Gillis, A.J.; Ray, L.A. Novel agents for the pharmacological treatment of alcohol use disorder. Drugs 2022, 82, 251–274. [Google Scholar] [CrossRef] [PubMed]
- van den Brink, W.; Addolorato, G.; Aubin, H.; Benyamina, A.; Caputo, F.; Dematteis, M.; Gual, A.; Lesch, O.; Mann, K.; Maremmani, I.; et al. Efficacy and safety of sodium oxybate in alcohol-dependent patients with a very high drinking risk level. Addict. Biol. 2018, 23, 969–986. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chen, Y.; Chen, S.; Lee, C.; Cheng, C. Alcohol addiction, gut microbiota, and alcoholism treatment: A review. Int. J. Mol. Sci. 2020, 21, 6413. [Google Scholar] [CrossRef]
- Barson, J.R. The role of neuropeptides in drug and ethanol abuse: Medication targets for drug and alcohol use disorders. Brain Res. 2020, 1740, 146876. [Google Scholar] [CrossRef]
- Carraway, R.; Dobner, P. Neurotensin/Neuromedin N. In Handbook of Biologically Active Peptides; Academic Press: Boston, MA, USA, 2013; pp. 875–882. [Google Scholar]
- Coutant, J.; Curmi, P.A.; Toma, F.; Monti, J. NMR solution structure of neurotensin in membrane-mimetic environments: Molecular basis for neurotensin receptor recognition. Biochemistry 2007, 46, 5656–5663. [Google Scholar] [CrossRef]
- RCSB PDB: Homepage. Available online: https://www.rcsb.org/ (accessed on 30 January 2023).
- Mustain, W.C.; Rychahou, P.G.; Evers, B.M. The role of neurotensin in physiologic and pathologic processes. Curr. Opin. Endocrinol. Diabetes Obes. 2011, 18, 75–82. [Google Scholar] [CrossRef]
- Bugni, J.; Pothoulakis, C. Neurotensin. In Handbook of Biologically Active Peptides, 2nd ed.; Academic Press: Boston, MA, USA, 2013; pp. 1265–1270. [Google Scholar]
- Sánchez, M.L.; Coveñas, R. The Neurotensinergic system: A target for cancer treatment. Curr. Med. Chem. 2022, 29, 3231–3260. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Huang, Y.; Zhang, B.; Wang, Y.; Zhao, H.; Du, H.; Cong, Z.; Li, J.; Zhu, G. Association between neurotensin receptor 1 gene polymorphisms and alcohol dependence in a male Han Chinese population. J. Mol. Neurosci. 2013, 51, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.R.; Hinton, D.J.; Song, J.Y.; Lee, K.W.; Choo, C.; Johng, H.; Unal, S.S.; Richelson, E.; Choi, D. Neurotensin receptor type 1 regulates ethanol intoxication and consumption in mice. Pharmacol. Biochem. Behav. 2010, 95, 235–241. [Google Scholar] [CrossRef]
- Rock, S.; Li, X.; Song, J.; Townsend, C.M.J.; Weiss, H.L.; Rychahou, P.; Gao, T.; Li, J.; Evers, B.M. Kinase suppressor of Ras 1 and Exo70 promote fatty acid-stimulated neurotensin secretion through ERK1/2 signaling. PLoS ONE 2019, 14, e0211134. [Google Scholar] [CrossRef]
- Feng, Y.; Wang, J.; Dong, Y.; Wang, Y.; Li, Y. The roles of neurotensin and its analogs in pain. Curr. Pharm. Des. 2015, 21, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Namburi, P.; Olson, J.M.; Borio, M.; Lemieux, M.E.; Beyeler, A.; Calhoon, G.G.; Hitora-Imamura, N.; Coley, A.A.; Libster, A.; et al. Neurotensin orchestrates valence assignment in the amygdala. Nature 2022, 608, 586–592. [Google Scholar] [CrossRef]
- Arbogast, P.; Gauchotte, G.; Mougel, R.; Morel, O.; Ziyyat, A.; Agopiantz, M. Neurotensin and its involvement in reproductive functions: An exhaustive review of the literature. Int. J. Mol. Sci. 2023, 24, 4594. [Google Scholar] [CrossRef]
- Brun, P.; Mastrotto, C.; Beggiao, E.; Stefani, A.; Barzon, L.; Sturniolo, G.C.; Palu, G.; Castagliuolo, I. Neuropeptide neurotensin stimulates intestinal wound healing following chronic intestinal inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, 621. [Google Scholar] [CrossRef]
- Ferraro, L.; Tiozzo Fasiolo, L.; Beggiato, S.; Borelli, A.C.; Pomierny-Chamiolo, L.; Frankowska, M.; Antonelli, T.; Tomasini, M.C.; Fuxe, K.; Filip, M. Neurotensin: A role in substance use disorder? J. Psychopharmacol. 2016, 30, 112–127. [Google Scholar] [CrossRef]
- Cáceda, R.; Kinkead, B.; Nemeroff, C.B. Neurotensin: Role in psychiatric and neurological diseases. Peptides 2006, 27, 2385–2404. [Google Scholar] [CrossRef]
- Cuber, J.C.; Herrmann, C.; Kitabgi, P.; Bosshard, A.; Bernard, C.; De Nadai, F.; Chayvialle, J.A. Neuromedin-N is not released with neurotensin from rat ileum. Endocrinology 1990, 126, 1584–1592. [Google Scholar] [CrossRef] [PubMed]
- Kitabgi, P.; De Nadai, F.; Cuber, J.C.; Dubuc, I.; Nouel, D.; Costentin, J. Calcium-dependent release of neuromedin N and neurotensin from mouse hypothalamus. Neuropeptides 1990, 15, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Torruella-Suárez, M.L.; Vandenberg, J.R.; Cogan, E.S.; Tipton, G.J.; Teklezghi, A.; Dange, K.; Patel, G.K.; Mchenry, J.A.; Hardaway, J.A.; Kantak, P.A.; et al. Manipulations of central amygdala neurotensin neurons alter the consumption of ethanol and sweet fluids in mice. J. Neurosci. 2019, 40, 632. [Google Scholar] [CrossRef]
- Torruella-Suárez, M.L.; McElligott, Z.A. Neurotensin in reward processes. Neuropharmacology 2020, 167, 108005. [Google Scholar] [CrossRef]
- Vincent, J.; Mazella, J.; Kitabgi, P. Neurotensin, and neurotensin receptors. Trends Pharmacol. Sci. 1999, 20, 302–307. [Google Scholar] [CrossRef]
- Vincent, J.P. Neurotensin receptors: Binding properties, transduction pathways, and structure. Cell. Mol. Neurobiol. 1995, 15, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Jordan, B.A.; Devi, L.A. G-protein-coupled receptor heterodimerization modulates receptor function. Nature 1999, 399, 697–700. [Google Scholar] [CrossRef]
- Ullmann, T.; Gienger, M.; Budzinski, J.; Hellmann, J.; Hübner, H.; Gmeiner, P.; Weikert, D. Homobivalent dopamine D 2 receptor ligands modulate the dynamic equilibrium of D 2 monomers and homo- and heterodimers. ACS Chem. Biol. 2021, 16, 371–379. [Google Scholar] [CrossRef]
- Budzinski, J.; Maschauer, S.; Kobayashi, H.; Couvineau, P.; Vogt, H.; Gmeiner, P.; Roggenhofer, A.; Prante, O.; Bouvier, M.; Weikert, D. Bivalent ligands promote endosomal trafficking of the dopamine D3 receptor-neurotensin receptor 1 heterodimer. Commun. Biol. 2021, 4, 1062. [Google Scholar] [CrossRef]
- UniProt Database. Available online: https://www.uniprot.org/ (accessed on 26 January 2023).
- GPCRdb Database. Available online: https://gpcrdb.org/protein/ntr1_human/ (accessed on 30 January 2023).
- Deluigi, M.; Klipp, A.; Klenk, C.; Merklinger, L.; Eberle, S.A.; Morstein, L.; Heine, P.; Mittl, P.R.E.; Ernst, P.; Kamenecka, T.M.; et al. Complexes of the neurotensin receptor 1 with small-molecule ligands reveal structural determinants of full, partial, and inverse agonism. Sci. Adv. 2021, 7, eabe5504. [Google Scholar] [CrossRef]
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Svobodova, R.; Berka, K.; Bazgier, V.; Velankar, S.; Burley, S.K.; Koca, J.; Rose, A.S. Mol* Viewer: Modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic Acids Res. 2021, 49, W431–W437. [Google Scholar] [CrossRef] [PubMed]
- Robertson, M.J.; Papasergi-Scott, M.M.; He, F.; Seven, A.B.; Meyerowitz, J.G.; Panova, O.; Peroto, M.C.; Che, T.; Skiniotis, G. Structure determination of inactive-state GPCRs with a universal nanobody. Nat. Struct. Mol. Biol. 2022, 29, 1188. [Google Scholar] [CrossRef] [PubMed]
- White, J.F.; NoinaJ, N.; Grusshammer, R.; Shibata, Y.; Love, J.; KLoss, B.; Feng, X.U.; Gvozdenovic-Jeremic, J.; Shah, P.; Shiloach, J.; et al. Structure of the agonist-bound neurotensin receptor. Nature 2012, 490, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.E.; Zhang, Y.; Hu, H.; Suomivuori, C.; Kadji, F.M.N.; Aoki, J.; Krishna Kumar, K.; Fonseca, R.; Hilger, D.; Huang, W.; et al. Conformational transitions of a neurotensin receptor 1–Gi1 complex. Nature 2019, 572, 80–85. [Google Scholar] [CrossRef]
- Zhang, M.; Gui, M.; Wang, Z.; Gorgulla, C.; Yu, J.J.; Wu, H.; Sun, Z.J.; Klenk, C.; Merklinger, L.; Morstein, L.; et al. Cryo-EM structure of an activated GPCR–G protein complex in lipid nanodiscs. Nat. Struct. Mol. Biol. 2021, 28, 258. [Google Scholar] [CrossRef]
- Huang, W.; Masureel, M.; Qu, Q.; Janetzko, J.; Inoue, A.; Kato, H.E.; Robertson, M.J.; Nguyen, K.C.; Glenn, J.S.; Skiniotis, G.; et al. Structure of the neurotensin receptor 1 in complex with β-arrestin 1. Nature 2020, 579, 303. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Labbé-Jullié, C.; Barroso, S.; Nicolas-Etève, D.; Reversat, J.L.; Botto, J.M.; Mazella, J.; Bernassau, J.M.; Kitabgi, P. Mutagenesis and modeling of the neurotensin receptor NTR1. Identification of residues that are critical for binding SR 48692, a nonpeptide neurotensin antagonist. J. Biol. Chem. 1998, 273, 16351–16357. [Google Scholar] [CrossRef]
- Talbot, H.; Saada, S.; Naves, T.; Gallet, P.; Fauchais, A.; Jauberteau, M. Regulatory Roles of Sortilin and SorLA in Immune-Related Processes. Front. Pharmacol. 2019, 9, 1507. [Google Scholar] [CrossRef]
- Leloup, N.; Lössl, P.; Meijer, D.H.; Brennich, M.; Heck, A.J.R.; Thies-Weesie, D.M.E.; Janssen, B.J. C Low pH-induced conformational change and dimerization of sortilin triggers endocytosed ligand release. Nat. Commun. 2017, 8, 1708–1716. [Google Scholar] [CrossRef]
- Ghaemimanesh, F.; Mehravar, M.; Milani, S.; Poursani, E.M.; Saliminejad, K. The multifaceted role of sortilin/neurotensin receptor 3 in human cancer development. J. Cell. Physiol. 2021, 236, 6271–6281. [Google Scholar] [CrossRef]
- Blondeau, N.; Beraud-Dufour, S.; Lebrun, P.; Hivelin, C.; Coppola, T. Sortilin in glucose homeostasis: From accessory protein to key player? Front. Pharmacol. 2019, 9, 1561. [Google Scholar] [CrossRef] [PubMed]
- Petersen, C.M.; Nielsen, M.S.; Nykjær, A.; Jacobsen, L.; Tommerup, N.; Rasmussen, H.H.; RØigaard, H.; Gliemann, J.; Madsen, P.; Moestrup, S.K. Molecular identification of a novel candidate sorting receptor purified from the human brain by receptor-associated protein affinity chromatography. J. Biol. Chem. 1997, 272, 3599–3605. [Google Scholar] [CrossRef] [PubMed]
- Petersen, C.M.; Thirup, S.S.; Quistgaard, E.M.; Madsen, P.; Grøftehauge, M.K.; Nissen, P. Ligands bind to Sortilin in the tunnel of a ten-bladed β-propeller domain. Nat. Struct. Mol. Biol. 2009, 16, 96–98. [Google Scholar] [CrossRef]
- Moody, T.W.; Ramos-Alvarez, I.; Jensen, R.T. Bombesin, endothelin, neurotensin, and pituitary adenylate cyclase-activating polypeptide cause tyrosine phosphorylation of receptor tyrosine kinases. Peptides 2021, 137, 170480. [Google Scholar] [CrossRef]
- Fields, H.L.; Margolis, E.B. Understanding opioid reward. Trends Neurosci. 2015, 38, 217–225. [Google Scholar] [CrossRef]
- Russo, S.J.; Nestler, E.J. The brain reward circuitry in mood disorders. Nat. Rev. Neurosci. 2013, 14, 609–625. [Google Scholar] [CrossRef]
- Cooper, S.; Robison, A.J.; Mazei-Robison, M.S. Reward circuitry in addiction. Neurotherapeutics 2017, 14, 687–697. [Google Scholar] [CrossRef]
- Normandeau, C.P.; Ventura-Silva, A.P.; Hawken, E.R.; Angelis, S.; Sjaarda, C.; Liu, X.; Pêgo, J.M.; Dumont, É.C. A Key Role for neurotensin in chronic-stress-induced anxiety-like behavior in rats. Neuropsychopharmacol 2018, 43, 285. [Google Scholar] [CrossRef]
- Soden, M.E.; Yee, J.X.; Cuevas, B.; Rastani, A.; Elum, J.; Zweifel, L.S. Distinct encoding of reward and aversion by peptidergic BNST inputs to the VTA. Front. Neural Circuits 2022, 16, 918839. [Google Scholar] [CrossRef]
- Gao, C.; Leng, Y.; Ma, J.; Rooke, V.; Rodriguez-Gonzalez, S.; Ramakrishnan, C.; Deisseroth, K.; Penzo, M.A. Two genetically, anatomically, and functionally distinct cell types segregate across anteroposterior axis of paraventricular thalamus. Nat. Neurosci. 2020, 23, 217. [Google Scholar] [CrossRef]
- Chen, W. Neural circuits provide insights into reward and aversion. Front. Neural Circuits 2022, 16, 1002485. [Google Scholar] [CrossRef] [PubMed]
- Hackleman, A.; Ibrahim, M.; Shim, K.; Sangha, S. Interaction of stress and alcohol on discriminating fear from safety and reward in male and female rats. Psychopharmacology 2022, 240, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Avegno, E.M.; Gilpin, N.W. Reciprocal midbrain-extended amygdala circuit activity in preclinical models of alcohol use and misuse. Neuropharmacology 2022, 202, 108856. [Google Scholar] [CrossRef] [PubMed]
- Dugré, J.R.; Orban, P.; Potvin, S. Disrupted functional connectivity of the brain reward system in substance use problems: A meta-analysis of functional neuroimaging studies. Addict. Biol. 2022, 28, e13257. [Google Scholar] [CrossRef]
- Woodworth, H.L.; Brown, J.A.; Batchelor, H.M.; Bugescu, R.; Leinninger, G.M. Determination of neurotensin projections to the ventral tegmental area in mice. Neuropeptides 2018, 68, 57–74. [Google Scholar] [CrossRef]
- Levine, O.B.; Skelly, M.J.; Miller, J.D.; Rivera-Irizarry, J.K.; Rowson, S.A.; DiBerto, J.F.; Rinker, J.A.; Thiele, T.E.; Kash, T.L.; Pleil, K.E. The paraventricular thalamus provides a polysynaptic brake on limbic CRF neurons to sex-dependently blunt binge alcohol drinking and avoidance behavior in mice. Nat. Commun. 2021, 12, 5080. [Google Scholar] [CrossRef] [PubMed]
- Salgado, S.; Kaplitt, M.G. The nucleus accumbens: A comprehensive review. Stereotact. Funct. Neurosurg. 2015, 93, 75–93. [Google Scholar] [CrossRef]
- Castro, D.C.; Bruchas, M.R. A motivational and neuropeptidergic hub: Anatomical and functional diversity within the nucleus accumbens shell. Neuron 2019, 102, 529–552. [Google Scholar] [CrossRef]
- Sah, P.; Faber, E.S.L.; Lopez de Armentia, M.; Power, J. The amygdaloid complex: Anatomy and physiology. Physiol. Rev. 2003, 83, 803–834. [Google Scholar] [CrossRef]
- Avecillas-Chasin, J.M.; Levinson, S.; Kuhn, T.; Omidbeigi, M.; Langevin, J.; Pouratian, N.; Bari, A. Connectivity-based parcellation of the amygdala and identification of its main white matter connections. Sci. Rep. 2023, 13, 1305–1306. [Google Scholar] [CrossRef] [PubMed]
- de Guglielmo, G.; Simpson, S.; Kimbrough, A.; Conlisk, D.; Baker, R.; Cantor, M.; Kallupi, M.; George, O. Voluntary and forced exposure to ethanol vapor produces similar escalation of alcohol drinking but differential recruitment of brain regions related to stress, habit, and reward in male rats. Neuropharmacology 2023, 222, 109309. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.B.; Yorgason, J.T.; Nelson, A.C.; Lewis, N.; Nufer, T.M.; Edwards, J.G.; Steffensen, S.C. Glutamate transmission to ventral tegmental area GABA neurons is altered by acute and chronic ethanol. Alcohol. Clin. Exp. Res. 2018, 42, 2186–2195. [Google Scholar] [CrossRef] [PubMed]
- Becker, H.C. Influence of stress associated with chronic alcohol exposure on drinking. Neuropharmacology 2017, 122, 115–126. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, W.; Huq, S.N.; Allen, K.; Scally, L.; Petri, A.; Wujek, M.; Sachs, B.D. Chronic, but not sub-chronic, stress increases binge-like alcohol consumption in male and female c57BL6 mice. Front. Behav. Neurosci. 2022, 16, 958342. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, D.E.; Lippard, E.T.C. Early life stress and substance use disorders: The critical role of adolescent substance use. Pharmacol. Biochem. Behav. 2022, 215, 173360. [Google Scholar] [CrossRef]
- Gilpin, N.W. Corticotropin-releasing factor (CRF) and neuropeptide Y (NPY): Effects on inhibitory transmission in central amygdala, and anxiety- & alcohol-related behaviors. Alcohol 2012, 46, 329–337. [Google Scholar] [CrossRef]
- Lycas, M.D.; Ejdrup, A.L.; Sørensen, A.T.; Haahr, N.O.; Jørgensen, S.H.; Guthrie, D.A.; Støier, J.F.; Werner, C.; Newman, A.H.; Sauer, M.; et al. Nanoscopic dopamine transporter distribution and conformation are inversely regulated by excitatory drive and D2 autoreceptor activity. Cell Rep. 2022, 40, 111431. [Google Scholar] [CrossRef]
- Kolpakova, J.; van der Vinne, V.; Gimenez-Gomez, P.; Le, T.; Martin, G.E. Binge alcohol drinking alters the differential control of cholinergic interneurons over nucleus accumbens D1 and D2 medium spiny neurons. Front. Cell Neurosci. 2022, 16, 1010121. [Google Scholar] [CrossRef]
- Zinsmaier, A.K.; Dong, Y.; Huang, Y.H. Cocaine-induced projection-specific and cell type-specific adaptations in the nucleus accumbens. Mol. Psychiatry 2022, 27, 669–686. [Google Scholar] [CrossRef]
- Sala-Bayo, J.; Fiddian, L.; Nilsson, S.R.O.; Hervig, M.E.; McKenzie, C.; Mareschi, A.; Boulos, M.; Zhukovsky, P.; Nicholson, J.; Dalley, J.W.; et al. Dorsal and ventral striatal dopamine D1 and D2 receptors differentially modulate distinct phases of serial visual reversal learning. Neuropsychopharmacology 2020, 45, 736–744. [Google Scholar] [CrossRef] [PubMed]
- Strong, C.E.; Hagarty, D.P.; Brea Guerrero, A.; Schoepfer, K.J.; Cajuste, S.M.; Kabbaj, M. Chemogenetic selective manipulation of nucleus accumbens medium spiny neurons bidirectionally controls alcohol intake in male and female rats. Sci. Rep. 2020, 10, 19178. [Google Scholar] [CrossRef] [PubMed]
- Jennes, L.; Stumpf, W.E.; Kalivas, P.W. Neurotensin: Topographical distribution in rat brain by immunohistochemistry. J. Comp. Neurol. 1982, 210, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Vadnie, C.A.; Park, J.H.; Abdel Gawad, N.; Ho, A.M.C.; Hinton, D.J.; Choi, D. Gut-brain peptides in corticostriatal-limbic circuitry and alcohol use disorders. Front. Neurosci. 2014, 8, 288. [Google Scholar] [CrossRef]
- Dobbs, L.K.; Morikawa, H. Biasing Neurotensin Receptor Signaling to arrest psychostimulant abuse. Cell 2020, 181, 1205–1206. [Google Scholar] [CrossRef]
- Li, B.; Chang, L.; Xi, K. Neurotensin 1 receptor in the prelimbic cortex regulates anxiety-like behavior in rats. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 104, 110011. [Google Scholar] [CrossRef]
- Keller, B.N.; Hajnal, A.; Browning, K.N.; Arnold, A.C.; Silberman, Y. Involvement of the dorsal vagal complex in alcohol-related behaviors. Front. Behav. Neurosci. 2022, 16, 801825. [Google Scholar] [CrossRef]
- Erwin, V.G.; Jones, B.C. Comparison of neurotensin levels, receptors, and actions in LS/Ibg and SS/Ibg mice. Peptides 1989, 10, 435–440. [Google Scholar] [CrossRef]
- Erwin, V.G.; Jones, B.C.; Myers, R. Effects of acute and chronic ethanol administration on neurotensinergic systems. Ann. N. Y. Acad. Sci. 1994, 739, 185–196. [Google Scholar] [CrossRef]
- Widdowson, P.S. The effect of neurotensin, TRH, and the δ-opioid receptor antagonist ICI 174864 on alcohol-induced narcosis in rats. Brain Res. 1987, 424, 281. [Google Scholar] [CrossRef]
- Campbell, A.D.; Gene Erwin, V. Chronic ethanol administration downregulates neurotensin receptors in long- and short-sleep mice. Pharmacol. Biochem. Behav. 1993, 45, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.R.; Hinton, D.J.; Unal, S.S.; Richelson, E.; Choi, D. Increased ethanol consumption and preference in mice lacking neurotensin receptor Type 2. Alcohol. Clin. Exp. Res. 2011, 35, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Barson, J.R. Heightened exploratory behavior following chronic excessive ethanol drinking: Mediation by neurotensin receptor type 2 in the anterior paraventricular thalamus. Alcohol. Clin. Exp. Res. 2020, 44, 1747–1759. [Google Scholar] [CrossRef] [PubMed]
- Borroto-Escuela, D.O.; Fuxe, K. Diversity and bias through dopamine D2R heteroreceptor complexes. Curr. Opin. Pharmacol. 2017, 32, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Borroto-Escuela, D.O.; Ravani, A.; Tarakanov, A.O.; Brito, I.; Narvaez, M.; Romero-Fernandez, W.; Corrales, F.; Agnati, L.F.; Tanganelli, S.; Ferraro, L.; et al. Dopamine D2 receptor signaling dynamics of dopamine D2-neurotensin 1 receptor heteromers. Biochem. Biophys. Res. Commun. 2013, 435, 140–146. [Google Scholar] [CrossRef]
- Ferraro, L.; Beggiato, S.; Tomasini, M.C.; Fuxe, K.; Tanganelli, S.; Antonelli, T. Neurotensin regulates cortical glutamate transmission by modulating N-methyl-D-aspartate receptor functional activity: An in vivo microdialysis study. J. Neurosci. Res. 2011, 89, 1618–1626. [Google Scholar] [CrossRef]
- Li, Z.; Boules, M.; Richelson, E. NT69L blocks ethanol-induced increase of dopamine and glutamate levels in striatum of mouse. Neurosci. Lett. 2011, 487, 322–324. [Google Scholar] [CrossRef]
- Vanakoski, J.; Mazzanti, C.; Naukkarinen, H.; Virkkunen, M.; Goldman, D. An abundant proneurotensin polymorphism, 479A>G, and a test of its association with alcohol dependence in a Finnish population. Alcohol. Clin. Exp. Res. 2000, 24, 762–765. [Google Scholar] [CrossRef]
- Erwin, V.G.; Radcliffe, R.A.; Gehle, V.M.; Jones, B.C. Common quantitative trait loci for alcohol-related behaviors and central nervous system neurotensin measures: Locomotor activation. J. Pharmacol. Exp. Ther. 1997, 280, 919–926. [Google Scholar]
- Erwin, V.G.; Markel, P.D.; Johnson, T.E.; Gehle, V.M.; Jones, B.C. Common quantitative trait loci for alcohol-related behaviors and central nervous system neurotensin measures: Hypnotic and hypothermic effects. J. Pharmacol. Exp. Ther. 1997, 280, 911–918. [Google Scholar]
- Erwin, V.G.; Gehle, V.M.; Davidson, K.; Radcliffe, R.A. Confirmation of correlations and common quantitative trait loci between neurotensin receptor density and hypnotic sensitivity to ethanol. Alcohol. Clin. Exp. Res. 2001, 25, 1699–1707. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.M.; Glukhova, A.; Sexton, P.M.; Christopoulos, A. Structural insights into G-protein-coupled receptor allostery. Nature 2018, 559, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Dixon, A.D.; Inoue, A.; Robson, S.A.; Culhane, K.J.; Trinidad, J.C.; Sivaramakrishnan, S.; Bumbak, F.; Ziarek, J.J. Effect of Ligands and Transducers on the Neurotensin Receptor 1 Conformational Ensemble. J. Am. Chem. Soc. 2022, 144, 10241. [Google Scholar] [CrossRef] [PubMed]
- Bumbak, F.; Pons, M.; Inoue, A.; Paniagua, J.C.; Yan, F.; Wu, H.; Robson, S.A.; Bathgate, R.A.D.; Scott, D.J.; Gooley, P.R.; et al. Ligands selectively tune the local and global motions of neurotensin receptor 1 (NTS1). Cell Rep. 2023, 42, 112015. [Google Scholar] [CrossRef]
- Carrion-Antoli, A.; Mallor-Franco, J.; Arroyo-Urea, S.; Garcia-Nafria, J. Structural insights into promiscuous GPCR-G protein coupling. Prog. Mol. Biol. Transl. Sci. 2023, 195, 137–152. [Google Scholar] [CrossRef]
- Sadler, F.; Ma, N.; Ritt, M.; Sharma, Y.; Vaidehi, N.; Sivaramakrishnan, S. Autoregulation of GPCR signaling through the third intracellular loop. Nature 2023, 615, 734–741. [Google Scholar] [CrossRef]
- Krumm, B.E.; DiBerto, J.F.; Olsen, R.H.J.; Kang, H.J.; Slocum, S.T.; Zhang, S.; Strachan, R.T.; Huang, X.; Slosky, L.M.; Pinkerton, A.B.; et al. Neurotensin receptor allosterism revealed in complex with a biased allosteric modulator. Biochemistry 2023, 62, 1233–1248. [Google Scholar] [CrossRef]
- Li, A.; Liu, S.; Huang, R.; Ahn, S.; Lefkowitz, R.J. Loss of biased signaling at a G protein-coupled receptor in overexpressed systems. PLoS ONE 2023, 18, e0283477. [Google Scholar] [CrossRef]
- Cusack, B.; Mccormick, D.J.; Pangl, Y.; Terrance Souder, T.; Garcia, R.; Eauq, A.; Richelson, E. Pharmacological and biochemical profiles of unique neurotensin 8–13 analogs exhibiting species selectivity, stereoselectivity, and superagonism. J. Biol. Chem. 1995, 270, 18359–18366. [Google Scholar] [CrossRef]
- Previti, S.; Desgagne, M.; Tourwe, D.; Cavelier, F.; Sarret, P.; Ballet, S. Opening the amino acid toolbox for peptide-based NTS2-selective ligands as promising lead compounds for pain management. J. Pept. Sci. 2023, 29, e3471. [Google Scholar] [CrossRef]
- Schindler, L.; Bernhardt, G.; Keller, M. Modifications at Arg and Ile give neurotensin (8–13) derivatives with high stability and retained NTS1 receptor affinity. ACS Med. Chem. Lett. 2019, 10, 960–965. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.B.; Navarro, H.; Warner, K.R.; Gilmour, B. The identification of nonpeptide neurotensin receptor partial agonists from the potent antagonist SR48692 using a calcium mobilization assay. Bioorg. Med. Chem. Lett. 2009, 19, 1438–1441. [Google Scholar] [CrossRef] [PubMed]
- Gully, D.; Labeeuw, B.; Boigegrain, R.; Oury-Donat, F.; Bachy, A.; Poncelet, M.; Steinberg, R.; Suaud-Chagny, M.F.; Santucci, V.; Vita, N.; et al. Biochemical and pharmacological activities of SR 142948A, a new potent neurotensin receptor antagonist. J. Pharmacol. Exp. Ther. 1997, 280, 802–812. [Google Scholar] [PubMed]
- Gully, D.; Canton, M.; Boigegrain, R.; Jeanjean, F.; Molimard, J.C.; Poncelet, M.; Gueudet, C.; Heaulme, M.; Leyris, R.; Brouard, A.; et al. Biochemical and Pharmacological Profile of a Potent and Selective Nonpeptide Antagonist of the Neurotensin Receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 65–69. [Google Scholar] [CrossRef]
- Barroso, S.; Richard, F.; Nicolas-Ethève, D.; Kitabgi, P.; Labbé-Jullié, C. Constitutive activation of the neurotensin receptor 1 by mutation of Phe (358) in Helix seven. Br. J. Pharmacol. 2002, 135, 997–1002. [Google Scholar] [CrossRef]
- Peddibhotla, S.; Hedrick, M.P.; Hershberger, P.; Maloney, P.R.; Li, Y.; Milewski, M.; Gosalia, P.; Gray, W.; Mehta, A.; Sugarman, E.; et al. Discovery of ML314, a brain penetrant nonpeptidic β-arrestin biased agonist of the neurotensin NTR1 receptor. ACS Med. Chem. Lett. 2013, 4, 846–851. [Google Scholar] [CrossRef]
- Barak, L.S.; Bai, Y.; Peterson, S.; Evron, T.; Urs, N.M.; Peddibhotla, S.; Hedrick, M.P.; Hershberger, P.; Maloney, P.R.; Chung, T.D.Y.; et al. ML314: A biased neurotensin receptor ligand for methamphetamine abuse. ACS Chem. Biol. 2016, 11, 1880–1890. [Google Scholar] [CrossRef]
- Pinkerton, A.B.; Peddibhotla, S.; Yamamoto, F.; Slosky, L.M.; Bai, Y.; Maloney, P.; Hershberger, P.; Hedrick, M.P.; Falter, B.; Ardecky, R.J.; et al. Discovery of β-arrestin biased, orally bioavailable, and CNS penetrant neurotensin receptor 1 (NTR1) allosteric modulators. J. Med. Chem. 2019, 62, 8357–8363. [Google Scholar] [CrossRef]
- Slosky, L.M.; Bai, Y.; Toth, K.; Ray, C.; Rochelle, L.K.; Badea, A.; Chandrasekhar, R.; Pogorelov, V.M.; Abraham, D.M.; Atluri, N.; et al. β-Arrestin-biased allosteric modulator of NTSR1 selectively attenuates addictive behaviors. Cell 2020, 181, 1364–1379. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, G.; Zhong, J.; Jiang, S.; Lai, S.; Xu, H.; Deng, X.; Li, F.; Lu, S.; Zhou, K.; et al. A circuit from lateral septum neurotensin neurons to tuberal nucleus controls hedonic feeding. Mol. Psychiatry 2022, 27, 4843–4860. [Google Scholar] [CrossRef]
- Li, L.; Durand-De Cuttoli, R.; Aubry, A.V.; Burnett, C.J.; Cathomas, F.; Parise, L.F.; Chan, K.L.; Morel, C.; Yuan, C.; Shimo, Y.; et al. Social trauma engages lateral septum circuitry to occlude social reward. Nature 2023, 613, 696. [Google Scholar] [CrossRef] [PubMed]
- Younes, M.; Wu, Z.; Dupouy, S.; Lupo, A.M.; Mourra, N.; Takahashi, T.; Flejou, J.F.; Tredaniel, J.; Regnard, J.F.; Damotte, D.; et al. Neurotensin (NTS) and its receptor (NTSR1) causes EGFR, HER2, and HER3 over-expression and their autocrine/paracrine activation in lung tumors, confirming responsiveness to erlotinib. Oncotarget 2014, 5, 8252–8269. [Google Scholar] [CrossRef] [PubMed]
- Bird, J.L.; Simpson, R.; Vllasaliu, D.; Goddard, A.D. Neurotensin receptor 1 facilitates intracellular and transepithelial delivery of macromolecules. Eur. J. Pharm. Biopharm. 2017, 119, 300–309. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Experimental Model | In Vitro Studies | References |
| Hetero bivalents DR2-NTR1 (agonist/antagonist properties) | Cultured CHO cells expressing receptors | Radioligand saturation and BRET assays to determine affinities and promotion of D2R homodimerization and heterodimerization. | [55,56] |
| NT8–13 analogs | Cultured CHO-K1 cells expressing rat and human NTR | Radioligand saturation analysis for determination of crucial amino acid positions related to receptor affinity and PI turnover and stability. | [128,129,130] |
| Small non-peptide ligands of NTR: SRI-9829, RTI-3a, SR-48692, SR142948A | Transfected cultured HEK293, COS-7 cultured cells | Structural, ligand binding, and signaling assays. | [59] |
| NT8–13 and SR142948A | Transfected cultured HEK293 cells | Structural dynamics of NTR1 with analysis of its global motions. | [123] |
| Derivatives of SR-48692 antagonist | CHO-K1 cell line expressing NTR1 | Calcium mobilization assays | [131] |
| SR-48692 antagonist | Cultured transfected COS-7 cells, and brain tissue, form different species, | Radioligand binding assays | [132,133] |
| Nonpeptide ML314 biased agonist of NTR1 | β-arrestin conjugated (GFP) reporter expressed in a U2OS cell line | Activates the β-arrestin pathway and blocks G-protein -dependent signaling. | [135,136] |
| Quinazoline NTR1 modulator, SBI-553 | In vitro radioligand binding, NTR1-β-arrestin-GFP, and calcium flux assays in HEK-293 transfected cells | Activates β-arrestin signaling and inhibits Gq-protein-dependent calcium signaling. | [137] |
| Quinazoline NTR1 modulator, SBI-553 | In vitro assays in HEK 293 and U2OS transfected cells | Promotes receptor phosphorylation, receptor internalization, and β-arrestin activation. | [138] |
| Compounds | Experimental Model | In Vivo Studies | References |
| Nonpeptide ML314 biased agonist of NTR1 | Injection in rats | Inhibits amphetamine self-administration. | [136] |
| Silencing of lateral septum neurotensin-positive neurons | In vivo suppression of NT neurons in LS in C57BL/6J mice | Promotion of palatable feeding. | [139] |
| In vivo genetic ablation septum neurotensin-positive neurons (LSNT) | Male and female C57BL/6J mice | LSNT modulates social interaction and social reward (stress susceptibility). | [140] |
| NTR1 agonist PD- 149,163, and antagonist SR-48692 | Brain microinjections in male Sprague–Dawley rats | PD-149163 induces anxiogenic effects when injected into the prelimbic region of the medial prefrontal cortex. | [105] |
| Nonpeptide ML314 biased agonist of NTR1 | Intraperitoneal injection in C57BL/6J mice | Attenuates methamphetamine-induced hyperlocomotion and methamphetamine-associated conditioned place preference. | [136] |
| Quinazoline NTR1 modulator, SBI-553 | Intraperitoneal injection and oral administration to DAT KO C57BL/6J mice (dopamine-depleted dopamine transporter knockout) | Attenuates hyperdopaminergic activity. | [137] |
| Quinazoline NTR1 modulator, SBI-55 | Intraperitoneal injection in cocaine self-administration in C57BL/6J mice | Attenuates cocaine-induced hyperlocomotion without producing non-desired side effects dependent on Gq protein activation. | [138] |
| Selective NTR2 agonist JMV-431 | Brain injection in rats consuming alcohol (chronic consumption) | Chronic and excessive alcohol consumption induces behavioral changes, partly mediated by the NTR2. | [112] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez, F.D.; Sánchez, M.L.; Coveñas, R. Neurotensin and Alcohol Use Disorders: Towards a Pharmacological Treatment. Int. J. Mol. Sci. 2023, 24, 8656. https://doi.org/10.3390/ijms24108656
Rodríguez FD, Sánchez ML, Coveñas R. Neurotensin and Alcohol Use Disorders: Towards a Pharmacological Treatment. International Journal of Molecular Sciences. 2023; 24(10):8656. https://doi.org/10.3390/ijms24108656
Chicago/Turabian StyleRodríguez, Francisco D., Manuel Lisardo Sánchez, and Rafael Coveñas. 2023. "Neurotensin and Alcohol Use Disorders: Towards a Pharmacological Treatment" International Journal of Molecular Sciences 24, no. 10: 8656. https://doi.org/10.3390/ijms24108656
APA StyleRodríguez, F. D., Sánchez, M. L., & Coveñas, R. (2023). Neurotensin and Alcohol Use Disorders: Towards a Pharmacological Treatment. International Journal of Molecular Sciences, 24(10), 8656. https://doi.org/10.3390/ijms24108656