

Nuclear DJ-1 Regulates DNA Damage Repair via the Regulation of PARP1 Activity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. DJ-1 Responds to DNA Damage and Is Recruited to Sites of DSBs
2.2. DJ-1 Enhances DNA Damage Repair
2.3. DJ-1 Promotes Both HR- and NHEJ-Mediated DNA Repair
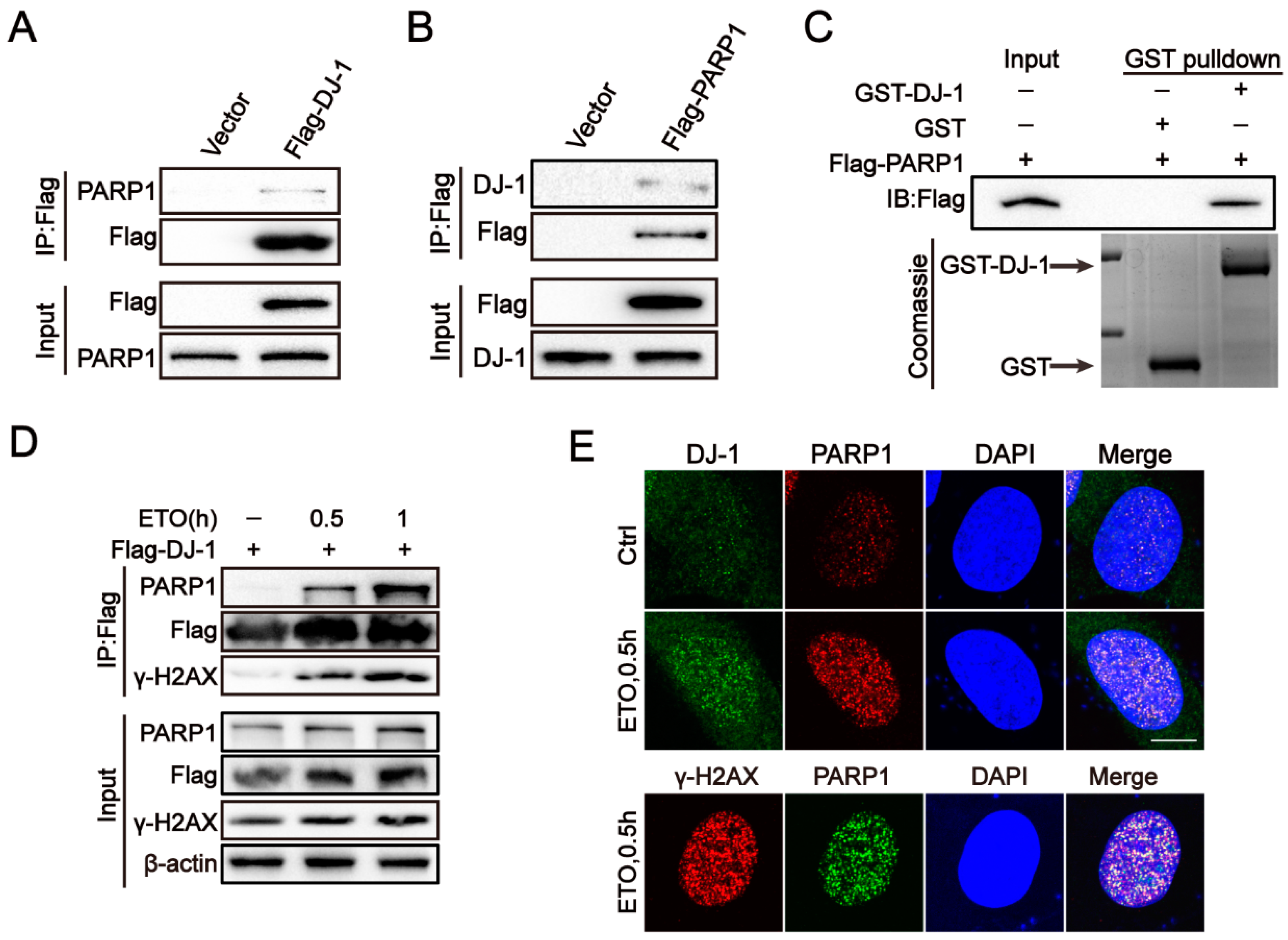
2.4. DJ-1 Interacts with PARP1
2.5. DJ-1 Directly Regulates PARP1 Activity
2.6. DNA Damage Accumulation and Defective PARP1 Activity in DJ-1 Mutant Fibroblasts
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Human Fibroblast Cultures
4.2. Antibodies and Chemical Reagents
4.3. Plasmids, siRNA Transfection, and Lentiviral Infection
4.4. Western Blot
4.5. Co-Immunoprecipitation (Co-IP)
4.6. GST Pull-Down Assay
4.7. Immunofluorescence
4.8. Proximity Ligation Assays (PLA)
4.9. Cell Viability Assay
4.10. Real-Time Quantitative PCR (qPCR)
4.11. Comet Assay
4.12. HR and NHEJ Assays
4.13. Subcellular Fractionation
4.14. PARP1 Activity Assay
4.15. Cell Cycle Analysis
4.16. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Armstrong, M.J.; Okun, M.S. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA 2020, 323, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef]
- Ascherio, A.; Schwarzschild, M.A. The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurol. 2016, 15, 1257–1272. [Google Scholar] [CrossRef] [PubMed]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020, 19, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Vélez, G.E.; Zoghbi, H.Y. Parkinson’s Disease Genetics and Pathophysiology. Annu. Rev. Neurosci. 2021, 44, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ma, X.; Fujioka, H.; Liu, J.; Chen, S.; Zhu, X. DJ-1 regulates the integrity and function of ER-mitochondria association through interaction with IP3R3-Grp75-VDAC1. Proc. Natl. Acad. Sci. USA 2019, 116, 25322–25328. [Google Scholar] [CrossRef] [PubMed]
- Madabhushi, R.; Pan, L.; Tsai, L.-H. DNA Damage and Its Links to Neurodegeneration. Neuron 2014, 83, 266–282. [Google Scholar] [CrossRef]
- Coppedè, F.; Migliore, L. DNA damage in neurodegenerative diseases. Mutat. Res. Mol. Mech. Mutagen. 2015, 776, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-L.; Wang, Z.-X.; Ying, C.-Z.; Zhang, B.-R.; Pu, J.-L. Decoding the Role of Familial Parkinson’s Disease-Related Genes in DNA Damage and Repair. Aging Dis. 2022, 13, 1405–1412. [Google Scholar] [CrossRef]
- Gonzalez-Hunt, C.P.; Sanders, L.H. DNA damage and repair in Parkinson’s disease: Recent advances and new opportunities. J. Neurosci. Res. 2020, 99, 180–189. [Google Scholar] [CrossRef]
- Jeppesen, D.K.; Bohr, V.A.; Stevnsner, T. DNA repair deficiency in neurodegeneration. Prog. Neurobiol. 2011, 94, 166–200. [Google Scholar] [CrossRef]
- Wang, H.; Lautrup, S.; Caponio, D.; Zhang, J.; Fang, E. DNA Damage-Induced Neurodegeneration in Accelerated Ageing and Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 6748. [Google Scholar] [CrossRef]
- Welch, G.; Tsai, L. Mechanisms of DNA damage-mediated neurotoxicity in neurodegenerative disease. EMBO Rep. 2022, 23, e54217. [Google Scholar] [CrossRef]
- Schumacher, B.; Pothof, J.; Vijg, J.; Hoeijmakers, J.H.J. The central role of DNA damage in the ageing process. Nature 2021, 592, 695–703. [Google Scholar] [CrossRef]
- Wang, Z.-X.; Li, Y.-L.; Pu, J.-L.; Zhang, B.-R. DNA Damage-Mediated Neurotoxicity in Parkinson’s Disease. Int. J. Mol. Sci. 2023, 24, 6313. [Google Scholar] [CrossRef]
- Kam, T.-I.; Mao, X.; Park, H.; Chou, S.-C.; Karuppagounder, S.S.; Umanah, G.E.; Yun, S.P.; Brahmachari, S.; Panicker, N.; Chen, R.; et al. Poly(ADP-ribose) drives pathologic α-synuclein neurodegeneration in Parkinson’s disease. Science 2018, 362, eaat8407. [Google Scholar] [CrossRef] [PubMed]
- Milanese, C.; Cerri, S.; Ulusoy, A.; Gornati, S.V.; Plat, A.; Gabriels, S.; Blandini, F.; Di Monte, D.A.; Hoeijmakers, J.H.; Mastroberardino, P.G. Activation of the DNA damage response in vivo in synucleinopathy models of Parkinson’s disease. Cell Death Dis. 2018, 9, 818. [Google Scholar] [CrossRef]
- Sepe, S.; Milanese, C.; Gabriels, S.; Derks, K.W.; Payan-Gomez, C.; van Ijcken, W.F.; Rijksen, Y.M.; Nigg, A.L.; Moreno, S.; Cerri, S.; et al. Inefficient DNA Repair Is an Aging-Related Modifier of Parkinson’s Disease. Cell Rep. 2016, 15, 1866–1875. [Google Scholar] [CrossRef] [PubMed]
- Eilam, R.; Peter, Y.; Elson, A.; Rotman, G.; Shiloh, Y.; Groner, Y.; Segal, M. Selective loss of dopaminergic nigro-striatal neurons in brains of Atm-deficient mice. Proc. Natl. Acad. Sci. USA 1998, 95, 12653–12656. [Google Scholar] [CrossRef]
- Kirshner, M.; Galron, R.; Frenkel, D.; Mandelbaum, G.; Shiloh, Y.; Wang, Z.-Q.; Barzilai, A. Malfunctioning DNA Damage Response (DDR) Leads to the Degeneration of Nigro-Striatal Pathway in Mouse Brain. J. Mol. Neurosci. 2011, 46, 554–568. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Paull, T.T. Cellular functions of the protein kinase ATM and their relevance to human disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 796–814. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.E.; Dollé, M.E.T.; Vermeij, W.P.; Gyenis, A.; Vogel, K.; Hoeijmakers, J.H.J.; Wiley, C.D.; Davalos, A.R.; Hasty, P.; Desprez, P.; et al. Deficiency in the DNA repair protein ERCC1 triggers a link between senescence and apoptosis in human fibroblasts and mouse skin. Aging Cell 2019, 19, e13072. [Google Scholar] [CrossRef]
- Huang, M.; Chen, S. DJ-1 in neurodegenerative diseases: Pathogenesis and clinical application. Prog. Neurobiol. 2021, 204, 102114. [Google Scholar] [CrossRef]
- Neves, M.; Grãos, M.; Anjo, S.I.; Manadas, B. Modulation of signaling pathways by DJ-1: An updated overview. Redox Biol. 2022, 51, 102283. [Google Scholar] [CrossRef]
- Dolgacheva, L.P.; Berezhnov, A.V.; Fedotova, E.I.; Zinchenko, V.P.; Abramov, A.Y. Role of DJ-1 in the mechanism of pathogenesis of Parkinson’s disease. J. Bioenerg. Biomembr. 2019, 51, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-J.; Park, Y.-J.; Hwang, I.-Y.; Youdim, M.B.; Park, K.-S.; Oh, Y.J. Nuclear translocation of DJ-1 during oxidative stress-induced neuronal cell death. Free. Radic. Biol. Med. 2012, 53, 936–950. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Kim, H.-J.; Jung, C.-W.; Choi, B.-I.; Lee, D.-H.; Park, M.-J. PARK7 maintains the stemness of glioblastoma stem cells by stabilizing epidermal growth factor receptor variant III. Oncogene 2020, 40, 508–521. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, C. Quantitative reactive cysteinome profiling reveals a functional link between ferroptosis and proteasome-mediated degradation. Cell Death Differ. 2022, 30, 125–136. [Google Scholar] [CrossRef]
- Kosmider, B.; Lin, C.-R.; Vlasenko, L.; Marchetti, N.; Bolla, S.; Criner, G.J.; Messier, E.; Reisdorph, N.; Powell, R.L.; Madesh, M.; et al. Impaired non-homologous end joining in human primary alveolar type II cells in emphysema. Sci. Rep. 2019, 9, 920. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef]
- Zhao, B.; Zhang, W.-D.; Duan, Y.-L.; Lu, Y.-Q.; Cun, Y.-X.; Li, C.-H.; Guo, K.; Nie, W.-H.; Li, L.; Zhang, R.; et al. Filia Is an ESC-Specific Regulator of DNA Damage Response and Safeguards Genomic Stability. Cell Stem Cell 2015, 16, 684–698. [Google Scholar] [CrossRef]
- Konopka, A.; Whelan, D.R.; Jamali, S.; Perri, E.; Shahheydari, H.; Toth, R.P.; Parakh, S.; Robinson, T.; Cheong, A.; Mehta, P.; et al. Impaired NHEJ repair in amyotrophic lateral sclerosis is associated with TDP-43 mutations. Mol. Neurodegener. 2020, 15, 51. [Google Scholar] [CrossRef]
- Bolderson, E.; Burgess, J.T.; Li, J.; Gandhi, N.S.; Boucher, D.; Croft, L.V.; Beard, S.; Plowman, J.J.; Suraweera, A.; Adams, M.N.; et al. Barrier-to-autointegration factor 1 (Banf1) regulates poly [ADP-ribose] polymerase 1 (PARP1) activity following oxidative DNA damage. Nat. Commun. 2019, 10, 55011. [Google Scholar] [CrossRef]
- Shahi, A.; Lee, J.-H.; Kang, Y.; Lee, S.H.; Hyun, J.-W.; Chang, I.-Y.; Jun, J.-Y.; You, H.J. Mismatch-repair protein MSH6 is associated with Ku70 and regulates DNA double-strand break repair. Nucleic Acids Res. 2010, 39, 2130–2143. [Google Scholar] [CrossRef]
- Wang, W.-Y.; Pan, L.; Su, S.C.; Quinn, E.J.; Sasaki, M.; Jimenez, J.C.; A Mackenzie, I.R.; Huang, E.; Tsai, L.-H. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat. Neurosci. 2013, 16, 1383–1391. [Google Scholar] [CrossRef]
- Zhang, Z.; Samsa, W.; De, Y.; Zhang, F.; Reizes, O.; Almasan, A.; Gong, Z. HDGFRP3 interaction with 53BP1 promotes DNA double-strand break repair. Nucleic Acids Res. 2023, 51, 2238–2256. [Google Scholar] [CrossRef]
- Gupta, A.; Hunt, C.R.; Hegde, M.L.; Chakraborty, S.; Udayakumar, D.; Horikoshi, N.; Singh, M.; Ramnarain, D.B.; Hittelman, W.N.; Namjoshi, S.; et al. MOF Phosphorylation by ATM Regulates 53BP1-Mediated Double-Strand Break Repair Pathway Choice. Cell Rep. 2014, 8, 177–189. [Google Scholar] [CrossRef]
- Chen, Y.; Jiang, T.; Zhang, H.; Gou, X.; Han, C.; Wang, J.; Chen, A.T.; Ma, J.; Liu, J.; Chen, Z.; et al. LRRC31 inhibits DNA repair and sensitizes breast cancer brain metastasis to radiation therapy. Nature 2020, 22, 1276–1285. [Google Scholar] [CrossRef]
- Jiang, H.; Xue, X.; Panda, S.; Kawale, A.; Hooy, R.M.; Liang, F.; Sohn, J.; Sung, P.; Gekara, N. Chromatin-bound cGAS is an inhibitor of DNA repair and hence accelerates genome destabilization and cell death. EMBO J. 2019, 38, e102718. [Google Scholar] [CrossRef]
- Batenburg, N.L.; Thompson, E.L.; Hendrickson, E.A.; Zhu, X.D. Cockayne syndrome group B protein regulates DNA double-strand break repair and checkpoint activation. EMBO J. 2015, 34, 1399–1416. [Google Scholar] [CrossRef]
- Chen, J.-K.; Lin, W.-L.; Chen, Z.; Liu, H.-W. PARP-1–dependent recruitment of cold-inducible RNA-binding protein promotes double-strand break repair and genome stability. Proc. Natl. Acad. Sci. USA 2018, 115, E1759–E1768. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.R.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Repici, M.; Hassanjani, M.; Maddison, D.C.; Garção, P.; Cimini, S.; Patel, B.; Szegö, M.; Straatman, K.R.; Lilley, K.S.; Borsello, T.; et al. The Parkinson’s Disease-Linked Protein DJ-1 Associates with Cytoplasmic mRNP Granules During Stress and Neurodegeneration. Mol. Neurobiol. 2018, 56, 61–77. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Bonkowski, M.S.; Moniot, S.; Zhang, D.; Hubbard, B.P.; Ling, A.J.Y.; Rajman, L.A.; Qin, B.; Lou, Z.; Gorbunova, V.; et al. A conserved NAD(+) binding pocket that regulates protein-protein interactions during aging. Science 2017, 355, 1312–1317. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Hine, C.; Tian, X.; Van Meter, M.; Au, M.; Vaidya, A.; Seluanov, A.; Gorbunova, V. SIRT6 Promotes DNA Repair Under Stress by Activating PARP1. Science 2011, 332, 1443–1446. [Google Scholar] [CrossRef]
- Lin, R.-R.; Tao, Q.-Q.; Wu, Z.-Y. Early-Onset Parkinson’s Disease and Brain Iron Accumulation Caused by a Novel Homozygous DJ-1 Mutation. J. Park. Dis. 2022, 12, 813–819. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Ariga, H.; Takahashi-Niki, K.; Kato, I.; Maita, H.; Niki, T.; Iguchi-Ariga, S.M.M. Neuroprotective Function of DJ-1 in Parkinson’s Disease. Oxidative Med. Cell. Longev. 2013, 2013, 6839202013. [Google Scholar] [CrossRef]
- Abou-Sleiman, P.M.; Healy, D.G.; Quinn, N.; Lees, A.J.; Wood, N.W. The role of pathogenicDJ-1 mutations in Parkinson’s disease. Ann. Neurol. 2003, 54, 283–286. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.J.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 Gene Associated with Autosomal Recessive Early-Onset Parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef]
- Dobbin, M.M.; Madabhushi, R.; Pan, L.; Chen, Y.; Kim, D.; Gao, J.; Ahanonu, B.; Pao, P.-C.; Qiu, Y.; Zhao, Y.; et al. SIRT1 collaborates with ATM and HDAC1 to maintain genomic stability in neurons. Nat. Neurosci. 2013, 16, 1008–1015. [Google Scholar] [CrossRef]
- He, L.; Liang, J.; Chen, C.; Chen, J.; Shen, Y.; Sun, S.; Li, L. C9orf72 functions in the nucleus to regulate DNA damage repair. Cell Death Differ. 2023, 30, 716–730. [Google Scholar] [CrossRef]
- Groelly, F.J.; Fawkes, M.; Dagg, R.A.; Blackford, A.N.; Tarsounas, M. Targeting DNA damage response pathways in cancer. Nat. Rev. Cancer 2022, 23, 78–94. [Google Scholar] [CrossRef]
- Huang, D.; Kraus, W.L. The expanding universe of PARP1-mediated molecular and therapeutic mechanisms. Mol. Cell 2022, 82, 2315–2334. [Google Scholar] [CrossRef]
- Richarme, G.; Liu, C.; Mihoub, M.; Abdallah, J.; Leger, T.; Joly, N.; Liebart, J.-C.; Jurkunas, U.V.; Nadal, M.; Bouloc, P.; et al. Guanine glycation repair by DJ-1/Park7 and its bacterial homologs. Science 2017, 357, 208–211. [Google Scholar] [CrossRef]
- Takahashi-Niki, K.; Ganaha, Y.; Niki, T.; Nakagawa, S.; Kato-Ose, I.; Iguchi-Ariga, S.M.; Ariga, H. DJ-1 activates SIRT1 through its direct binding to SIRT1. Biochem. Biophys. Res. Commun. 2016, 474, 131–136. [Google Scholar] [CrossRef]
- Onn, L.; Portillo, M.; Ilic, S.; Cleitman, G.; Stein, D.; Kaluski, S.; Shirat, I.; Slobodnik, Z.; Einav, M.; Erdel, F.; et al. SIRT6 is a DNA double-strand break sensor. eLife 2020, 9, 9. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.-X.; Liu, Y.; Li, Y.-L.; Wei, Q.; Lin, R.-R.; Kang, R.; Ruan, Y.; Lin, Z.-H.; Xue, N.-J.; Zhang, B.-R.; et al. Nuclear DJ-1 Regulates DNA Damage Repair via the Regulation of PARP1 Activity. Int. J. Mol. Sci. 2023, 24, 8651. https://doi.org/10.3390/ijms24108651
Wang Z-X, Liu Y, Li Y-L, Wei Q, Lin R-R, Kang R, Ruan Y, Lin Z-H, Xue N-J, Zhang B-R, et al. Nuclear DJ-1 Regulates DNA Damage Repair via the Regulation of PARP1 Activity. International Journal of Molecular Sciences. 2023; 24(10):8651. https://doi.org/10.3390/ijms24108651
Chicago/Turabian StyleWang, Zhong-Xuan, Yi Liu, Yao-Lin Li, Qiao Wei, Rong-Rong Lin, Ruiqing Kang, Yang Ruan, Zhi-Hao Lin, Nai-Jia Xue, Bao-Rong Zhang, and et al. 2023. "Nuclear DJ-1 Regulates DNA Damage Repair via the Regulation of PARP1 Activity" International Journal of Molecular Sciences 24, no. 10: 8651. https://doi.org/10.3390/ijms24108651
APA StyleWang, Z.-X., Liu, Y., Li, Y.-L., Wei, Q., Lin, R.-R., Kang, R., Ruan, Y., Lin, Z.-H., Xue, N.-J., Zhang, B.-R., & Pu, J.-L. (2023). Nuclear DJ-1 Regulates DNA Damage Repair via the Regulation of PARP1 Activity. International Journal of Molecular Sciences, 24(10), 8651. https://doi.org/10.3390/ijms24108651