

Metabolic Cardiomyopathies and Cardiac Defects in Inherited Disorders of Carbohydrate Metabolism: A Systematic Review



Abstract
1. Introduction
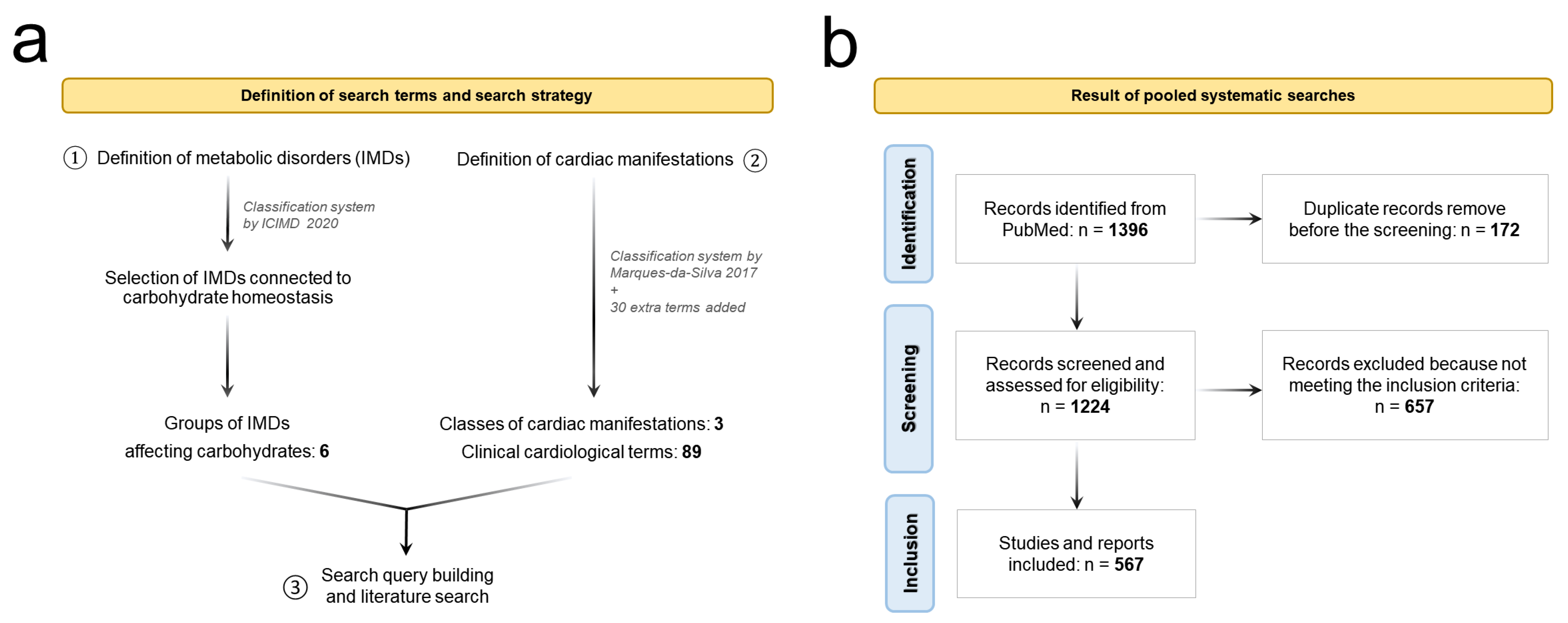
2. Method and Search Strategies
2.1. Definition of the Search Terms
2.2. Systematic Literature Search
3. Results
3.1. Disorders of Plasma Membrane Transporters of Sugars and Linked Metabolites
3.1.1. GLUT3 Deficiency
3.1.2. GLUT10 Deficiency
3.1.3. THTR1 Deficiency

{kind=link}
{kind=link}
| Affected Gene | Affected Protein | Inheritance | Heart Defects & Manifestations | No. Patients Identified by Our Search | Ref. | ||
|---|---|---|---|---|---|---|---|
| Cardiomyopathies | Structural Defects | Arrhythmogenic Disorders | |||||
| SLC2A3 | Glucose transporter type 3 (GLUT3) | AR | - | ASD, PVSD | TC, HF | 6 | [22] |
| SLC2A10 | Glucose transporter type 10 (GLUT10) | AR | CM, ICM, RDA | DAR, MVP, PPS | - | 4 | [28,29,30] |
| SLC19A2 | THTR1 transporter | AR | BVH, DCM | DC, ECD, RBBB, VSD, ASD, EA, TI (Ebstein) | TC, AF, HF | 24 | [32,33,34,35,36,37,38,39,40,41,42,43,45,46,47] |
3.2. Disorders of the Pentose Phosphate Pathway
3.2.1. TALDO Deficiency
| Affected Gene | Affected Protein | Inheritance | Heart Defects & Manifestations | No. Patients Identified by Our Search | Ref. | ||
|---|---|---|---|---|---|---|---|
| Cardiomyopathies | Structural Defects | Arrhythmogenic Disorders | |||||
| TALDO | Transaldolase (TALDO) | AR | CM, LVH | VSD, ASD, BAV, DC, MVP, TR | HF | 35 | [53,55,56,57] |
| G6PDH | Glucose-6-phosphate dehydrogenase (G6PDH) | XL | CD | PDA, PVSD, MVS, TR | HF | >300 | [58,59,60,61,62,63,64,65,66,67] (selected) * |
3.2.2. G6PDH Deficiency
3.3. Disorders of Glycogen Metabolism
3.3.1. GAA-GSD
3.3.2. GBE-GSD
3.3.3. GDE-GSD
3.3.4. GYG1-GSD
3.3.5. GYS1-GSD
3.3.6. LAMP2-GSD
3.3.7. PRKAG2-GSD
3.3.8. RBCK1-GSD
3.3.9. SLC37A4-GSD
| Affected Gene | Affected Protein | Inheritance | Heart Defects & Manifestations | No. Patients Identified by Our Search | Ref. | ||
|---|---|---|---|---|---|---|---|
| Cardiomyopathies | Structural Defects | Arrhythmogenic Disorders | |||||
| GAA | α-1,4-glucosidase (GAA) | AR | DCM, HCM, LVH, VEFR, BVH | VLD, LAE, LBBB, RBBB, AVB, MVP | AT, AF, TC, HF, LQTS | >300 | [72,73,74,75] (selected) * |
| GBE1 | Glycogen branching enzyme (GBE1) | AR | DCM, HCM, LVH | CMG | AT, HF | 35 | [76,77,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101] |
| AGL | Glycogen debranching enzyme (GDE) | AR | DCM/HCM, LVH, RDH, LVD | SD | - | 204 | [80,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146] |
| GYG1 | Glycogenitype 1 (GYG1) | AR | HCM | - | VF, TC, HF | 7 | [147,148,149,150] |
| GYS1 | Muscle glycogen synthase | AR | HCM, LVH | LAE | HF | 4 | [152,153] |
| LAMP2 | Lysosome-associated membrane protein-2 (LAMP2) | XLD | DCM, HCM, CH | - | - | 200 | [110,157,158,159,160,161,162,163,164,165,166,167,168,169,170,171,172,173,174,175,176,177,178,179,180,181,182,183,184,185,186,187,188,189,190,191,192] |
| PRKAG2 | AMP-activated protein kinase (AMPK) | AD | HCM, LVH, DI | AVB, LBBB, RBBB | HF, TC | 103 | [195,196,197,198,199,200,201,202,203,204,205] |
| RBCK1 | E3 Ubiquitin ligase | AR | DCM | - | HF | 16 | [206,209,210,211] |
| SCL37A4 | Glucose 6-phosphate translocase type I (G6PT1) | AD | RVH | ASD, VSD, ToF, PPS | HF | 4 | [213,214] |
3.4. Congenital Disorders of Glycosylation
3.4.1. Disorders Affecting N-Glycosylation
ALG3-CDG
ALG6-CDG
ALG9-CDG
ALG12-CDG
GMPPB-CDG
NPL-CDG
PGM1-CDG
PMM2-CDG
3.4.2. Disorders Affecting O-Glycosylation
B3GALTL-CDG
B3GAT3-CDG
FKRP-CDG
FKTN-CDG
POMT1-CDG
POMT2-CDG
XYLT2-CDG
3.4.3. Dolichol-Phosphate Synthesis Defects
DOLK-CDG
DPM3-CDG
MPDU1-CDG
SRD5A3-CDG
3.4.4. Glycosylphosphatidylinositol (GPI)-Anchor Biosynthesis Defects
PIGA-CDG
PIGL-CDG
PIGN-CDG
PIGT-CDG
PIGV-CDG & PIGO-CDG
3.4.5. COG Complex Defects
COG1-CDG & COG7-CDG
3.4.6. V-ATPase Complex Defects
ATP6V1A-CDG & ATP6V1E1-CDG
| Affected Gene | Affected Protein | Inheritance | Heart Defects & Manifestations | No. Patients Identified by Our Search | Ref. | ||
|---|---|---|---|---|---|---|---|
| Cardiomyopathies | Structural Defects | Arrhythmogenic Disorders | |||||
| Defects of N-Glycosilation | |||||||
| ALG3 | Dolichol-P-mannose: Man5GlcNAc2-PP-dolichol mannosyltransferase | AR | HOCM, RDA, TR | VSD, AVD, PFO, PDA, MVS | - | 15 | [200,201,202,203,204,205,206,207,208,209,210,211,212,213,214,215,216,216,217,218] |
| ALG6 | α-1,3-glucosyltransferase | AR | DCM, LVD | - | - | 1 | 221 |
| ALG9 | α-1,2-mannosyltransferase | AR | RVD | ASD, BAV, SAI, PDA, TVR | - | 12 | [14,219,222,223,224,225,226] |
| ALG12 | Dolichol-P-mannose: Man7GlcNAc2-PP-dolichol mannosyltransferase | AR | HCM, PVSD | PDA, SD, VSD, PFO | AT, HF | 9 | [14,227,228,230] |
| GMPPB | GDP-mannose pyrophosphorylase B (GMPPB) | AR | VD, LVD, VEFR | RBBB | - | 5 | [232,233,234] |
| NPL | N-acetylneuraminate pyruvate lyase (NPL) | AR | DCM, LVH, VEFR | - | AT, HF | 1 | 236 |
| PGM1 | Phosphoglucomutase 1 (PGM1) | AR | DCM, RCM, LVD, LVH, AC, CMG | VSD, MP | AT, AF, HF | 30 | [14,239,241,243,244,245,246,247,248,249,250,251,252,253,254,255,256,257] |
| PMM2 | Phosphomannomutase 2 (PMM2) | AR | HCM, ToF | ASD, PDA, PFO, PPS | - | 70 | [14,230,243,259,260,261,262,263,264,265,266,267,268,269,270,271,272,273,274,275,276,277,278,279,280,281,282,283,284] |
| Defects of O-Glycosilation | |||||||
| B3GALTL | O-fucose-specific β-1,3-N-glucosyltransferasen (B3GALTL) | AR | HoLV | ASD, VSD, BVP | - | 19 | [14,286,287,288,289,290,291,292] |
| B3GAT3 | β-1,3-glucuronyltransferase 3 (B3GAT3) | AR | BAV, VSD, ASD, MVP, PDA, PPS, DAR | - | 15 | [14,293,294,295,296,297,298,299,300,301] | |
| FKRP | Fukutin-related protein (FKRP) | AR | LVD, LVRWMA, VEFR, DCM | VSD, TI, RBBB, TGA | HF | 220 | [14,304,305,306,307,308,309,310,311,312,313,314,315,316,317,318,319,320,321,322,323,324,325,326,327,328,329] |
| FKTN | Fukutin (FKTN) | AR | DCM, VEFR, CD | PPS, TGA, CMG | - | 77 | [14,183,330,331,332,333,334,335,336,337,338,339,340,341,342,343] |
| POMT1 | O-mannosyltransferase 1 (POMT1) | AR | DCM, VD, VEFR, LVH, SI | - | - | 5 | [14,321,345,346] |
| POMT2 | O-mannosyltransferase 2 (POMT2) | AR | LVH, DCM, VEFR | AD, DAR | - | 7 | [14,321,346,348,349] |
| XYLT2 | Xylosyltransferase 2 (XYLT2) | AR | - | ASD, AVD, MVP | - | 3 | [14,353,354] |
| Dolichol-phosphate synthesis defects | |||||||
| DOLK | Dolichol kinase (DOLK) | AR | DCM, HCM, BVD, CD, LVD | CMG, PDA, VSD | HF, TC, BR, AT | 26 | [14,227,355,356,357,358,359,360] |
| DPM3 | Dolichol-phosphate mannose synthase subunit 3 (DPM3) | AR | DCM, LVD, LVRWMA | - | - | 3 | [361,362,363] |
| MPDU1 | Mannose-phosphate-dolichol utilization defect 1 (MPDU1) | AR | DCM, NCM | - | - | 4 | [364,365,366] |
| SRD5A3 | Steroid 5 α-reductase 3 (SRD5A3) | AR | CM | ASD, TGA, PFO | AT, LQTS | 7 | [14,368,369,370,371] |
| Glycosylphosphatidylinositol anchor olichol-phosphate synthesis defects | |||||||
| PIGA | Phosphatidylinositol glycan anchor biosynthesis class A protein (PIGA) | XL | HCM | AD, BAV, PFO, ASD | AR. HF | 19 | [14,372,373,374,375,376,377] |
| PIGL | Phosphatidylinositol glycan anchor biosynthesis class L protein (PIGL) | AR | - | HSS, TGA, ToF, VSD, DOV, PPS | - | 8 | [14,378,379,380,381] |
| PIGN | Phosphatidylinositol glycan anchor biosynthesis class N protein (PIGN) | AR | NCM, RVD | DAR, PVSD, DC, PDA, PFO, ToF, PPS, DC, VSD, ASD, OA | - | 18 | [14,383,384,385,386,387,388] |
| PIGT | Phosphatidylinositol glycan anchor biosynthesis class T protein (PIGT) | AR | RCM | PDA, PFO, VSD, ASD | - | 8 | [14,389,391,392] |
| PIGV | Phosphatidylinositol glycan anchor biosynthetic enzymes class V (PIGV) | AR | - | ASD | - | 1 | [393] |
| PIGO | Phosphatidylinositol glycan anchor biosynthetic enzymes class O (PIGO) | AR | - | ASD, ToF | TC | 2 | [395,396,397] |
| COG complex defects | |||||||
| COG1 | Subunit 1 of the COG complex in Golgi trafficking (COG1) | AR | HCM | PFO, ASH, AI, PMV | - | 4 | [14,398,399,400,401] |
| COG7 | Subunit 7 of the COG complex in Golgi trafficking (COG7) | AR | - | PVSD, TI, ASD | - | 6 | [14,398,399,400,401] |
| V-ATPase complex defects | |||||||
| ATP6V1A | V-ATPase A subunit 1 (ATP6V1A) | AR | HCM | SD, RBBB | HF, LQTS | 4 | [14,405,406] |
| ATP6V1E1 | ATPase subunit E (ATP6V1E1) | AR | HoRV, RVD, HCM | AD, MVR, TVR, PFO, SD, TI, AI, RHHS, TVS, PDA, MVP, RBBB | - | 5 | [14,405,407] |
3.5. Disorders of Lysosomal Carbohydrate-Processing
3.5.1. CTSA-LSD
3.5.2. GBA1-LSD
3.5.3. GLA-LSD
3.5.4. GLB1-LSD
3.5.5. HEXB-LSD
3.5.6. IDUA-LSD
3.5.7. IDS-LSD
3.5.8. SGSH-LSD
3.5.9. NAGLU-LSD
3.5.10. HGSNAT-LSD and GNS-LSD
3.5.11. GALNS-LSD
3.5.12. ARSB-LSD
3.5.13. GUSB-LSD
3.5.14. ARSK-LSD
| Affected Gene | Affected Protein | Inheritance | Heart Defects & Manifestations | No. Patients Identified by Our Search | Ref. | ||
|---|---|---|---|---|---|---|---|
| Cardiomyopathies | Structural Defects | Arrhythmogenic Disorders | |||||
| CTSA | Cathepsin A (CTSA) | AR | CM, LVRWMA | DC, SD, VLD | HF | 10 | [411,412,413,414,415,416,417,418,419,420] |
| GBA1 | Glucocerebrosidase (GBA) | AR | LVH, HCM, DCM | VC, ARe, MVR, CMG, MF, AVS, MVS, LAE, RAE, AVB | AF, TC, BR, HF | 141 | [415,422,423,424,425,426,427,428,429,430,431,432,433,434,435,436,437,438,439,440,441,442,443,444,445,446,447,448,449,450,451,452,453,454,455,456,457,458,459,460,461,462,463,464] |
| GLA | α-galactosidase (GLA) | XL | LVH, LVD, RVH, RCM | ARe, MVR, AD, MF, DI, MVP, AVP, AVB | AT, BR | >300 | [467,468,469,470] * selected |
| GLB1 | β-galactosidase (GLB1) | AR (GM1 gangliosidosis) | DCM, LVH, LVD, HCM | RBBB, HSS | HF | 25 | [472,473,474,475,476,477,478,479,480] |
| GLB1 | β-galactosidase (GLB1) | AR (mucopolysaccharidosis) | LVH, SH | ARe, DAR, MVR, MVS, AVS, MF, VF | - | 8 | [481,482,483] |
| HEXB | β-hexosaminidase B (HEXB) | AR | HCM, LVD | CMG, ARe, MVR, MVP, MVS, AVP, VLD | HF | 9 | [484,485,486,487,488,489,490] |
| IDUA | α-L-iduronidase (IDUA) | AR | LVD, LVH | ARe, MVR, MVS, AVS, SI, DI | HF | 440 | [492,493,494,495,496,497,498,499,500,501,502,503,504,505,506,507,508,509,510,511,512,513,514,515,516,517,518,519,520,521,522,523,524,525,526,527,528,529,530,531,532,533,534,535,536,537,538,539,540,541,542,543,544,545,546,547,548,549,550,551] |
| IDS | 2-sulfatase (IDS) | XLR | HCM, SH | Are, MVR, DAR, MVS, MI, AVS, AI, TI | AF, AT | 742 | [497,509,512,540,544,553,554,555,556,557,558,559,560,561,562,563,564,565,566,567,568,569,570,571,572,573,574,575,576,577,578,579,580,581,582,583,584,585,586,587,588,589,590,591,592,593,594,595,596,597,598] |
| SGSH | N-sulfoglucosamine sulfohydrolase | AD | SH, CH, RVHCM | CVD, ARe, MVR, MVS, AVS, VSD | BR, AT, HF | 47 | [532,589,604,605,606,607,608,609] |
| NAGLU | N-α-acetylglucosaminidase (NAGLU) | AD | SH, CH, RVHCM | ARe, MVR, CVD, MVS, AVS, VSD | BR, AT | 39 | [589,604,605,606,609,612,613,614] |
| HGSNAT | heparan acetyl-CoA:alpha-glucosaminide N-acetyltransferase (HGSNAT) | AD | SH, CH RVHCM | ARe, MVR, CVD, MVS, AVS, VSD | BR, AT, HF | 10 | [589,606,607,617] |
| GNS | glucosamine (N-acetyl)-6-sulfatase (GNS) | AD | SH | MVR | - | 2 | [609] |
| GALNS | galactosamine (N-acetyl)-6-sulfatase (GALNS) | AD | CD | ARe, MVR, TVR, CVD, MF, VF, AVS, MVS, TVS | - | >300 | [482,618,619,620,621,622] *selected |
| ARSB | N-acetylgalactosamine 4-sulfatase (ARSB) | AD | VSD, SH, HCM | DAR, MVR, ARe, TVR, VLD, MVP, AVP, TVP, AI, MI, MVS, TVS, LVCR | HF | 520 | [497,512,532,540,544,556,564,570,574,579,589,590,591,596,625,626,627,628,629,630,631,632,633,634,635,636,637,638,639,640,641,642,643,644,645,646,647,648,649,650,651,652,653,654,655,656,657,658,659,660,661,662,663] |
| GUSB | β-glucuronidase (GUSB) | AD | LVD, LVH, CH | ARe, DAR, MVR, TVR, CVD, AVI, AVS, MVS, MI, MF | HF | 46 | [540,564,590,654,666,667,668,669,670,671,672,673,674,675] |
| ARSK | arylsulfatase K (ARSK) | AD | LVH | ARe, MVR, AVS, MVS | = | 1 | [678] |
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Tayal, U.; Prasad, S.; Cook, S.A. Genetics and genomics of dilated cardiomyopathy and systolic heart failure. Genome Med. 2017, 9, 20. [Google Scholar] [CrossRef]
- Urbich, M.; Globe, G.; Pantiri, K.; Heisen, M.; Bennison, C.; Wirtz, H.S.; Di Tanna, G.L. A Systematic Review of Medical Costs Associated with Heart Failure in the USA (2014–2020). PharmacoEconomics 2020, 38, 1219–1236. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, D.; McKenna, W.J. Genetics of inherited cardiomyopathy. Eur. Heart J. 2012, 33, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Czepluch, F.S.; Wollnik, B.; Hasenfuß, G. Genetic determinants of heart failure: Facts and numbers. ESC Heart Fail. 2018, 5, 211–217. [Google Scholar] [CrossRef]
- Sacks, D.; Baxter, B.; Campbell, B.C.V.; Carpenter, J.S.; Cognard, C.; Dippel, D.; Eesa, M.; Fischer, U.; Hausegger, K.; Hirsch, J.A.; et al. Multisociety Consensus Quality Improvement Revised Consensus Statement for Endovascular Therapy of Acute Ischemic Stroke. Int. J. Stroke Off. J. Int. Stroke Soc. 2018, 13, 612–632. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.R.; van Karnebeek, C.D.M. Inborn errors of metabolism. Handb. Clin. Neurol. 2019, 162, 449–481. [Google Scholar] [CrossRef]
- Ferreira, C.R.; Rahman, S.; Keller, M.; Zschocke, J. An international classification of inherited metabolic disorders (ICIMD). J. Inherit. Metab. Dis. 2021, 44, 164–177. [Google Scholar] [CrossRef]
- Jeanmonod, R.; Asuka, E.; Jeanmonod, D. Inborn Errors Of Metabolism; StatPearls Publishing: Copyright © 2022; StatPearls Publishing LLC: Treasure Island, FL, USA, 2022. [Google Scholar]
- Waters, D.; Adeloye, D.; Woolham, D.; Wastnedge, E.; Patel, S.; Rudan, I. Global birth prevalence and mortality from inborn errors of metabolism: A systematic analysis of the evidence. J. Glob. Health 2018, 8, 021102. [Google Scholar] [CrossRef]
- Elliott, P.; Limongelli, G. Cardiac Aspects of Inherited Metabolic Diseases. In Inherited Metabolic Disease in Adults: A Clinical Guide; Hollak, C.E.M., Lachmann, R., Eds.; Oxford University Press: Oxford, UK, 2016. [Google Scholar]
- Kassiotis, C.; Rajabi, M.; Taegtmeyer, H. Metabolic reserve of the heart: The forgotten link between contraction and coronary flow. Prog. Cardiovasc. Dis. 2008, 51, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, E.Y.; Seo, Y.M.; Yoon, T.K.; Lee, W.S.; Lee, K.A. Function of the pentose phosphate pathway and its key enzyme, transketolase, in the regulation of the meiotic cell cycle in oocytes. Clin. Exp. Reprod. Med. 2012, 39, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Loaeza-Reyes, K.J.; Zenteno, E.; Moreno-Rodríguez, A.; Torres-Rosas, R.; Argueta-Figueroa, L.; Salinas-Marín, R.; Castillo-Real, L.M.; Pina-Canseco, S.; Cervera, Y.P. An Overview of Glycosylation and its Impact on Cardiovascular Health and Disease. Front. Mol. Biosci. 2021, 8, 751637. [Google Scholar] [CrossRef] [PubMed]
- Marques-da-Silva, D.; Francisco, R.; Webster, D.; Dos Reis Ferreira, V.; Jaeken, J.; Pulinilkunnil, T. Cardiac complications of congenital disorders of glycosylation (CDG): A systematic review of the literature. J. Inherit. Metab. Dis. 2017, 40, 657–672. [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ (Clin. Res. Ed.) 2021, 372, n71. [Google Scholar] [CrossRef]
- Chen, L.Q.; Cheung, L.S.; Feng, L.; Tanner, W.; Frommer, W.B. Transport of sugars. Annu. Rev. Biochem. 2015, 84, 865–894. [Google Scholar] [CrossRef] [PubMed]
- Kalra, J.; Mangali, S.B.; Dasari, D.; Bhat, A.; Goyal, S.; Dhar, I.; Sriram, D.; Dhar, A. SGLT1 inhibition boon or bane for diabetes-associated cardiomyopathy. Fundam. Clin. Pharmacol. 2020, 34, 173–188. [Google Scholar] [CrossRef]
- Banerjee, S.K.; McGaffin, K.R.; Pastor-Soler, N.M.; Ahmad, F. SGLT1 is a novel cardiac glucose transporter that is perturbed in disease states. Cardiovasc. Res. 2009, 84, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Packer, M. Potential Interactions When Prescribing SGLT2 Inhibitors and Intravenous Iron in Combination in Heart Failure. JACC Heart Fail. 2022, 11, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Jhund, P.S. SGLT2 Inhibitors and Heart Failure with Preserved Ejection Fraction. Heart Fail. Clin. 2022, 18, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Santer, R.; Klepper, J. Disorders of Glucose Transport. In Inborn Metabolic Diseases: Diagnosis and Treatment; Saudubray, J.-M., Baumgartner, M.R., Walter, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2016; pp. 175–183. [Google Scholar]
- Ma, L.; Xu, J.; Tang, Q.; Cao, Y.; Kong, R.; Li, K.; Liu, J.; Jiang, L. SLC2A3 variants in familial and sporadic congenital heart diseases in a Chinese Yunnan population. J. Clin. Lab. Anal. 2022, 36, e24456. [Google Scholar] [CrossRef] [PubMed]
- Grover-McKay, M.; Walsh, S.A.; Thompson, S.A. Glucose transporter 3 (GLUT3) protein is present in human myocardium. Biochim. Biophys. Acta 1999, 1416, 145–154. [Google Scholar] [CrossRef]
- Willaert, A.; Khatri, S.; Callewaert, B.L.; Coucke, P.J.; Crosby, S.D.; Lee, J.G.; Davis, E.C.; Shiva, S.; Tsang, M.; De Paepe, A.; et al. GLUT10 is required for the development of the cardiovascular system and the notochord and connects mitochondrial function to TGFβ signaling. Hum. Mol. Genet. 2012, 21, 1248–1259. [Google Scholar] [CrossRef] [PubMed]
- Coucke, P.J.; Willaert, A.; Wessels, M.W.; Callewaert, B.; Zoppi, N.; De Backer, J.; Fox, J.E.; Mancini, G.M.; Kambouris, M.; Gardella, R.; et al. Mutations in the facilitative glucose transporter GLUT10 alter angiogenesis and cause arterial tortuosity syndrome. Nat. Genet. 2006, 38, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.H.; Meyer, S.; Peltekova, V.D.; Lindinger, A.; Teebi, A.S.; Faiyaz-Ul-Haque, M. A novel non-sense mutation in the SLC2A10 gene of an arterial tortuosity syndrome patient of Kurdish origin. Eur. J. Pediatr. 2009, 168, 867–870. [Google Scholar] [CrossRef] [PubMed]
- Zoppi, N.; Chiarelli, N.; Cinquina, V.; Ritelli, M.; Colombi, M. GLUT10 deficiency leads to oxidative stress and non-canonical αvβ3 integrin-mediated TGFβ signalling associated with extracellular matrix disarray in arterial tortuosity syndrome skin fibroblasts. Hum. Mol. Genet. 2015, 24, 6769–6787. [Google Scholar] [CrossRef]
- Castori, M.; Ritelli, M.; Zoppi, N.; Molisso, L.; Chiarelli, N.; Zaccagna, F.; Grammatico, P.; Colombi, M. Adult presentation of arterial tortuosity syndrome in a 51-year-old woman with a novel homozygous c.1411+1G>A mutation in the SLC2A10 gene. Am. J. Med. Genet. Part A 2012, 158, 1164–1169. [Google Scholar] [CrossRef]
- Kocova, M.; Kacarska, R.; Kuzevska-Maneva, K.; Prijic, S.; Lazareska, M.; Dordoni, C.; Ritelli, M.; Colombi, M. Clinical Variability in Two Macedonian Families with Arterial Tortuosity Syndrome. Balk. J. Med. Genet. BJMG 2018, 21, 47–52. [Google Scholar] [CrossRef]
- Boel, A.; Veszelyi, K.; Németh, C.E.; Beyens, A.; Willaert, A.; Coucke, P.; Callewaert, B.; Margittai, É. Arterial Tortuosity Syndrome: An Ascorbate Compartmentalization Disorder? Antioxid. Redox Signal. 2021, 34, 875–889. [Google Scholar] [CrossRef] [PubMed]
- Beltramo, E.; Berrone, E.; Tarallo, S.; Porta, M. Effects of thiamine and benfotiamine on intracellular glucose metabolism and relevance in the prevention of diabetic complications. Acta Diabetol. 2008, 45, 131–141. [Google Scholar] [CrossRef]
- Zhang, S.; Qiao, Y.; Wang, Z.; Zhuang, J.; Sun, Y.; Shang, X.; Li, G. Identification of novel compound heterozygous variants in SLC19A2 and the genotype-phenotype associations in thiamine-responsive megaloblastic anemia. Clin. Chim. Acta Int. J. Clin. Chem. 2021, 516, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Bay, A.; Keskin, M.; Hizli, S.; Uygun, H.; Dai, A.; Gumruk, F. Thiamine-responsive megaloblastic anemia syndrome. Int. J. Hematol. 2010, 92, 524–526. [Google Scholar] [CrossRef] [PubMed]
- Viana, M.B.; Carvalho, R.I. Thiamine-responsive megaloblastic anemia, sensorineural deafness, and diabetes mellitus: A new syndrome? J. Pediatr. 1978, 93, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Mandel, H.; Berant, M.; Hazani, A.; Naveh, Y. Thiamine-dependent beriberi in the “thiamine-responsive anemia syndrome”. N. Engl. J. Med. 1984, 311, 836–838. [Google Scholar] [CrossRef] [PubMed]
- Abboud, M.R.; Alexander, D.; Najjar, S.S. Diabetes mellitus, thiamine-dependent megaloblastic anemia, and sensorineural deafness associated with deficient alpha-ketoglutarate dehydrogenase activity. J. Pediatr. 1985, 107, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Poggi, V.; Rindi, G.; Patrini, C.; De Vizia, B.; Longo, G.; Andria, G. Studies on thiamine metabolism in thiamine-responsive megaloblastic anaemia. Eur. J. Pediatr. 1989, 148, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Scharfe, C.; Hauschild, M.; Klopstock, T.; Janssen, A.J.; Heidemann, P.H.; Meitinger, T.; Jaksch, M. A novel mutation in the thiamine responsive megaloblastic anaemia gene SLC19A2 in a patient with deficiency of respiratory chain complex I. J. Med. Genet. 2000, 37, 669–673. [Google Scholar] [CrossRef]
- Gritli, S.; Omar, S.; Tartaglini, E.; Guannouni, S.; Fleming, J.C.; Steinkamp, M.P.; Berul, C.I.; Hafsia, R.; Jilani, S.B.; Belhani, A.; et al. A novel mutation in the SLC19A2 gene in a Tunisian family with thiamine-responsive megaloblastic anaemia, diabetes and deafness syndrome. Br. J. Haematol. 2001, 113, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Lorber, A.; Gazit, A.Z.; Khoury, A.; Schwartz, Y.; Mandel, H. Cardiac manifestations in thiamine-responsive megaloblastic anemia syndrome. Pediatr. Cardiol. 2003, 24, 476–481. [Google Scholar] [CrossRef]
- Lagarde, W.H.; Underwood, L.E.; Moats-Staats, B.M.; Calikoglu, A.S. Novel mutation in the SLC19A2 gene in an African-American female with thiamine-responsive megaloblastic anemia syndrome. Am. J. Med. Genet. Part A 2004, 125, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Saedi, S.; Maleki, M.; Pezeshki, S. Right ventricular dysfunction in thiamine-responsive megaloblastic anaemia syndrome: A case report. Heart Asia 2011, 3, 140–142. [Google Scholar]
- Aycan, Z.; Baş, V.N.; Cetinkaya, S.; Ağladioğlu, S.Y.; Kendirci, H.N.; Senocak, F. Thiamine-responsive megaloblastic anemia syndrome with atrial standstill: A case report. J. Pediatr. Hematol./Oncol. 2011, 33, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Xian, X.; Liao, L.; Shu, W.; Li, H.; Qin, Y.; Yan, J.; Luo, J.; Lin, F.Q. A Novel Mutation of SLC19A2 in a Chinese Zhuang Ethnic Family with Thiamine-Responsive Megaloblastic Anemia. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 47, 1989–1997. [Google Scholar] [CrossRef] [PubMed]
- Argun, M.; Baykan, A.; Hatipoğlu, N.; Akın, L.; Şahin, Y.; Narin, N.; Kurtoğlu, S. Arrhythmia in thiamine responsive megaloblastic anemia syndrome. Turk. J. Pediatr. 2018, 60, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Akbari, M.T.; Zare Karizi, S.; Mirfakhraie, R.; Keikhaei, B. Thiamine-responsive megaloblastic anemia syndrome with Ebstein anomaly: A case report. Eur. J. Pediatr. 2014, 173, 1663–1665. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Cheng, Q.; Ding, Y.; Li, Q.; Yao, R.; Wang, J.; Wang, X. TRMA syndrome with a severe phenotype, cerebral infarction, and novel compound heterozygous SLC19A2 mutation: A case report. BMC Pediatr. 2019, 19, 233. [Google Scholar] [CrossRef]
- Rahman, M.; Hasan, M. Pentose Phosphate Pathway in Disease and Therapy. Adv. Mater. Res. 2014, 995, 1–27. [Google Scholar] [CrossRef]
- Debasis, B.; Sreejayan, N. Nutritional and therapeutic interventions for diabetes and metabolic syndrome. Acta Endocrinol. 2018, 14, 438. [Google Scholar] [CrossRef]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef]
- Verhoeven, N.M.; Wallot, M.; Huck, J.H.; Dirsch, O.; Ballauf, A.; Neudorf, U.; Salomons, G.S.; van der Knaap, M.S.; Voit, T.; Jakobs, C. A newborn with severe liver failure, cardiomyopathy and transaldolase deficiency. J. Inherit. Metab. Dis. 2005, 28, 169–179. [Google Scholar] [CrossRef]
- Verhoeven, N.M.; Jakobs, C. Disorders of the Pentose Phosphate Pathway. In Inborn Metabolic Diseases: Diagnosis and Treatment; Fernandes, J., Saudubray, J.-M., van den Berghe, G., Walter, J.H., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; pp. 131–134. [Google Scholar]
- Williams, M.; Valayannopoulos, V.; Altassan, R.; Chung, W.K.; Heijboer, A.C.; Keng, W.T.; Lapatto, R.; McClean, P.; Mulder, M.F.; Tylki-Szymańska, A.; et al. Clinical, biochemical, and molecular overview of transaldolase deficiency and evaluation of the endocrine function: Update of 34 patients. J. Inherit. Metab. Dis. 2019, 42, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Kishnani, P.S.; Chen, Y.-T. Chapter 156. Disorders of Pentose Phosphate Pathway. In Rudolph′s Pediatrics, 22e; Rudolph, C.D., Rudolph, A.M., Lister, G.E., First, L.R., Gershon, A.A., Eds.; The McGraw-Hill Companies: New York, NY, USA, 2011. [Google Scholar]
- Verhoeven, N.M.; Huck, J.H.; Roos, B.; Struys, E.A.; Salomons, G.S.; Douwes, A.C.; Van der Knaap, M.S.; Jakobs, C. Transaldolase deficiency: Liver cirrhosis associated with a new inborn error in the pentose phosphate pathway. Am. J. Hum. Genet. 2001, 68, 1086–1092. [Google Scholar] [CrossRef]
- Eyaid, W.; Al Harbi, T.; Anazi, S.; Wamelink, M.M.; Jakobs, C.; Al Salammah, M.; Al Balwi, M.; Alfadhel, M.; Alkuraya, F.S. Transaldolase deficiency: Report of 12 new cases and further delineation of the phenotype. J. Inherit. Metab. Dis. 2013, 36, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Jassim, N.; Alghaihab, M.; Saleh, S.A.; Alfadhel, M.; Wamelink, M.M.; Eyaid, W. Pulmonary manifestations in a patient with transaldolase deficiency. JIMD Rep. 2014, 12, 47–50. [Google Scholar] [CrossRef]
- Feghaly, J.; Al Hout, A.R.; Mercieca Balbi, M. Aspirin safety in glucose-6-phosphate dehydrogenase deficiency patients with acute coronary syndrome undergoing percutaneous coronary intervention. BMJ Case Rep. 2017, 2017, bcr2017220483. [Google Scholar] [CrossRef]
- Rawat, D.K.; Hecker, P.; Watanabe, M.; Chettimada, S.; Levy, R.J.; Okada, T.; Edwards, J.G.; Gupte, S.A. Glucose-6-phosphate dehydrogenase and NADPH redox regulates cardiac myocyte L-type calcium channel activity and myocardial contractile function. PLoS ONE 2012, 7, e45365. [Google Scholar] [CrossRef]
- Rigattieri, S.; Silvestri, P.; Minucci, A.; Di Russo, C.; Ferraiuolo, G.; Giardina, B.; Capoluongo, E.; Loschiavo, P. Drug-eluting stents in a patient with favism: Is the aspirin administration safe? J. Cardiovasc. Med. 2008, 9, 1159–1162. [Google Scholar] [CrossRef]
- Dogra, N.; Puri, G.D.; Rana, S.S. Glucose-6-phosphate dehydrogenase deficiency and cardiac surgery. Perfusion 2010, 25, 417–421. [Google Scholar] [CrossRef]
- Porto, I.; Leo, A.; Crea, F. Glucose-6-phosphate dehydrogenase (G6PDH) deficiency in a patient with ST-segment elevation acute myocardial infarction successfully treated by simple thrombectomy. J. Atheroscler. Thromb. 2011, 18, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Chowdhry, V.; Bisoyi, S.; Mishra, B. Perioperative challenges in a patient of severe G6PD deficiency undergoing open heart surgery. Ann. Card. Anaesth. 2012, 15, 50–53. [Google Scholar] [CrossRef] [PubMed]
- Albertsen, J.; Ommen, H.B.; Wandler, A.; Munk, K. Fatal haemolytic crisis with microvascular pulmonary obstruction mimicking a pulmonary embolism in a young African man with glucose-6-phosphate dehydrogenase deficiency. BMJ Case Rep. 2014, 2014, bcr2013201432. [Google Scholar] [CrossRef] [PubMed]
- Balderia, P.G.; Wongrakpanich, S.; Patel, M.; Stanek, M. Healing the orphaned heart: Heart failure in a patient with glucose-6-phosphate dehydrogenase deficiency. BMJ Case Rep. 2015, 2015, bcr2015209365. [Google Scholar] [CrossRef] [PubMed]
- Padakanti, A.; Shenoy, A.; Kamath, A.; Chakrapani, M. Drug-induced Hemolysis in G6PD Deficiency: An Unusual Presentation of a Common Clinical Condition. Acta Med. 2019, 62, 166–169. [Google Scholar] [CrossRef][Green Version]
- Dore, M.P.; Portoghese, M.; Pes, G.M. The Elderly with Glucose-6-Phosphate Dehydrogenase Deficiency are More Susceptible to Cardiovascular Disease. J. Atheroscler. Thromb. 2021, 28, 604–610. [Google Scholar] [CrossRef]
- Meloni, L.; Manca, M.R.; Loddo, I.; Cioglia, G.; Cocco, P.; Schwartz, A.; Muntoni, S.; Muntoni, S. Glucose-6-phosphate dehydrogenase deficiency protects against coronary heart disease. J. Inherit. Metab. Dis. 2008, 31, 412–417. [Google Scholar] [CrossRef]
- Hecker, P.A.; Leopold, J.A.; Gupte, S.A.; Recchia, F.A.; Stanley, W.C. Impact of glucose-6-phosphate dehydrogenase deficiency on the pathophysiology of cardiovascular disease. Am. J. Physiology. Heart Circ. Physiol. 2013, 304, H491–H500. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Brenner, D.A.; Cui, L.; Lim, C.C.; Wang, B.; Pimentel, D.R.; Koh, S.; Sawyer, D.B.; Leopold, J.A.; Handy, D.E.; et al. Glucose-6-phosphate dehydrogenase modulates cytosolic redox status and contractile phenotype in adult cardiomyocytes. Circ. Res. 2003, 93, e9–e16. [Google Scholar] [CrossRef] [PubMed]
- Brown, A. 457Glycogen and Energy Metabolism. In Neuroglia; Kettenmann, H., Ransom, B.R., Eds.; Oxford University Press: Oxford, UK, 2012. [Google Scholar]
- Di Rocco, M.; Buzzi, D.; Tarò, M. Glycogen storage disease type II: Clinical overview. Acta Myol. Myopathies Cardiomyopathies Off. J. Mediterr. Soc. Myol. 2007, 26, 42–44. [Google Scholar]
- Lim, J.A.; Li, L.; Raben, N. Pompe disease: From pathophysiology to therapy and back again. Front. Aging Neurosci. 2014, 6, 177. [Google Scholar] [CrossRef] [PubMed]
- Matsuishi, T.; Yoshino, M.; Terasawa, K.; Nonaka, I. Childhood acid maltase deficiency. A clinical, biochemical, and morphologic study of three patients. Arch. Neurol. 1984, 41, 47–52. [Google Scholar] [CrossRef]
- Van Kooten, H.A.; Roelen, C.H.A.; Brusse, E.; Van der Beek, N.; Michels, M.; Van der Ploeg, A.T.; Wagenmakers, M.; Van Doorn, P.A. Cardiovascular disease in non-classic Pompe disease: A systematic review. Neuromuscul. Disord. NMD 2021, 31, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Szymańska, E.; Szymańska, S.; Truszkowska, G.; Ciara, E.; Pronicki, M.; Shin, Y.S.; Podskarbi, T.; Kępka, A.; Śpiewak, M.; Płoski, R.; et al. Variable clinical presentation of glycogen storage disease type IV: From severe hepatosplenomegaly to cardiac insufficiency. Some discrepancies in genetic and biochemical abnormalities. Arch. Med. Sci. AMS 2018, 14, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Ndugga-Kabuye, M.K.; Maleszewski, J.; Chanprasert, S.; Smith, K.D. Glycogen storage disease type IV: Dilated cardiomyopathy as the isolated initial presentation in an adult patient. BMJ Case Rep. 2019, 12, e230068. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.S. Glycogen storage disease: Clinical, biochemical, and molecular heterogeneity. Semin. Pediatr. Neurol. 2006, 13, 115–120. [Google Scholar] [CrossRef]
- Ishihara, T.; Uchino, F.; Adachi, H.; Takahashi, M.; Watanabe, S.; Tsunetoshi, S.; Fuji, T.; Ikee, Y. Type IV glycogenosis—A study of two cases. Acta Pathol. Jpn. 1975, 25, 613–633. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Shirasawa, K.; Yotsumoto, S.; Nagahara, S. Type III glycogenosis with deposition of urate and amyloid. Acta Pathol. Jpn. 1980, 30, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Servidei, S.; Riepe, R.E.; Langston, C.; Tani, L.Y.; Bricker, J.T.; Crisp-Lindgren, N.; Travers, H.; Armstrong, D.; DiMauro, S. Severe cardiopathy in branching enzyme deficiency. J. Pediatr. 1987, 111, 51–56. [Google Scholar] [CrossRef]
- Hemsrichart, V.; Karalak, S.; Thakerngpol, K.; Stitnimankarn, T. Type IV glycogen storage disease: First reported case in Thailand. J. Med. Assoc. Thail. Chotmaihet Thangphaet 1989, 72, 697–700. [Google Scholar]
- Sokal, E.M.; Van Hoof, F.; Alberti, D.; De Ville de Goyet, J.; De Barsy, T.; Otte, J.B. Progressive cardiac failure following orthotopic liver transplantation for type IV glycogenosis. Eur. J. Pediatr. 1992, 151, 200–203. [Google Scholar] [CrossRef] [PubMed]
- Schröder, J.M.; May, R.; Shin, Y.S.; Sigmund, M.; Nase-Hüppmeier, S. Juvenile hereditary polyglucosan body disease with complete branching enzyme deficiency (type IV glycogenosis). Acta Neuropathol. 1993, 85, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Van Noort, G.; Straks, W.; Van Diggelen, O.P.; Hennekam, R.C. A congenital variant of glycogenosis type IV. Pediatr. Pathol. 1993, 13, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Starzl, T.E.; Demetris, A.J.; Trucco, M.; Ricordi, C.; Ildstad, S.; Terasaki, P.I.; Murase, N.; Kendall, R.S.; Kocova, M.; Rudert, W.A.; et al. Chimerism after liver transplantation for type IV glycogen storage disease and type 1 Gaucher′s disease. N. Engl. J. Med. 1993, 328, 745–749. [Google Scholar] [CrossRef]
- Herrick, M.K.; Twiss, J.L.; Vladutiu, G.D.; Glasscock, G.F.; Horoupian, D.S. Concomitant branching enzyme and phosphorylase deficiencies. An unusual glycogenosis with extensive neuronal polyglucosan storage. J. Neuropathol. Exp. Neurol. 1994, 53, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Alshak, N.S.; Cocjin, J.; Podesta, L.; Van de Velde, R.; Makowka, L.; Rosenthal, P.; Geller, S.A. Hepatocellular adenoma in glycogen storage disease type IV. Arch. Pathol. Lab. Med. 1994, 118, 88–91. [Google Scholar] [PubMed]
- Nase, S.; Kunze, K.P.; Sigmund, M.; Schroeder, J.M.; Shin, Y.; Hanrath, P. A new variant of type IV glycogenosis with primary cardiac manifestation and complete branching enzyme deficiency. In vivo detection by heart muscle biopsy. Eur. Heart J. 1995, 16, 1698–1704. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, P.; Podesta, L.; Grier, R.; Said, J.W.; Sher, L.; Cocjin, J.; Watanabe, F.; Vasiliauskas, E.; Van de Velde, R.; Makowka, L. Failure of liver transplantation to diminish cardiac deposits of amylopectin and leukocyte inclusions in type IV glycogen storage disease. Liver Transplant. Surg. Off. Publ. Am. Assoc. Study Liver Dis. Int. Liver Transplant. Soc. 1995, 1, 373–376. [Google Scholar] [CrossRef]
- Giuffrè, B.; Parini, R.; Rizzuti, T.; Morandi, L.; Van Diggelen, O.P.; Bruno, C.; Giuffrè, M.; Corsello, G.; Mosca, F. Severe neonatal onset of glycogenosis type IV: Clinical and laboratory findings leading to diagnosis in two siblings. J. Inherit. Metab. Dis. 2004, 27, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Das, B.B.; Narkewicz, M.R.; Sokol, R.J.; Chen, Y.T.; Bali, D.; Li, S.C.; Matthews, M.R.; Mierau, G.W.; Ivy, D.D. Amylopectinosis disease isolated to the heart with normal glycogen branching enzyme activity and gene sequence. Pediatr. Transplant. 2005, 9, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Raju, G.P.; Li, H.C.; Bali, D.S.; Chen, Y.T.; Urion, D.K.; Lidov, H.G.; Kang, P.B. A case of congenital glycogen storage disease type IV with a novel GBE1 mutation. J. Child Neurol. 2008, 23, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Shandling, A.H.; Safani, M. Coexistent manifestations of the Andersen-Tawil and Brugada syndromes. J. Electrocardiol. 2008, 41, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Eminoglu, T.F.; Tumer, L.; Okur, I.; Olgunturk, R.; Hasanoglu, A.; Gonul, I.I.; Dalgic, B. Multisystem involvement in a patient due to accumulation of amylopectin-like material with diminished branching enzyme activity. J. Inherit. Metab. Dis. 2008, 31 (Suppl. S2), S255–S259. [Google Scholar] [CrossRef] [PubMed]
- Lamperti, C.; Salani, S.; Lucchiari, S.; Bordoni, A.; Ripolone, M.; Fagiolari, G.; Fruguglietti, M.E.; Crugnola, V.; Colombo, C.; Cappellini, A.; et al. Neuropathological study of skeletal muscle, heart, liver, and brain in a neonatal form of glycogen storage disease type IV associated with a new mutation in GBE1 gene. J. Inherit. Metab. Dis. 2009, 32 (Suppl. 1), S161–S168. [Google Scholar] [CrossRef]
- Willot, S.; Marchand, V.; Rasquin, A.; Alvarez, F.; Martin, S.R. Systemic progression of type IV glycogen storage disease after liver transplantation. J. Pediatr. Gastroenterol. Nutr. 2010, 51, 661–664. [Google Scholar] [CrossRef] [PubMed]
- Magoulas, P.L.; El-Hattab, A.W.; Roy, A.; Bali, D.S.; Finegold, M.J.; Craigen, W.J. Diffuse reticuloendothelial system involvement in type IV glycogen storage disease with a novel GBE1 mutation: A case report and review. Hum. Pathol. 2012, 43, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Aksu, T.; Colak, A.; Tufekcioglu, O. Cardiac Involvement in Glycogen Storage Disease Type IV: Two Cases and the Two Ends of a Spectrum. Case Rep. Med. 2012, 2012, 764286. [Google Scholar] [CrossRef]
- Yu, W.; Brundler, M.A.; Wright, J.R., Jr. Polyglucosan Bodies in Placental Extravillious Trophoblast for the Diagnosis of Fatal Perinatal Neuromuscular-type Glycogen Storage Disease Type IV. Pediatr. Dev. Pathol. Off. J. Soc. Pediatr. Pathol. Paediatr. Pathol. Soc. 2018, 21, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Lyo, S.; Miles, J.; Meisner, J.; Guelfguat, M. Case report: Adult-onset manifesting heterozygous glycogen storage disease type IV with dilated cardiomyopathy and absent late gadolinium enhancement on cardiac magnetic resonance imaging. Eur. Heart J. Case Rep. 2020, 4, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Cenacchi, G.; Papa, V.; Costa, R.; Pegoraro, V.; Marozzo, R.; Fanin, M.; Angelini, C. Update on polyglucosan storage diseases. Virchows Arch. Int. J. Pathol. 2019, 475, 671–686. [Google Scholar] [CrossRef] [PubMed]
- Ellingwood, S.S.; Cheng, A. Biochemical and clinical aspects of glycogen storage diseases. J. Endocrinol. 2018, 238, R131–R141. [Google Scholar] [CrossRef]
- Mili, A.; Ben Charfeddine, I.; Mamaï, O.; Abdelhak, S.; Adala, L.; Amara, A.; Pagliarani, S.; Lucchiarri, S.; Ayadi, A.; Tebib, N.; et al. Molecular and biochemical characterization of Tunisian patients with glycogen storage disease type III. J. Hum. Genet. 2012, 57, 170–175. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S.; Hartwig, G.B.; Hays, A.; Eastwood, A.B.; Franco, R.; Olarte, M.; Chang, M.; Roses, A.D.; Fetell, M.; Schoenfeldt, R.S.; et al. Debrancher deficiency: Neuromuscular disorder in 5 adults. Ann. Neurol. 1979, 5, 422–436. [Google Scholar] [CrossRef]
- Moses, S.W.; Gadoth, N.; Bashan, N.; Ben-David, E.; Slonim, A.; Wanderman, K.L. Neuromuscular involvement in glycogen storage disease type III. Acta Paediatr. Scand. 1986, 75, 289–296. [Google Scholar] [CrossRef]
- Moses, S.W.; Wanderman, K.L.; Myroz, A.; Frydman, M. Cardiac involvement in glycogen storage disease type III. Eur. J. Pediatr. 1989, 148, 764–766. [Google Scholar] [CrossRef]
- Labrune, P.; Huguet, P.; Odievre, M. Cardiomyopathy in glycogen-storage disease type III: Clinical and echographic study of 18 patients. Pediatr. Cardiol. 1991, 12, 161–163. [Google Scholar] [CrossRef]
- Carvalho, J.S.; Matthews, E.E.; Leonard, J.V.; Deanfield, J. Cardiomyopathy of glycogen storage disease type III. Heart Vessel. 1993, 8, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Tada, H.; Kurita, T.; Ohe, T.; Shimomura, K.; Ishihara, T.; Yamada, Y.; Osawa, N. Glycogen storage disease type III associated with ventricular tachycardia. Am. Heart J. 1995, 130, 911–912. [Google Scholar] [CrossRef] [PubMed]
- Akazawa, H.; Kuroda, T.; Kim, S.; Mito, H.; Kojo, T.; Shimada, K. Specific heart muscle disease associated with glycogen storage disease type III: Clinical similarity to the dilated phase of hypertrophic cardiomyopathy. Eur. Heart J. 1997, 18, 532–533. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.J.; Deanfield, J.E.; Burch, M.; Baig, K.; McKenna, W.J.; Leonard, J.V. Comparison of the functional significance of left ventricular hypertrophy in hypertrophic cardiomyopathy and glycogenosis type III. Am. J. Cardiol. 1997, 79, 834–838. [Google Scholar] [CrossRef]
- Cuspidi, C.; Sampieri, L.; Pelizzoli, S.; Pontiggia, G.; Zanchetti, A.; Nappo, A.; Caputo, V.; Matturri, L. Obstructive hypertrophic cardiomyopathy in type III glycogen-storage disease. Acta Cardiol. 1997, 52, 117–123. [Google Scholar]
- Hashimoto, M.; Watanabe, G.; Yokoyama, T.; Tsutsumi, K.; Dohi, T.; Matsuda, M.; Okubo, M.; Nakamura, N.; Tsurumaru, M. Case report: Rupture of a gastric varix in liver cirrhosis associated with glycogen storage disease type III. J. Gastroenterol. Hepatol. 1998, 13, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Okuda, S.; Kanda, F.; Takahashi, K.; Kawanami, C.; Kinoshita, Y.; Fujita, M.; Maeda, S.; Jinnai, K.; Matsushita, T.; Sugio, T.; et al. Fatal liver cirrhosis and esophageal variceal hemorrhage in a patient with type IIIa glycogen storage disease. Intern. Med. 1998, 37, 1055–1057. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sugie, H.; Fukuda, T.; Ito, M.; Sugie, Y.; Kojoh, T.; Nonaka, I. Novel exon 11 skipping mutation in a patient with glycogen storage disease type IIId. J. Inherit. Metab. Dis. 2001, 24, 535–545. [Google Scholar] [CrossRef]
- Toda, G.; Yoshimuta, T.; Kawano, H.; Yano, K. Glycogen storage disease associated with left ventricular aneurysm in an elderly patient. Jpn. Circ. J. 2001, 65, 462–464. [Google Scholar] [CrossRef][Green Version]
- Mohart, D.; Russo, P.; Tobias, J.D. Perioperative management of a child with glycogen storage disease type III undergoing cardiopulmonary bypass and repair of an atrial septal defect. Paediatr. Anaesth. 2002, 12, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.C.; Mundy, H.R.; Lee, P.J.; Mohiaddin, R.H.; Pennell, D.J. Images in cardiovascular medicine. Myocardial fibrosis in glycogen storage disease type III. Circulation 2003, 107, e47. [Google Scholar] [CrossRef]
- Ogimoto, A.; Okubo, M.; Okayama, H.; Shin, Y.S.; Endo, Y.; Ebara, T.; Inoue, K.; Ohtsuka, T.; Tahara, H.; Murase, T.; et al. A Japanese patient with cardiomyopathy caused by a novel mutation R285X in the AGL gene. Circ. J. Off. J. Jpn. Circ. Soc. 2007, 71, 1653–1656. [Google Scholar] [CrossRef]
- Dagli, A.I.; Zori, R.T.; McCune, H.; Ivsic, T.; Maisenbacher, M.K.; Weinstein, D.A. Reversal of glycogen storage disease type IIIa-related cardiomyopathy with modification of diet. J. Inherit. Metab. Dis. 2009, 32 (Suppl. S1), S103–S106. [Google Scholar] [CrossRef]
- Vertilus, S.M.; Austin, S.L.; Foster, K.S.; Boyette, K.E.; Bali, D.S.; Li, J.S.; Kishnani, P.S.; Wechsler, S.B. Echocardiographic manifestations of Glycogen Storage Disease III: Increase in wall thickness and left ventricular mass over time. Genet. Med. Off. J. Am. Coll. Med. Genet. 2010, 12, 413–423. [Google Scholar] [CrossRef] [PubMed]
- LaBarbera, M.; Milechman, G.; Dulbecco, F. Premature coronary artery disease in a patient with glycogen storage disease III. J. Invasive Cardiol. 2010, 22, E156–E158. [Google Scholar]
- Clemente, M.; Gussinyer, M.; Arranz, J.A.; Riudor, E.; Yeste, D.; Albisu, M.; Carrascosa, A. Glycogen storage disease type III with hypoketosis. J. Pediatr. Endocrinol. Metab. JPEM 2010, 23, 833–836. [Google Scholar] [CrossRef] [PubMed]
- Valayannopoulos, V.; Bajolle, F.; Arnoux, J.B.; Dubois, S.; Sannier, N.; Baussan, C.; Petit, F.; Labrune, P.; Rabier, D.; Ottolenghi, C.; et al. Successful treatment of severe cardiomyopathy in glycogen storage disease type III with D,L-3-hydroxybutyrate, ketogenic and high-protein diet. Pediatr. Res. 2011, 70, 638–641. [Google Scholar] [CrossRef]
- Lee, T.M.; Berman-Rosenzweig, E.S.; Slonim, A.E.; Chung, W.K. Two Cases of Pulmonary Hypertension Associated with Type III Glycogen Storage Disease. JIMD Rep. 2011, 1, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, R.; Wedatilake, Y.; Coats, C.; Walker, F.; Elliott, P.; Lee, P.J.; Lachmann, R.H.; Murphy, E. Pregnancy and its management in women with GSD type III—A single centre experience. J. Inherit. Metab. Dis. 2012, 35, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Austin, S.L.; Proia, A.D.; Spencer-Manzon, M.J.; Butany, J.; Wechsler, S.B.; Kishnani, P.S. Cardiac Pathology in Glycogen Storage Disease Type III. JIMD Rep. 2012, 6, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Sentner, C.P.; Vos, Y.J.; Niezen-Koning, K.N.; Mol, B.; Smit, G.P. Mutation Analysis in Glycogen Storage Disease Type III Patients in the Netherlands: Novel Genotype-Phenotype Relationships and Five Novel Mutations in the AGL Gene. JIMD Rep. 2013, 7, 19–26. [Google Scholar] [CrossRef]
- Preisler, N.; Pradel, A.; Husu, E.; Madsen, K.L.; Becquemin, M.H.; Mollet, A.; Labrune, P.; Petit, F.; Hogrel, J.Y.; Jardel, C.; et al. Exercise intolerance in Glycogen Storage Disease Type III: Weakness or energy deficiency? Mol. Genet. Metab. 2013, 109, 14–20. [Google Scholar] [CrossRef]
- Brambilla, A.; Mannarino, S.; Pretese, R.; Gasperini, S.; Galimberti, C.; Parini, R. Improvement of Cardiomyopathy After High-Fat Diet in Two Siblings with Glycogen Storage Disease Type III. JIMD Rep. 2014, 17, 91–95. [Google Scholar] [CrossRef]
- Mayorandan, S.; Meyer, U.; Hartmann, H.; Das, A.M. Glycogen storage disease type III: Modified Atkins diet improves myopathy. Orphanet J. Rare Dis. 2014, 9, 196. [Google Scholar] [CrossRef]
- Rousseau-Nepton, I.; Okubo, M.; Grabs, R.; Mitchell, J.; Polychronakos, C.; Rodd, C. A founder AGL mutation causing glycogen storage disease type IIIa in Inuit identified through whole-exome sequencing: A case series. Can. Med. Assoc. J. J. L′Assoc. Med. Can. 2015, 187, E68–E73. [Google Scholar] [CrossRef] [PubMed]
- Mogahed, E.A.; Girgis, M.Y.; Sobhy, R.; Elhabashy, H.; Abdelaziz, O.M.; El-Karaksy, H. Skeletal and cardiac muscle involvement in children with glycogen storage disease type III. Eur. J. Pediatr. 2015, 174, 1545–1548. [Google Scholar] [CrossRef]
- Sentner, C.P.; Hoogeveen, I.J.; Weinstein, D.A.; Santer, R.; Murphy, E.; McKiernan, P.J.; Steuerwald, U.; Beauchamp, N.J.; Taybert, J.; Laforêt, P.; et al. Glycogen storage disease type III: Diagnosis, genotype, management, clinical course and outcome. J. Inherit. Metab. Dis. 2016, 39, 697–704. [Google Scholar] [CrossRef]
- Ben Chehida, A.; Ben Messaoud, S.; Ben Abdelaziz, R.; Mansouri, H.; Boudabous, H.; Hakim, K.; Ben Ali, N.; Ben Ameur, Z.; Sassi, Y.; Kaabachi, N.; et al. A lower energetic, protein and uncooked cornstarch intake is associated with a more severe outcome in glycogen storage disease type III: An observational study of 50 patients. J. Pediatr. Endocrinol. Metab. JPEM 2018, 31, 979–986. [Google Scholar] [CrossRef]
- Nazari, F.; Sinaei, F.; Nilipour, Y.; Petit, F.; Oveisgharan, S.; Nassiri-Toosi, M.; Razzaghy-Azar, M.; Mahmoudi, M.; Nafissi, S. Distinct Clinical and Genetic Findings in Iranian Patients with Glycogen Storage Disease Type 3. J. Clin. Neuromuscul. Dis. 2018, 19, 203–210. [Google Scholar] [CrossRef]
- Francini-Pesenti, F.; Tresso, S.; Vitturi, N. Modified Atkins ketogenic diet improves heart and skeletal muscle function in glycogen storage disease type III. Acta Myol. Myopathies Cardiomyopathies Off. J. Mediterr. Soc. Myol. 2019, 38, 17–20. [Google Scholar]
- Laforêt, P.; Inoue, M.; Goillot, E.; Lefeuvre, C.; Cagin, U.; Streichenberger, N.; Leonard-Louis, S.; Brochier, G.; Madelaine, A.; Labasse, C.; et al. Deep morphological analysis of muscle biopsies from type III glycogenesis (GSDIII), debranching enzyme deficiency, revealed stereotyped vacuolar myopathy and autophagy impairment. Acta Neuropathol. Commun. 2019, 7, 167. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Wei, H.; Zhang, M.; Hu, M.; Li, Z.; Zhang, C.; Luo, X.; Liang, Y. Genetic analysis and long-term treatment monitoring of 11 children with glycogen storage disease type IIIa. J. Pediatr. Endocrinol. Metab. JPEM 2020, 33, 923–930. [Google Scholar] [CrossRef]
- Olgac, A.; İnci, A.; Okur, İ.; Biberoğlu, G.; Oğuz, D.; Ezgü, F.S.; Kasapkara, Ç.S.; Aktaş, E.; Tümer, L. Beneficial Effects of Modified Atkins Diet in Glycogen Storage Disease Type IIIa. Ann. Nutr. Metab. 2020, 76, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Hoogeveen, I.J.; Bastek, V.B.; De Boer, F.; Montanari, C.; Meyer, U.; Maiorana, A.; Bordugo, A.; Dianin, A.; Campana, C.; et al. Dietary lipids in glycogen storage disease type III: A systematic literature study, case studies, and future recommendations. J. Inherit. Metab. Dis. 2020, 43, 770–777. [Google Scholar] [CrossRef]
- Focardi, M.; Bosco, A.; Bugelli, V.; Defraia, B.; Donati, M.A.; Pinchi, V. “On air” diagnosis of sudden cardiac death with dynamic Holter ECG in glycogen storage disease type III young female. Minerva Pediatr. 2020, 72, 142–144. [Google Scholar] [CrossRef]
- Marusic, T.; Zerjav Tansek, M.; Sirca Campa, A.; Mezek, A.; Berden, P.; Battelino, T.; Groselj, U. Data highlighting effects of Ketogenic diet on cardiomyopathy and hepatopathy in Glycogen storage disease Type IIIA. Data Brief 2020, 32, 106205. [Google Scholar] [CrossRef] [PubMed]
- Hijazi, G.; Paschall, A.; Young, S.P.; Smith, B.; Case, L.E.; Boggs, T.; Amarasekara, S.; Austin, S.L.; Pendyal, S.; El-Gharbawy, A.; et al. A retrospective longitudinal study and comprehensive review of adult patients with glycogen storage disease type III. Mol. Genet. Metab. Rep. 2021, 29, 100821. [Google Scholar] [CrossRef] [PubMed]
- Kumru Akin, B.; Ozturk Hismi, B.; Daly, A. Improvement in hypertrophic cardiomyopathy after using a high-fat, high-protein and low-carbohydrate diet in a non-adherent child with glycogen storage disease type IIIa. Mol. Genet. Metab. Rep. 2022, 32, 100904. [Google Scholar] [CrossRef]
- Moslemi, A.R.; Lindberg, C.; Nilsson, J.; Tajsharghi, H.; Andersson, B.; Oldfors, A. Glycogenin-1 deficiency and inactivated priming of glycogen synthesis. N. Engl. J. Med. 2010, 362, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Hedberg-Oldfors, C.; Glamuzina, E.; Ruygrok, P.; Anderson, L.J.; Elliott, P.; Watkinson, O.; Occleshaw, C.; Abernathy, M.; Turner, C.; Kingston, N.; et al. Cardiomyopathy as presenting sign of glycogenin-1 deficiency-report of three cases and review of the literature. J. Inherit. Metab. Dis. 2017, 40, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Hedberg-Oldfors, C.; De Ridder, W.; Kalev, O.; Böck, K.; Visuttijai, K.; Caravias, G.; Töpf, A.; Straub, V.; Baets, J.; Oldfors, A. Functional characterization of GYG1 variants in two patients with myopathy and glycogenin-1 deficiency. Neuromuscul. Disord. NMD 2019, 29, 951–960. [Google Scholar] [CrossRef]
- Visuttijai, K.; Hedberg-Oldfors, C.; Thomsen, C.; Glamuzina, E.; Kornblum, C.; Tasca, G.; Hernandez-Lain, A.; Sandstedt, J.; Dellgren, G.; Roach, P.; et al. Glycogenin is Dispensable for Glycogen Synthesis in Human Muscle, and Glycogenin Deficiency Causes Polyglucosan Storage. J. Clin. Endocrinol. Metab. 2020, 105, 557–566. [Google Scholar] [CrossRef]
- Malfatti, E.; Nilsson, J.; Hedberg-Oldfors, C.; Hernandez-Lain, A.; Michel, F.; Dominguez-Gonzalez, C.; Viennet, G.; Akman, H.O.; Kornblum, C.; Van den Bergh, P.; et al. A new muscle glycogen storage disease associated with glycogenin-1 deficiency. Ann. Neurol. 2014, 76, 891–898. [Google Scholar] [CrossRef]
- Kollberg, G.; Tulinius, M.; Gilljam, T.; Ostman-Smith, I.; Forsander, G.; Jotorp, P.; Oldfors, A.; Holme, E. Cardiomyopathy and exercise intolerance in muscle glycogen storage disease 0. N. Engl. J. Med. 2007, 357, 1507–1514. [Google Scholar] [CrossRef]
- Sukigara, S.; Liang, W.C.; Komaki, H.; Fukuda, T.; Miyamoto, T.; Saito, T.; Saito, Y.; Nakagawa, E.; Sugai, K.; Hayashi, Y.K.; et al. Muscle glycogen storage disease 0 presenting recurrent syncope with weakness and myalgia. Neuromuscul. Disord. NMD 2012, 22, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Cameron, J.M.; Levandovskiy, V.; MacKay, N.; Utgikar, R.; Ackerley, C.; Chiasson, D.; Halliday, W.; Raiman, J.; Robinson, B.H. Identification of a novel mutation in GYS1 (muscle-specific glycogen synthase) resulting in sudden cardiac death, that is diagnosable from skin fibroblasts. Mol. Genet. Metab. 2009, 98, 378–382. [Google Scholar] [CrossRef]
- Cui, L.; Zhao, L.P.; Ye, J.Y.; Yang, L.; Huang, Y.; Jiang, X.P.; Zhang, Q.; Jia, J.Z.; Zhang, D.X.; Huang, Y. The Lysosomal Membrane Protein Lamp2 Alleviates Lysosomal Cell Death by Promoting Autophagic Flux in Ischemic Cardiomyocytes. Front. Cell Dev. Biol. 2020, 8, 31. [Google Scholar] [CrossRef]
- Eskelinen, E.L.; Illert, A.L.; Tanaka, Y.; Schwarzmann, G.; Blanz, J.; Von Figura, K.; Saftig, P. Role of LAMP-2 in lysosome biogenesis and autophagy. Mol. Biol. Cell 2002, 13, 3355–3368. [Google Scholar] [CrossRef] [PubMed]
- Arad, M.; Maron, B.J.; Gorham, J.M.; Johnson, W.H., Jr.; Saul, J.P.; Perez-Atayde, A.R.; Spirito, P.; Wright, G.B.; Kanter, R.J.; Seidman, C.E.; et al. Glycogen storage diseases presenting as hypertrophic cardiomyopathy. N. Engl. J. Med. 2005, 352, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Nishino, I.; Fu, J.; Tanji, K.; Yamada, T.; Shimojo, S.; Koori, T.; Mora, M.; Riggs, J.E.; Oh, S.J.; Koga, Y.; et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 2000, 406, 906–910. [Google Scholar] [CrossRef] [PubMed]
- Sugie, K.; Yamamoto, A.; Murayama, K.; Oh, S.J.; Takahashi, M.; Mora, M.; Riggs, J.E.; Colomer, J.; Iturriaga, C.; Meloni, A.; et al. Clinicopathological features of genetically confirmed Danon disease. Neurology 2002, 58, 1773–1778. [Google Scholar] [CrossRef]
- Takahashi, M.; Yamamoto, A.; Takano, K.; Sudo, A.; Wada, T.; Goto, Y.; Nishino, I.; Saitoh, S. Germline mosaicism of a novel mutation in lysosome-associated membrane protein-2 deficiency (Danon disease). Ann. Neurol. 2002, 52, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Lacoste-Collin, L.; Garcia, V.; Uro-Coste, E.; Arné-Bes, M.C.; Durand, D.; Levade, T.; Delisle, M.B. Danon′s disease (X-linked vacuolar cardiomyopathy and myopathy): A case with a novel Lamp-2 gene mutation. Neuromuscul. Disord. NMD 2002, 12, 882–885. [Google Scholar] [CrossRef]
- Sugie, K.; Koori, T.; Yamamoto, A.; Ogawa, M.; Hirano, M.; Inoue, K.; Nonaka, I.; Nishino, I. Characterization of Danon disease in a male patient and his affected mother. Neuromuscul. Disord. NMD 2003, 13, 708–711. [Google Scholar] [CrossRef]
- Charron, P.; Villard, E.; Sébillon, P.; Laforêt, P.; Maisonobe, T.; Duboscq-Bidot, L.; Romero, N.; Drouin-Garraud, V.; Frébourg, T.; Richard, P.; et al. Danon′s disease as a cause of hypertrophic cardiomyopathy: A systematic survey. Heart (Br. Card. Soc.) 2004, 90, 842–846. [Google Scholar] [CrossRef]
- Balmer, C.; Ballhausen, D.; Bosshard, N.U.; Steinmann, B.; Boltshauser, E.; Bauersfeld, U.; Superti-Furga, A. Familial X-linked cardiomyopathy (Danon disease): Diagnostic confirmation by mutation analysis of the LAMP2gene. Eur. J. Pediatr. 2005, 164, 509–514. [Google Scholar] [CrossRef]
- Echaniz-Laguna, A.; Mohr, M.; Epailly, E.; Nishino, I.; Charron, P.; Richard, P.; Guiraud-Chaumeil, C.; Tranchant, C. Novel Lamp-2 gene mutation and successful treatment with heart transplantation in a large family with Danon disease. Muscle Nerve 2006, 33, 393–397. [Google Scholar] [CrossRef]
- Fanin, M.; Nascimbeni, A.C.; Fulizio, L.; Spinazzi, M.; Melacini, P.; Angelini, C. Generalized lysosome-associated membrane protein-2 defect explains multisystem clinical involvement and allows leukocyte diagnostic screening in Danon disease. Am. J. Pathol. 2006, 168, 1309–1320. [Google Scholar] [CrossRef]
- Sugimoto, S.; Shiomi, K.; Yamamoto, A.; Nishino, I.; Nonaka, I.; Ohi, T. LAMP-2 positive vacuolar myopathy with dilated cardiomyopathy. Intern. Med. 2007, 46, 757–760. [Google Scholar] [CrossRef][Green Version]
- Taylor, M.R.G.; Ku, L.; Slavov, D.; Cavanaugh, J.; Boucek, M.; Zhu, X.; Graw, S.; Carniel, E.; Barnes, C.; Quan, D.; et al. Danon disease presenting with dilated cardiomyopathy and a complex phenotype. J. Hum. Genet. 2007, 52, 830–835. [Google Scholar] [CrossRef]
- Nadeau, A.; Therrien, C.; Karpati, G.; Sinnreich, M. Danon disease due to a novel splice mutation in the LAMP2 gene. Muscle Nerve 2008, 37, 338–342. [Google Scholar] [CrossRef]
- Dougu, N.; Joho, S.; Shan, L.; Shida, T.; Matsuki, A.; Uese, K.; Hirono, K.; Ichida, F.; Tanaka, K.; Nishino, I.; et al. Novel LAMP-2 mutation in a family with Danon disease presenting with hypertrophic cardiomyopathy. Circ. J. Off. J. Jpn. Circ. Soc. 2009, 73, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Roberts, W.C.; Arad, M.; Haas, T.S.; Spirito, P.; Wright, G.B.; Almquist, A.K.; Baffa, J.M.; Saul, J.P.; Ho, C.Y.; et al. Clinical outcome and phenotypic expression in LAMP2 cardiomyopathy. Jama 2009, 301, 1253–1259. [Google Scholar] [CrossRef] [PubMed]
- Regelsberger, G.; Höftberger, R.; Pickl, W.F.; Zlabinger, G.J.; Körmöczi, U.; Salzer-Muhar, U.; Luckner, D.; Bodamer, O.A.; Mayr, J.A.; Muss, W.H.; et al. Danon disease: Case report and detection of new mutation. J. Inherit. Metab. Dis. 2009, 32 (Suppl. S1), S115–S122. [Google Scholar] [CrossRef]
- Toib, A.; Grange, D.K.; Kozel, B.A.; Ewald, G.A.; White, F.V.; Canter, C.E. Distinct clinical and histopathological presentations of Danon cardiomyopathy in young women. J. Am. Coll. Cardiol. 2010, 55, 408–410. [Google Scholar] [CrossRef]
- Miani, D.; Taylor, M.; Mestroni, L.; D′Aurizio, F.; Finato, N.; Fanin, M.; Brigido, S.; Proclemer, A. Sudden death associated with danon disease in women. Am. J. Cardiol. 2012, 109, 406–411. [Google Scholar] [CrossRef]
- Cheng, Z.; Cui, Q.; Tian, Z.; Xie, H.; Chen, L.; Fang, L.; Zhu, K.; Fang, Q. Danon disease as a cause of concentric left ventricular hypertrophy in patients who underwent endomyocardial biopsy. Eur. Heart J. 2012, 33, 649–656. [Google Scholar] [CrossRef]
- Majer, F.; Vlaskova, H.; Krol, L.; Kalina, T.; Kubanek, M.; Stolnaya, L.; Dvorakova, L.; Elleder, M.; Sikora, J. Danon disease: A focus on processing of the novel LAMP2 mutation and comments on the beneficial use of peripheral white blood cells in the diagnosis of LAMP2 deficiency. Gene 2012, 498, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Fidzianska, A.; Madej-Pilarczyk, A.; Walczak, E.; Kuch, M. Morphologic and clinical aspects of Danon disease in a patient with a mutation c.137G > A in the LAMP-2 gene. Neuropediatrics 2013, 44, 276–280. [Google Scholar] [CrossRef]
- Majer, F.; Pelak, O.; Kalina, T.; Vlaskova, H.; Dvorakova, L.; Honzik, T.; Palecek, T.; Kuchynka, P.; Masek, M.; Zeman, J.; et al. Mosaic tissue distribution of the tandem duplication of LAMP2 exons 4 and 5 demonstrates the limits of Danon disease cellular and molecular diagnostics. J. Inherit. Metab. Dis. 2014, 37, 117–124. [Google Scholar] [CrossRef]
- Kim, J.; Parikh, P.; Mahboob, M.; Arrighi, J.A.; Atalay, M.K.; Rowin, E.J.; Maron, M.S. Asymptomatic young man with Danon disease. Tex. Heart Inst. J. 2014, 41, 332–334. [Google Scholar] [CrossRef]
- Sugie, K.; Yoshizawa, H.; Onoue, K.; Nakanishi, Y.; Eura, N.; Ogawa, M.; Nakano, T.; Sakaguchi, Y.; Hayashi, Y.K.; Kishimoto, T.; et al. Early onset of cardiomyopathy and intellectual disability in a girl with Danon disease associated with a de novo novel mutation of the LAMP2 gene. Neuropathol. Off. J. Jpn. Soc. Neuropathol. 2016, 36, 561–565. [Google Scholar] [CrossRef]
- Ng, K.M.; Mok, P.Y.; Butler, A.W.; Ho, J.C.; Choi, S.W.; Lee, Y.K.; Lai, W.H.; Au, K.W.; Lau, Y.M.; Wong, L.Y.; et al. Amelioration of X-Linked Related Autophagy Failure in Danon Disease with DNA Methylation Inhibitor. Circulation 2016, 134, 1373–1389. [Google Scholar] [CrossRef]
- Marino, M.; Musumeci, O.; Paleologo, G.; Cucinotta, M.; Migliorato, A.; Rodolico, C.; Toscano, A. Ischemic stroke due to hypoperfusion in a patient with a previously unrecognized Danon disease. Neuromuscul. Disord. NMD 2016, 26, 890–894. [Google Scholar] [CrossRef] [PubMed]
- Kitahara, H.; Nawata, K.; Kinoshita, O.; Itoda, Y.; Shintani, Y.; Fukayama, M.; Ono, M. Implantation of a Left Ventricular Assist Device for Danon Cardiomyopathy. Ann. Thorac. Surg. 2017, 103, e39–e41. [Google Scholar] [CrossRef] [PubMed]
- Samad, F.; Jain, R.; Jan, M.F.; Sulemanjee, N.Z.; Menaria, P.; Kalvin, L.; Bush, M.; Jahangir, A.; Khandheria, B.K.; Tajik, A.J. Malignant cardiac phenotypic expression of Danon disease (LAMP2 cardiomyopathy). Int. J. Cardiol. 2017, 245, 201–206. [Google Scholar] [CrossRef]
- Nguyen, T.V.; Tran Vu, M.T.; Do, T.N.P.; Tran, T.H.N.; Do, T.H.; Nguyen, T.M.H.; Tran Huynh, B.N.; Le, L.A.; Nguyen Pham, N.T.; Nguyen, T.D.A.; et al. Genetic Determinants and Genotype-Phenotype Correlations in Vietnamese Patients with Dilated Cardiomyopathy. Circ. J. Off. J. Jpn. Circ. Soc. 2021, 85, 1469–1478. [Google Scholar] [CrossRef] [PubMed]
- Sugie, K.; Komaki, H.; Eura, N.; Shiota, T.; Onoue, K.; Tsukaguchi, H.; Minami, N.; Ogawa, M.; Kiriyama, T.; Kataoka, H.; et al. A Nationwide Survey on Danon Disease in Japan. Int. J. Mol. Sci. 2018, 19, 3507. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, X.; Wang, F.; Liang, Y.; Deng, H.; Liao, H.; Zhang, Q.; Zhang, B.; Zhan, X.; Fang, X.; et al. Prevalence and clinical characteristics of Danon disease among patients with left ventricular hypertrophy and concomitant electrocardiographic preexcitation. Mol. Genet. Genom. Med. 2019, 7, e638. [Google Scholar] [CrossRef]
- Di Nora, C.; Miani, D.; D′Elia, A.V.; Poli, S.; Iascone, M.; Nucifora, G.; Finato, N.; Sponga, S.; Proclemer, A.; Livi, U. Heart transplantation in Danon disease: Long term single centre experience and review of the literature. Eur. J. Med. Genet. 2020, 63, 103645. [Google Scholar] [CrossRef]
- Meinert, M.; Englund, E.; Hedberg-Oldfors, C.; Oldfors, A.; Kornhall, B.; Lundin, C.; Wittström, E. Danon disease presenting with early onset of hypertrophic cardiomyopathy and peripheral pigmentary retinal dystrophy in a female with a de novo novel mosaic mutation in the LAMP2 gene. Ophthalmic Genet. 2019, 40, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.; Noguchi, S.; Sugie, K.; Matsuo, Y.; Nguyen, C.T.H.; Koito, H.; Shiojima, I.; Nishino, I.; Tsukaguchi, H. Small-Vessel Vasculopathy Due to Aberrant Autophagy in LAMP-2 Deficiency. Sci. Rep. 2018, 8, 3326. [Google Scholar] [CrossRef]
- Miliou, A.; Antonopoulos, A.S.; Kouris, N.; Lazaros, G.; Tsioufis, K.; Vlachopoulos, C. Danon Cardiomyopathy: Specific Imaging Signs. JACC Case Rep. 2022, 4, 1496–1500. [Google Scholar] [CrossRef] [PubMed]
- Hashida, Y.; Wada, T.; Saito, T.; Ohta, K.; Kasahara, Y.; Yachie, A. Early diagnosis of Danon disease: Flow cytometric detection of lysosome-associated membrane protein-2-negative leukocytes. J. Cardiol. 2015, 66, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Gollob, M.H. Glycogen storage disease as a unifying mechanism of disease in the PRKAG2 cardiac syndrome. Biochem. Soc. Trans. 2003, 31, 228–231. [Google Scholar] [CrossRef]
- Blair, E.; Redwood, C.; Ashrafian, H.; Oliveira, M.; Broxholme, J.; Kerr, B.; Salmon, A.; Ostman-Smith, I.; Watkins, H. Mutations in the gamma(2) subunit of AMP-activated protein kinase cause familial hypertrophic cardiomyopathy: Evidence for the central role of energy compromise in disease pathogenesis. Hum. Mol. Genet. 2001, 10, 1215–1220. [Google Scholar] [CrossRef]
- Arad, M.; Benson, D.W.; Perez-Atayde, A.R.; McKenna, W.J.; Sparks, E.A.; Kanter, R.J.; McGarry, K.; Seidman, J.G.; Seidman, C.E. Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy. J. Clin. Investig. 2002, 109, 357–362. [Google Scholar] [CrossRef]
- Vaughan, C.J.; Hom, Y.; Okin, D.A.; McDermott, D.A.; Lerman, B.B.; Basson, C.T. Molecular genetic analysis of PRKAG2 in sporadic Wolff-Parkinson-White syndrome. J. Cardiovasc. Electrophysiol. 2003, 14, 263–268. [Google Scholar] [CrossRef]
- Murphy, R.T.; Mogensen, J.; McGarry, K.; Bahl, A.; Evans, A.; Osman, E.; Syrris, P.; Gorman, G.; Farrell, M.; Holton, J.L.; et al. Adenosine monophosphate-activated protein kinase disease mimicks hypertrophic cardiomyopathy and Wolff-Parkinson-White syndrome: Natural history. J. Am. Coll. Cardiol. 2005, 45, 922–930. [Google Scholar] [CrossRef]
- Burwinkel, B.; Scott, J.W.; Bührer, C.; Van Landeghem, F.K.; Cox, G.F.; Wilson, C.J.; Grahame Hardie, D.; Kilimann, M.W. Fatal congenital heart glycogenosis caused by a recurrent activating R531Q mutation in the gamma 2-subunit of AMP-activated protein kinase (PRKAG2), not by phosphorylase kinase deficiency. Am. J. Hum. Genet. 2005, 76, 1034–1049. [Google Scholar] [CrossRef]
- Laforêt, P.; Richard, P.; Said, M.A.; Romero, N.B.; Lacene, E.; Leroy, J.P.; Baussan, C.; Hogrel, J.Y.; Lavergne, T.; Wahbi, K.; et al. A new mutation in PRKAG2 gene causing hypertrophic cardiomyopathy with conduction system disease and muscular glycogenosis. Neuromuscul. Disord. NMD 2006, 16, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Akman, H.O.; Sampayo, J.N.; Ross, F.A.; Scott, J.W.; Wilson, G.; Benson, L.; Bruno, C.; Shanske, S.; Hardie, D.G.; Dimauro, S. Fatal infantile cardiac glycogenosis with phosphorylase kinase deficiency and a mutation in the gamma2-subunit of AMP-activated protein kinase. Pediatr. Res. 2007, 62, 499–504. [Google Scholar] [CrossRef]
- Tan, H.L.; Van der Wal, A.C.; Campian, M.E.; Kruyswijk, H.H.; Ten Hove Jansen, B.; Van Doorn, D.J.; Oskam, H.J.; Becker, A.E.; Wilde, A.A. Nodoventricular accessory pathways in PRKAG2-dependent familial preexcitation syndrome reveal a disorder in cardiac development. Circ. Arrhythmia Electrophysiol. 2008, 1, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.Q.; Lu, C.X.; Zhang, Y.; Yang, Y.K.; Li, J.C.; Lan, T.; Meng, X.; Fan, P.; Tian, T.; Wang, L.P.; et al. A novel PRKAG2 mutation in a Chinese family with cardiac hypertrophy and ventricular pre-excitation. Sci. Rep. 2017, 7, 2407. [Google Scholar] [CrossRef]
- Thevenon, J.; Laurent, G.; Ader, F.; Laforêt, P.; Klug, D.; Duva Pentiah, A.; Gouya, L.; Maurage, C.A.; Kacet, S.; Eicher, J.C.; et al. High prevalence of arrhythmic and myocardial complications in patients with cardiac glycogenosis due to PRKAG2 mutations. EP Eur. 2017, 19, 651–659. [Google Scholar] [CrossRef]
- Hu, J.; Tang, B.; Wang, J.; Huang, K.; Wang, Y.; Lu, S.; Gowreesunkur, H.B.; Wang, Y.; Wu, D.; Mayala, H.A.; et al. Familial Atrial Enlargement, Conduction Disorder and Symmetric Cardiac Hypertrophy Are Early Signs of PRKAG2 R302Q. Curr. Med. Sci. 2020, 40, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Beyzaei, Z.; Ezgu, F.; Geramizadeh, B.; Imanieh, M.H.; Haghighat, M.; Dehghani, S.M.; Honar, N.; Zahmatkeshan, M.; Jassbi, A.; Mahboubifar, M.; et al. Clinical and genetic spectrum of glycogen storage disease in Iranian population using targeted gene sequencing. Sci. Rep. 2021, 11, 7040. [Google Scholar] [CrossRef]
- Nilsson, J.; Schoser, B.; Laforet, P.; Kalev, O.; Lindberg, C.; Romero, N.B.; Dávila López, M.; Akman, H.O.; Wahbi, K.; Iglseder, S.; et al. Polyglucosan body myopathy caused by defective ubiquitin ligase RBCK1. Ann. Neurol. 2013, 74, 914–919. [Google Scholar] [CrossRef]
- Boisson, B.; Laplantine, E.; Prando, C.; Giliani, S.; Israelsson, E.; Xu, Z.; Abhyankar, A.; Israël, L.; Trevejo-Nunez, G.; Bogunovic, D.; et al. Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nat. Immunol. 2012, 13, 1178–1186. [Google Scholar] [CrossRef]
- Yamanaka, K.; Ishikawa, H.; Megumi, Y.; Tokunaga, F.; Kanie, M.; Rouault, T.A.; Morishima, I.; Minato, N.; Ishimori, K.; Iwai, K. Identification of the ubiquitin-protein ligase that recognizes oxidized IRP2. Nat. Cell Biol. 2003, 5, 336–340. [Google Scholar] [CrossRef]
- Wang, K.; Kim, C.; Bradfield, J.; Guo, Y.; Toskala, E.; Otieno, F.G.; Hou, C.; Thomas, K.; Cardinale, C.; Lyon, G.J.; et al. Whole-genome DNA/RNA sequencing identifies truncating mutations in RBCK1 in a novel Mendelian disease with neuromuscular and cardiac involvement. Genome Med. 2013, 5, 67. [Google Scholar] [CrossRef] [PubMed]
- Krenn, M.; Salzer, E.; Simonitsch-Klupp, I.; Rath, J.; Wagner, M.; Haack, T.B.; Strom, T.M.; Schänzer, A.; Kilimann, M.W.; Schmidt, R.L.J.; et al. Mutations outside the N-terminal part of RBCK1 may cause polyglucosan body myopathy with immunological dysfunction: Expanding the genotype-phenotype spectrum. J. Neurol. 2018, 265, 394–401. [Google Scholar] [CrossRef]
- Phadke, R.; Hedberg-Oldfors, C.; Scalco, R.S.; Lowe, D.M.; Ashworth, M.; Novelli, M.; Vara, R.; Merwick, A.; Amer, H.; Sofat, R.; et al. RBCK1-related disease: A rare multisystem disorder with polyglucosan storage, auto-inflammation, recurrent infections, skeletal, and cardiac myopathy-Four additional patients and a review of the current literature. J. Inherit. Metab. Dis. 2020, 43, 1002–1013. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, A.; Copland, I.B.; Massillon, D.; Annabi, B. Silencing of the human microsomal glucose-6-phosphate translocase induces glioma cell death: Potential new anticancer target for curcumin. FEBS Lett. 2006, 580, 3746–3752. [Google Scholar] [CrossRef]
- Ng, B.G.; Sosicka, P.; Fenaille, F.; Harroche, A.; Vuillaumier-Barrot, S.; Porterfield, M.; Xia, Z.J.; Wagner, S.; Bamshad, M.J.; Vergnes-Boiteux, M.C.; et al. A mutation in SLC37A4 causes a dominantly inherited congenital disorder of glycosylation characterized by liver dysfunction. Am. J. Hum. Genet. 2021, 108, 1040–1052. [Google Scholar] [CrossRef] [PubMed]
- Meimand, S.E.; Azizi, G.; Yazdani, R.; Sanadgol, N.; Rezaei, N. Novel mutation of SLC37A4 in a glycogen storage disease type Ib patient with neutropenia, horseshoe kidney, and arteriovenous malformation: A case report. Immunol. Res. 2022, 71, 107–111. [Google Scholar] [CrossRef]
- Lefeber, D.J.; Freeze, H.H.; Steet, R.; Kinoshita, T. Congenital Disorders of Glycosylation. In Essentials of Glycobiology, 4th ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Mohnen, D., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor: Long Island, NY, USA, 2022; pp. 599–614. [Google Scholar]
- Alsharhan, H.; Ng, B.G.; Daniel, E.J.P.; Friedman, J.; Pivnick, E.K.; Al-Hashem, A.; Faqeih, E.A.; Liu, P.; Engelhardt, N.M.; Keller, K.N.; et al. Expanding the phenotype, genotype and biochemical knowledge of ALG3-CDG. J. Inherit. Metab. Dis. 2021, 44, 987–1000. [Google Scholar] [CrossRef]
- Bian, Y.; Qiao, C.; Zheng, S.; Qiu, H.; Li, H.; Zhang, Z.; Yin, S.; Jiang, H.; Li-Ling, J.; Liu, C.; et al. ALG3-CDG: Lethal phenotype and novel variants in Chinese siblings. J. Hum. Genet. 2020, 65, 1129–1134. [Google Scholar] [CrossRef]
- Farolfi, M.; Cechova, A.; Ondruskova, N.; Zidkova, J.; Kousal, B.; Hansikova, H.; Honzik, T.; Liskova, P. ALG3-CDG: A patient with novel variants and review of the genetic and ophthalmic findings. BMC Ophthalmol. 2021, 21, 249. [Google Scholar] [CrossRef]
- Alsubhi, S.; Alhashem, A.; Faqeih, E.; Alfadhel, M.; Alfaifi, A.; Altuwaijri, W.; Alsahli, S.; Aldhalaan, H.; Alkuraya, F.S.; Hundallah, K.; et al. Congenital disorders of glycosylation: The Saudi experience. Am. J. Med. Genet. Part A 2017, 173, 2614–2621. [Google Scholar] [CrossRef] [PubMed]
- Himmelreich, N.; Dimitrov, B.; Geiger, V.; Zielonka, M.; Hutter, A.M.; Beedgen, L.; Hüllen, A.; Breuer, M.; Peters, V.; Thiemann, K.C.; et al. Novel variants and clinical symptoms in four new ALG3-CDG patients, review of the literature, and identification of AAGRP-ALG3 as a novel ALG3 variant with alanine and glycine-rich N-terminus. Hum. Mutat. 2019, 40, 938–951. [Google Scholar] [CrossRef]
- Al-Owain, M.; Mohamed, S.; Kaya, N.; Zagal, A.; Matthijs, G.; Jaeken, J. A novel mutation and first report of dilated cardiomyopathy in ALG6-CDG (CDG-Ic): A case report. Orphanet J. Rare Dis. 2010, 5, 7. [Google Scholar] [CrossRef]
- Himmelreich, N.; Dimitrov, B.; Zielonka, M.; Hüllen, A.; Hoffmann, G.F.; Juenger, H.; Müller, H.; Lorenz, I.; Busse, B.; Marschall, C.; et al. Missense variant c.1460 T > C (p.L487P) enhances protein degradation of ER mannosyltransferase ALG9 in two new ALG9-CDG patients presenting with West syndrome and review of the literature. Mol. Genet. Metab. 2022, 136, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, M.; Schollen, E.; Matthijs, G.; Neupert, C.; Hennet, T.; Grubenmann, C.E.; Frank, C.G.; Aebi, M.; Clarke, J.T.; Griffiths, A.; et al. CDG-IL: An infant with a novel mutation in the ALG9 gene and additional phenotypic features. Am. J. Med. Genet. Part A 2005, 136, 194–197. [Google Scholar] [CrossRef]
- Vleugels, W.; Keldermans, L.; Jaeken, J.; Butters, T.D.; Michalski, J.C.; Matthijs, G.; Foulquier, F. Quality control of glycoproteins bearing truncated glycans in an ALG9-defective (CDG-IL) patient. Glycobiology 2009, 19, 910–917. [Google Scholar] [CrossRef]
- Tham, E.; Eklund, E.A.; Hammarsjö, A.; Bengtson, P.; Geiberger, S.; Lagerstedt-Robinson, K.; Malmgren, H.; Nilsson, D.; Grigelionis, G.; Conner, P.; et al. A novel phenotype in N-glycosylation disorders: Gillessen-Kaesbach–Nishimura skeletal dysplasia due to pathogenic variants in ALG9. Eur. J. Hum. Genet. 2016, 24, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Davis, K.; Webster, D.; Smith, C.; Jackson, S.; Sinasac, D.; Seargeant, L.; Wei, X.C.; Ferreira, P.; Midgley, J.; Foster, Y.; et al. ALG9-CDG: New clinical case and review of the literature. Mol. Genet. Metab. Rep. 2017, 13, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Kranz, C.; Basinger, A.A.; Güçsavaş-Calikoğlu, M.; Sun, L.; Powell, C.M.; Henderson, F.W.; Aylsworth, A.S.; Freeze, H.H. Expanding spectrum of congenital disorder of glycosylation Ig (CDG-Ig): Sibs with a unique skeletal dysplasia, hypogammaglobulinemia, cardiomyopathy, genital malformations, and early lethality. Am. J. Med. Genet. Part A 2007, 143, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- De la Morena-Barrio, M.E.; Sabater, M.; De la Morena-Barrio, B.; Ruhaak, R.L.; Miñano, A.; Padilla, J.; Toderici, M.; Roldán, V.; Gimeno, J.R.; Vicente, V.; et al. ALG12-CDG: An unusual patient without intellectual disability and facial dysmorphism, and with a novel variant. Mol. Genet. Genom. Med. 2020, 8, e1304. [Google Scholar] [CrossRef]
- Tahata, S.; Gunderson, L.; Lanpher, B.; Morava, E. Complex phenotypes in ALG12-congenital disorder of glycosylation (ALG12-CDG): Case series and review of the literature. Mol. Genet. Metab. 2019, 128, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Kristiansson, B.; Stibler, H.; Conradi, N.; Eriksson, B.O.; Ryd, W. The heart and pericardial effusions in CDGS-I (carbohydrate-deficient glycoprotein syndrome type I). J. Inherit. Metab. Dis. 1998, 21, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Ning, B.; Elbein, A.D. Cloning, expression and characterization of the pig liver GDP-mannose pyrophosphorylase. Evidence that GDP-mannose and GDP-Glc pyrophosphorylases are different proteins. Eur. J. Biochem. 2000, 267, 6866–6874. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Serrano, M.; Ghaoui, R.; Ravenscroft, G.; Johnsen, R.D.; Davis, M.R.; Corbett, A.; Reddel, S.; Sue, C.M.; Liang, C.; Waddell, L.B.; et al. Expanding the phenotype of GMPPB mutations. Brain J. Neurol. 2015, 138, 836–844. [Google Scholar] [CrossRef]
- Carss, K.J.; Stevens, E.; Foley, A.R.; Cirak, S.; Riemersma, M.; Torelli, S.; Hoischen, A.; Willer, T.; Van Scherpenzeel, M.; Moore, S.A.; et al. Mutations in GDP-mannose pyrophosphorylase B cause congenital and limb-girdle muscular dystrophies associated with hypoglycosylation of α-dystroglycan. Am. J. Hum. Genet. 2013, 93, 29–41. [Google Scholar] [CrossRef]
- Oestergaard, S.T.; Stojkovic, T.; Dahlqvist, J.R.; Bouchet-Seraphin, C.; Nectoux, J.; Leturcq, F.; Cossée, M.; Solé, G.; Thomsen, C.; Krag, T.O.; et al. Muscle involvement in limb-girdle muscular dystrophy with GMPPB deficiency (LGMD2T). Neurol. Genet. 2016, 2, e112. [Google Scholar] [CrossRef] [PubMed]
- Willems, A.P.; Van Engelen, B.G.; Lefeber, D.J. Genetic defects in the hexosamine and sialic acid biosynthesis pathway. Biochim. Biophys. Acta 2016, 1860, 1640–1654. [Google Scholar] [CrossRef]
- Wen, X.Y.; Tarailo-Graovac, M.; Brand-Arzamendi, K.; Willems, A.; Rakic, B.; Huijben, K.; Da Silva, A.; Pan, X.; El-Rass, S.; Ng, R.; et al. Sialic acid catabolism by N-acetylneuraminate pyruvate lyase is essential for muscle function. JCI Insight 2018, 3, e122373. [Google Scholar] [CrossRef]
- Jin, G.Z.; Zhang, Y.; Cong, W.M.; Wu, X.; Wang, X.; Wu, S.; Wang, S.; Zhou, W.; Yuan, S.; Gao, H.; et al. Phosphoglucomutase 1 inhibits hepatocellular carcinoma progression by regulating glucose trafficking. PLoS Biol. 2018, 16, e2006483. [Google Scholar] [CrossRef]
- Conte, F.; van Buuringen, N.; Voermans, N.C.; Lefeber, D.J. Galactose in human metabolism, glycosylation and congenital metabolic diseases: Time for a closer look. Biochim. Biophys. Acta. Gen. Subj. 2021, 1865, 129898. [Google Scholar] [CrossRef]
- Conte, F.; Morava, E.; Bakar, N.A.; Wortmann, S.B.; Poerink, A.J.; Grunewald, S.; Crushell, E.; Al-Gazali, L.; de Vries, M.C.; Mørkrid, L.; et al. Phosphoglucomutase-1 deficiency: Early presentation, metabolic management and detection in neonatal blood spots. Mol. Genet. Metab. 2020, 131, 135–146. [Google Scholar] [CrossRef]
- Altassan, R.; Péanne, R.; Jaeken, J.; Barone, R.; Bidet, M.; Borgel, D.; Brasil, S.; Cassiman, D.; Cechova, A.; Coman, D.; et al. International clinical guidelines for the management of phosphomannomutase 2-congenital disorders of glycosylation: Diagnosis, treatment and follow up. J. Inherit. Metab. Dis. 2019, 42, 5–28. [Google Scholar] [CrossRef] [PubMed]
- Donoghue, S.E.; White, S.M.; Tan, T.Y.; Kowalski, R.; Morava, E.; Yaplito-Lee, J. Galactose treatment of a PGM1 patient presenting with restrictive cardiomyopathy. JIMD Rep. 2021, 57, 29–37. [Google Scholar] [CrossRef]
- Lipiński, P.; Bogdańska, A.; Tylki-Szymańska, A. Congenital disorders of glycosylation: Prevalence, incidence and mutational spectrum in the Polish population. Mol. Genet. Metab. Rep. 2021, 27, 100726. [Google Scholar] [CrossRef] [PubMed]
- Lipiński, P.; Cielecka-Kuszyk, J.; Czarnowska, E.; Bogdańska, A.; Socha, P.; Tylki-Szymańska, A. Congenital disorders of glycosylation in children–Histopathological and ultrastructural changes in the liver. Pediatr. Neonatol. 2021, 62, 278–283. [Google Scholar] [CrossRef]
- Timal, S.; Hoischen, A.; Lehle, L.; Adamowicz, M.; Huijben, K.; Sykut-Cegielska, J.; Paprocka, J.; Jamroz, E.; van Spronsen, F.J.; Körner, C.; et al. Gene identification in the congenital disorders of glycosylation type I by whole-exome sequencing. Hum. Mol. Genet. 2012, 21, 4151–4161. [Google Scholar] [CrossRef]
- Tegtmeyer, L.C.; Rust, S.; van Scherpenzeel, M.; Ng, B.G.; Losfeld, M.E.; Timal, S.; Raymond, K.; He, P.; Ichikawa, M.; Veltman, J.; et al. Multiple phenotypes in phosphoglucomutase 1 deficiency. N. Engl. J. Med. 2014, 370, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Küçükçongar, A.; Tümer, L.; Ezgü, F.S.; Kasapkara, Ç.S.; Jaeken, J.; Matthijs, G.; Rymen, D.; Dalgiç, B.; Bıdecı, A.; Hasanoğlu, A. A case with rare type of congenital disorder of glycosylation: PGM1-CDG. Genet. Couns. 2015, 26, 87–90. [Google Scholar]
- Loewenthal, N.; Haim, A.; Parvari, R.; Hershkovitz, E. Phosphoglucomutase-1 deficiency: Intrafamilial clinical variability and common secondary adrenal insufficiency. Am. J. Med. Genet. Part A 2015, 167, 3139–3143. [Google Scholar] [CrossRef]
- Wong, S.Y.; Beamer, L.J.; Gadomski, T.; Honzik, T.; Mohamed, M.; Wortmann, S.B.; Brocke Holmefjord, K.S.; Mork, M.; Bowling, F.; Sykut-Cegielska, J.; et al. Defining the Phenotype and Assessing Severity in Phosphoglucomutase-1 Deficiency. J. Pediatr. 2016, 175, 130–136.e138. [Google Scholar] [CrossRef]
- Schrapers, E.; Tegtmeyer, L.C.; Simic-Schleicher, G.; Debus, V.; Reunert, J.; Balbach, S.; Klingel, K.; Du Chesne, I.; Seelhöfer, A.; Fobker, M.; et al. News on Clinical Details and Treatment in PGM1-CDG. JIMD Rep. 2016, 26, 77–84. [Google Scholar] [CrossRef]
- Zeevaert, R.; Scalais, E.; Muino Mosquera, L.; De Meirleir, L.; De Beaufort, C.; Witsch, M.; Jaeken, J.; De Schepper, J. PGM1 deficiency diagnosed during an endocrine work-up of low IGF-1 mediated growth failure. Acta Clin. Belg. 2016, 71, 435–437. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.Y.; Gadomski, T.; van Scherpenzeel, M.; Honzik, T.; Hansikova, H.; Holmefjord, K.S.B.; Mork, M.; Bowling, F.; Sykut-Cegielska, J.; Koch, D.; et al. Oral D-galactose supplementation in PGM1-CDG. Genet. Med. Off. J. Am. Coll. Med. Genet. 2017, 19, 1226–1235. [Google Scholar] [CrossRef] [PubMed]
- Voermans, N.C.; Preisler, N.; Madsen, K.L.; Janssen, M.C.; Kusters, B.; Abu Bakar, N.; Conte, F.; Lamberti, V.M.; Nusman, F.; van Engelen, B.G.; et al. PGM1 deficiency: Substrate use during exercise and effect of treatment with galactose. Neuromuscul. Disord. NMD 2017, 27, 370–376. [Google Scholar] [CrossRef]
- Nolting, K.; Park, J.H.; Tegtmeyer, L.C.; Zühlsdorf, A.; Grüneberg, M.; Rust, S.; Reunert, J.; Du Chesne, I.; Debus, V.; Schulze-Bahr, E.; et al. Limitations of galactose therapy in phosphoglucomutase 1 deficiency. Mol. Genet. Metab. Rep. 2017, 13, 33–40. [Google Scholar] [CrossRef]
- Ding, Y.; Li, N.; Chang, G.; Li, J.; Yao, R.; Shen, Y.; Wang, J.; Huang, X.; Wang, X. Clinical and molecular genetic characterization of two patients with mutations in the phosphoglucomutase 1 (PGM1) gene. J. Pediatr. Endocrinol. Metab. JPEM 2018, 31, 781–788. [Google Scholar] [CrossRef]
- Tian, W.T.; Luan, X.H.; Zhou, H.Y.; Zhang, C.; Huang, X.J.; Liu, X.L.; Chen, S.D.; Tang, H.D.; Cao, L. Congenital disorder of glycosylation type 1T with a novel truncated homozygous mutation in PGM1 gene and literature review. Neuromuscul. Disord. NMD 2019, 29, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Fernlund, E.; Andersson, O.; Ellegård, R.; Årstrand, H.K.; Green, H.; Olsson, H.; Gunnarsson, C. The congenital disorder of glycosylation in PGM1 (PGM1-CDG) can cause severe cardiomyopathy and unexpected sudden cardiac death in childhood. Forensic Sci. Int. Genet. 2019, 43, 102111. [Google Scholar] [CrossRef]
- Perales-Clemente, E.; Liedtke, K.; Studinski, A.; Radenkovic, S.; Gavrilov, D.; Oglesbee, D.; Matern, D.; Rinaldo, P.; Tortorelli, S.; Morava, E.; et al. A new D-galactose treatment monitoring index for PGM1-CDG. J. Inherit. Metab. Dis. 2021, 44, 1263–1271. [Google Scholar] [CrossRef]
- Arimura, T.; Inagaki, N.; Hayashi, T.; Shichi, D.; Sato, A.; Hinohara, K.; Vatta, M.; Towbin, J.A.; Chikamori, T.; Yamashina, A.; et al. Impaired binding of ZASP/Cypher with phosphoglucomutase 1 is associated with dilated cardiomyopathy. Cardiovasc. Res. 2009, 83, 80–88. [Google Scholar] [CrossRef]
- Feldman, B.J.; Rosenthal, D. Carbohydrate-deficient glycoprotein syndrome-associated pericardial effusion treated with corticosteroids and salicylic acid. Pediatr. Cardiol. 2002, 23, 469–471. [Google Scholar] [CrossRef]
- Marquardt, T.; Hülskamp, G.; Gehrmann, J.; Debus, V.; Harms, E.; Kehl, H.G. Severe transient myocardial ischaemia caused by hypertrophic cardiomyopathy in a patient with congenital disorder of glycosylation type Ia. Eur. J. Pediatr. 2002, 161, 524–527. [Google Scholar] [CrossRef]
- Gehrmann, J.; Sohlbach, K.; Linnebank, M.; Böhles, H.J.; Buderus, S.; Kehl, H.G.; Vogt, J.; Harms, E.; Marquardt, T. Cardiomyopathy in congenital disorders of glycosylation. Cardiol. Young 2003, 13, 345–351. [Google Scholar] [CrossRef]
- Kusa, J.; Pyrkosz, A.; Skiba, A.; Szkutnik, M. Cardiac manifestations of carbohydrate-deficient glycoprotein syndrome. Pediatr. Cardiol. 2003, 24, 493–494. [Google Scholar] [CrossRef] [PubMed]
- Damen, G.; de Klerk, H.; Huijmans, J.; den Hollander, J.; Sinaasappel, M. Gastrointestinal and other clinical manifestations in 17 children with congenital disorders of glycosylation type Ia, Ib, and Ic. J. Pediatr. Gastroenterol. Nutr. 2004, 38, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Aronica, E.; van Kempen, A.A.; van der Heide, M.; Poll-The, B.T.; van Slooten, H.J.; Troost, D.; Rozemuller-Kwakkel, J.M. Congenital disorder of glycosylation type Ia: A clinicopathological report of a newborn infant with cerebellar pathology. Acta Neuropathol. 2005, 109, 433–442. [Google Scholar] [CrossRef]
- Noelle, V.; Knuepfer, M.; Pulzer, F.; Schuster, V.; Siekmeyer, W.; Matthijs, G.; Vogtmann, C. Unusual presentation of congenital disorder of glycosylation type 1a: Congenital persistent thrombocytopenia, hypertrophic cardiomyopathy and hydrops-like aspect due to marked peripheral oedema. Eur. J. Pediatr. 2005, 164, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Van de Kamp, J.M.; Lefeber, D.J.; Ruijter, G.J.; Steggerda, S.J.; den Hollander, N.S.; Willems, S.M.; Matthijs, G.; Poorthuis, B.J.; Wevers, R.A. Congenital disorder of glycosylation type Ia presenting with hydrops fetalis. J. Med. Genet. 2007, 44, 277–280. [Google Scholar] [CrossRef]
- Vermeer, S.; Kremer, H.P.; Leijten, Q.H.; Scheffer, H.; Matthijs, G.; Wevers, R.A.; Knoers, N.A.; Morava, E.; Lefeber, D.J. Cerebellar ataxia and congenital disorder of glycosylation Ia (CDG-Ia) with normal routine CDG screening. J. Neurol. 2007, 254, 1356–1358. [Google Scholar] [CrossRef]
- Schollen, E.; Keldermans, L.; Foulquier, F.; Briones, P.; Chabas, A.; Sánchez-Valverde, F.; Adamowicz, M.; Pronicka, E.; Wevers, R.; Matthijs, G. Characterization of two unusual truncating PMM2 mutations in two CDG-Ia patients. Mol. Genet. Metab. 2007, 90, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Truin, G.; Guillard, M.; Lefeber, D.J.; Sykut-Cegielska, J.; Adamowicz, M.; Hoppenreijs, E.; Sengers, R.C.A.; Wevers, R.A.; Morava, E. Pericardial and abdominal fluid accumulation in congenital disorder of glycosylation type Ia. Mol. Genet. Metab. 2008, 94, 481–484. [Google Scholar] [CrossRef]
- Coman, D.; Bostock, D.; Hunter, M.; Kannu, P.; Irving, M.; Mayne, V.; Fietz, M.; Jaeken, J.; Savarirayan, R. Primary skeletal dysplasia as a major manifesting feature in an infant with congenital disorder of glycosylation type Ia. Am. J. Med. Genet. Part A 2008, 146, 389–392. [Google Scholar] [CrossRef] [PubMed]
- Romano, S.; Bajolle, F.; Valayannopoulos, V.; Lyonnet, S.; Colomb, V.; de Baracé, C.; Vouhe, P.; Pouard, P.; Vuillaumier-Barrot, S.; Dupré, T.; et al. Conotruncal heart defects in three patients with congenital disorder of glycosylation type Ia (CDG Ia). J. Med. Genet. 2009, 46, 287–288. [Google Scholar] [CrossRef] [PubMed]
- Thong, M.K.; Fietz, M.; Nicholls, C.; Lee, M.H.; Asma, O. Congenital disorder of glycosylation type Ia in a Malaysian family: Clinical outcome and description of a novel PMM2 mutation. J. Inherit. Metab. Dis. 2009, 32 (Suppl. S1), S41–S44. [Google Scholar] [CrossRef]
- Verstegen, R.H.; Theodore, M.; van de Klerk, H.; Morava, E. Lymphatic edema in congenital disorders of glycosylation. JIMD Rep. 2012, 4, 113–116. [Google Scholar] [CrossRef]
- Rudaks, L.I.; Andersen, C.; Khong, T.Y.; Kelly, A.; Fietz, M.; Barnett, C.P. Hypertrophic cardiomyopathy with cardiac rupture and tamponade caused by congenital disorder of glycosylation type Ia. Pediatr. Cardiol. 2012, 33, 827–830. [Google Scholar] [CrossRef]
- Resende, C.; Carvalho, C.; Alegria, A.; Oliveira, D.; Quelhas, D.; Bandeira, A.; Proença, E. Congenital disorders of glycosylation with neonatal presentation. BMJ Case Rep. 2014, 2014, bcr2013010037. [Google Scholar] [CrossRef]
- Serrano, M.; de Diego, V.; Muchart, J.; Cuadras, D.; Felipe, A.; Macaya, A.; Velázquez, R.; Poo, M.P.; Fons, C.; O′Callaghan, M.M.; et al. Phosphomannomutase deficiency (PMM2-CDG): Ataxia and cerebellar assessment. Orphanet J. Rare Dis. 2015, 10, 138. [Google Scholar] [CrossRef]
- Al Teneiji, A.; Bruun, T.U.; Sidky, S.; Cordeiro, D.; Cohn, R.D.; Mendoza-Londono, R.; Moharir, M.; Raiman, J.; Siriwardena, K.; Kyriakopoulou, L.; et al. Phenotypic and genotypic spectrum of congenital disorders of glycosylation type I and type II. Mol. Genet. Metab. 2017, 120, 235–242. [Google Scholar] [CrossRef]
- Schiff, M.; Roda, C.; Monin, M.L.; Arion, A.; Barth, M.; Bednarek, N.; Bidet, M.; Bloch, C.; Boddaert, N.; Borgel, D.; et al. Clinical, laboratory and molecular findings and long-term follow-up data in 96 French patients with PMM2-CDG (phosphomannomutase 2-congenital disorder of glycosylation) and review of the literature. J. Med. Genet. 2017, 54, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.H.; Li, D.F.; Tang, W.T.; Qiu, K.Y.; Li, Y.; Liao, X.Y.; Tang, D.X.; Qin, L.J.; Deng, B.Q.; Luo, X.Y. Atrial septal defect in a patient with congenital disorder of glycosylation type 1a: A case report. J. Med. Case Rep. 2018, 12, 17. [Google Scholar] [CrossRef]
- Qian, Z.; Van den Eynde, J.; Heymans, S.; Mertens, L.; Morava, E. Vascular ring anomaly in a patient with phosphomannomutase 2 deficiency: A case report and review of the literature. JIMD Rep. 2020, 56, 27–33. [Google Scholar] [CrossRef]
- Görlacher, M.; Panagiotou, E.; Himmelreich, N.; Hüllen, A.; Beedgen, L.; Dimitrov, B.; Geiger, V.; Zielonka, M.; Peters, V.; Strahl, S.; et al. Fatal outcome after heart surgery in PMM2-CDG due to a rare homozygous gene variant with double effects. Mol. Genet. Metab. Rep. 2020, 25, 100673. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, A.; Pateman, A.; Chalmers, R.; Coman, D.; Menahem, S. Prenatal cardiac ultrasound finding in congenital disorder of glycosylation type 1a. Fetal Diagn. Ther. 2009, 25, 54–57. [Google Scholar] [CrossRef] [PubMed]
- Footitt, E.J.; Karimova, A.; Burch, M.; Yayeh, T.; Dupré, T.; Vuillaumier-Barrot, S.; Chantret, I.; Moore, S.E.; Seta, N.; Grunewald, S. Cardiomyopathy in the congenital disorders of glycosylation (CDG): A case of late presentation and literature review. J. Inherit. Metab. Dis. 2009, 32 (Suppl. S1), S313–S319. [Google Scholar] [CrossRef]
- Bogdańska, A.; Lipiński, P.; Szymańska-Rożek, P.; Jezela-Stanek, A.; Rokicki, D.; Socha, P.; Tylki-Szymańska, A. Clinical, biochemical and molecular phenotype of congenital disorders of glycosylation: Long-term follow-up. Orphanet J. Rare Dis. 2021, 16, 17. [Google Scholar] [CrossRef]
- Vasudevan, D.; Takeuchi, H.; Johar, S.S.; Majerus, E.; Haltiwanger, R.S. Peters plus syndrome mutations disrupt a noncanonical ER quality-control mechanism. Curr. Biol. CB 2015, 25, 286–295. [Google Scholar] [CrossRef]
- Lesnik Oberstein, S.A.; Kriek, M.; White, S.J.; Kalf, M.E.; Szuhai, K.; den Dunnen, J.T.; Breuning, M.H.; Hennekam, R.C. Peters Plus syndrome is caused by mutations in B3GALTL, a putative glycosyltransferase. Am. J. Hum. Genet. 2006, 79, 562–566. [Google Scholar] [CrossRef]
- Reis, L.M.; Tyler, R.C.; Abdul-Rahman, O.; Trapane, P.; Wallerstein, R.; Broome, D.; Hoffman, J.; Khan, A.; Paradiso, C.; Ron, N.; et al. Mutation analysis of B3GALTL in Peters Plus syndrome. Am. J. Med. Genet. Part A 2008, 146, 2603–2610. [Google Scholar] [CrossRef] [PubMed]
- Haldeman-Englert, C.R.; Naeem, T.; Geiger, E.A.; Warnock, A.; Feret, H.; Ciano, M.; Davidson, S.L.; Deardorff, M.A.; Zackai, E.H.; Shaikh, T.H. A 781-kb deletion of 13q12.3 in a patient with Peters plus syndrome. Am. J. Med. Genet. Part A 2009, 149, 1842–1845. [Google Scholar] [CrossRef]
- Dassie-Ajdid, J.; Causse, A.; Poidvin, A.; Granier, M.; Kaplan, J.; Burglen, L.; Doummar, D.; Teisseire, P.; Vigouroux, A.; Malecaze, F.; et al. Novel B3GALTL mutation in Peters-plus Syndrome. Clin. Genet. 2009, 76, 490–492. [Google Scholar] [CrossRef]
- Shimizu, R.; Saito, R.; Hoshino, K.; Ogawa, K.; Negishi, T.; Nishimura, J.; Mitsui, N.; Osawa, M.; Ohashi, H. Severe Peters Plus syndrome-like phenotype with anterior eye staphyloma and hypoplastic left heart syndrome: Proposal of a new syndrome. Congenit. Anom. 2010, 50, 197–199. [Google Scholar] [CrossRef]
- Faletra, F.; Athanasakis, E.; Minen, F.; Fornasier, F.; Marchetti, F.; Gasparini, P. Vertebral defects in patients with Peters plus syndrome and mutations in B3GALTL. Ophthalmic Genet. 2011, 32, 256–258. [Google Scholar] [CrossRef]
- Weh, E.; Reis, L.M.; Tyler, R.C.; Bick, D.; Rhead, W.J.; Wallace, S.; McGregor, T.L.; Dills, S.K.; Chao, M.C.; Murray, J.C.; et al. Novel B3GALTL mutations in classic Peters plus syndrome and lack of mutations in a large cohort of patients with similar phenotypes. Clin. Genet. 2014, 86, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, C.; Zhang, H.; Feng, W.; Wang, Q.; Fan, R. Severe phenotypes of B3GAT3-related disorder caused by two heterozygous variants: A case report and literature review. BMC Med. Genom. 2022, 15, 27. [Google Scholar] [CrossRef] [PubMed]
- Baasanjav, S.; Al-Gazali, L.; Hashiguchi, T.; Mizumoto, S.; Fischer, B.; Horn, D.; Seelow, D.; Ali, B.R.; Aziz, S.A.; Langer, R.; et al. Faulty initiation of proteoglycan synthesis causes cardiac and joint defects. Am. J. Hum. Genet. 2011, 89, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Von Oettingen, J.E.; Tan, W.H.; Dauber, A. Skeletal dysplasia, global developmental delay, and multiple congenital anomalies in a 5-year-old boy-report of the second family with B3GAT3 mutation and expansion of the phenotype. Am. J. Med. Genet. Part A 2014, 164, 1580–1586. [Google Scholar] [CrossRef]
- Jones, K.L.; Schwarze, U.; Adam, M.P.; Byers, P.H.; Mefford, H.C. A homozygous B3GAT3 mutation causes a severe syndrome with multiple fractures, expanding the phenotype of linkeropathy syndromes. Am. J. Med. Genet. Part A 2015, 167, 2691–2696. [Google Scholar] [CrossRef]
- Job, F.; Mizumoto, S.; Smith, L.; Couser, N.; Brazil, A.; Saal, H.; Patterson, M.; Gibson, M.I.; Soden, S.; Miller, N.; et al. Functional validation of novel compound heterozygous variants in B3GAT3 resulting in severe osteopenia and fractures: Expanding the disease phenotype. BMC Med. Genet. 2016, 17, 86. [Google Scholar] [CrossRef]
- Bloor, S.; Giri, D.; Didi, M.; Senniappan, S. Novel Splicing Mutation in B3GAT3 Associated with Short Stature, GH Deficiency, Hypoglycaemia, Developmental Delay, and Multiple Congenital Anomalies. Case Rep. Genet. 2017, 2017, 3941483. [Google Scholar] [CrossRef] [PubMed]
- Yauy, K.; Tran Mau-Them, F.; Willems, M.; Coubes, C.; Blanchet, P.; Herlin, C.; Taleb Arrada, I.; Sanchez, E.; Faure, J.M.; Le Gac, M.P.; et al. B3GAT3-related disorder with craniosynostosis and bone fragility due to a unique mutation. Genet. Med. Off. J. Am. Coll. Med. Genet. 2018, 20, 269–274. [Google Scholar] [CrossRef]
- Ritelli, M.; Cinquina, V.; Giacopuzzi, E.; Venturini, M.; Chiarelli, N.; Colombi, M. Further Defining the Phenotypic Spectrum of B3GAT3 Mutations and Literature Review on Linkeropathy Syndromes. Genes 2019, 10, 631. [Google Scholar] [CrossRef]
- Ranza, E.; Huber, C.; Levin, N.; Baujat, G.; Bole-Feysot, C.; Nitschke, P.; Masson, C.; Alanay, Y.; Al-Gazali, L.; Bitoun, P.; et al. Chondrodysplasia with multiple dislocations: Comprehensive study of a series of 30 cases. Clin. Genet. 2017, 91, 868–880. [Google Scholar] [CrossRef]
- Ortiz-Cordero, C.; Azzag, K.; Perlingeiro, R.C.R. Fukutin-Related Protein: From Pathology to Treatments. Trends Cell Biol. 2021, 31, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Van Reeuwijk, J.; Olderode-Berends, M.J.; Van den Elzen, C.; Brouwer, O.F.; Roscioli, T.; Van Pampus, M.G.; Scheffer, H.; Brunner, H.G.; Van Bokhoven, H.; Hol, F.A. A homozygous FKRP start codon mutation is associated with Walker-Warburg syndrome, the severe end of the clinical spectrum. Clin. Genet. 2010, 78, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Poppe, M.; Cree, L.; Bourke, J.; Eagle, M.; Anderson, L.V.; Birchall, D.; Brockington, M.; Buddles, M.; Busby, M.; Muntoni, F.; et al. The phenotype of limb-girdle muscular dystrophy type 2I. Neurology 2003, 60, 1246–1251. [Google Scholar] [CrossRef]
- Mercuri, E.; Brockington, M.; Straub, V.; Quijano-Roy, S.; Yuva, Y.; Herrmann, R.; Brown, S.C.; Torelli, S.; Dubowitz, V.; Blake, D.J.; et al. Phenotypic spectrum associated with mutations in the fukutin-related protein gene. Ann. Neurol. 2003, 53, 537–542. [Google Scholar] [CrossRef]
- Poppe, M.; Bourke, J.; Eagle, M.; Frosk, P.; Wrogemann, K.; Greenberg, C.; Muntoni, F.; Voit, T.; Straub, V.; Hilton-Jones, D.; et al. Cardiac and respiratory failure in limb-girdle muscular dystrophy 2I. Ann. Neurol. 2004, 56, 738–741. [Google Scholar] [CrossRef]
- Louhichi, N.; Triki, C.; Quijano-Roy, S.; Richard, P.; Makri, S.; Méziou, M.; Estournet, B.; Mrad, S.; Romero, N.B.; Ayadi, H.; et al. New FKRP mutations causing congenital muscular dystrophy associated with mental retardation and central nervous system abnormalities. Identification of a founder mutation in Tunisian families. Neurogenetics 2004, 5, 27–34. [Google Scholar] [CrossRef]
- Müller, T.; Krasnianski, M.; Witthaut, R.; Deschauer, M.; Zierz, S. Dilated cardiomyopathy may be an early sign of the C826A Fukutin-related protein mutation. Neuromuscul. Disord. NMD 2005, 15, 372–376. [Google Scholar] [CrossRef]
- Boito, C.A.; Melacini, P.; Vianello, A.; Prandini, P.; Gavassini, B.F.; Bagattin, A.; Siciliano, G.; Angelini, C.; Pegoraro, E. Clinical and molecular characterization of patients with limb-girdle muscular dystrophy type 2I. Arch. Neurol. 2005, 62, 1894–1899. [Google Scholar] [CrossRef]
- Schwartz, M.; Hertz, J.M.; Sveen, M.L.; Vissing, J. LGMD2I presenting with a characteristic Duchenne or Becker muscular dystrophy phenotype. Neurology 2005, 64, 1635–1637. [Google Scholar] [CrossRef] [PubMed]
- Frosk, P.; Greenberg, C.R.; Tennese, A.A.; Lamont, R.; Nylen, E.; Hirst, C.; Frappier, D.; Roslin, N.M.; Zaik, M.; Bushby, K.; et al. The most common mutation in FKRP causing limb girdle muscular dystrophy type 2I (LGMD2I) may have occurred only once and is present in Hutterites and other populations. Hum. Mutat. 2005, 25, 38–44. [Google Scholar] [CrossRef]
- Gaul, C.; Deschauer, M.; Tempelmann, C.; Vielhaber, S.; Klein, H.U.; Heinze, H.J.; Zierz, S.; Grothues, F. Cardiac involvement in limb-girdle muscular dystrophy 2I: Conventional cardiac diagnostic and cardiovascular magnetic resonance. J. Neurol. 2006, 253, 1317–1322. [Google Scholar] [CrossRef]
- Sveen, M.L.; Schwartz, M.; Vissing, J. High prevalence and phenotype-genotype correlations of limb girdle muscular dystrophy type 2I in Denmark. Ann. Neurol. 2006, 59, 808–815. [Google Scholar] [CrossRef]
- D′Amico, A.; Petrini, S.; Parisi, F.; Tessa, A.; Francalanci, P.; Grutter, G.; Santorelli, F.M.; Bertini, E. Heart transplantation in a child with LGMD2I presenting as isolated dilated cardiomyopathy. Neuromuscul. Disord. NMD 2008, 18, 153–155. [Google Scholar] [CrossRef]
- Wahbi, K.; Meune, C.; Hamouda, E.H.; Stojkovic, T.; Laforêt, P.; Bécane, H.M.; Eymard, B.; Duboc, D. Cardiac assessment of limb-girdle muscular dystrophy 2I patients: An echography, Holter ECG and magnetic resonance imaging study. Neuromuscul. Disord. NMD 2008, 18, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Kefi, M.; Amouri, R.; Chabrak, S.; Mechmeche, R.; Hentati, F. Variable cardiac involvement in Tunisian siblings harboring FKRP gene mutations. Neuropediatrics 2008, 39, 113–115. [Google Scholar] [CrossRef]
- Sveen, M.L.; Thune, J.J.; Køber, L.; Vissing, J. Cardiac involvement in patients with limb-girdle muscular dystrophy type 2 and Becker muscular dystrophy. Arch. Neurol. 2008, 65, 1196–1201. [Google Scholar] [CrossRef] [PubMed]
- Margeta, M.; Connolly, A.M.; Winder, T.L.; Pestronk, A.; Moore, S.A. Cardiac pathology exceeds skeletal muscle pathology in two cases of limb-girdle muscular dystrophy type 2I. Muscle Nerve 2009, 40, 883–889. [Google Scholar] [CrossRef]
- Bourteel, H.; Vermersch, P.; Cuisset, J.M.; Maurage, C.A.; Laforet, P.; Richard, P.; Stojkovic, T. Clinical and mutational spectrum of limb-girdle muscular dystrophy type 2I in 11 French patients. J. Neurol. Neurosurg. Psychiatry 2009, 80, 1405–1408. [Google Scholar] [CrossRef]
- Yilmaz, A.; Gdynia, H.J.; Ponfick, M.; Ludolph, A.C.; Rösch, S.; Sechtem, U. The proteoglycan-dystrophin complex in genetic cardiomyopathies—Lessons from three siblings with limb-girdle muscular dystrophy-2I (LGMD-2I). Clin. Res. Cardiol. Off. J. Ger. Card. Soc. 2011, 100, 611–615. [Google Scholar] [CrossRef]
- Pane, M.; Messina, S.; Vasco, G.; Foley, A.R.; Morandi, L.; Pegoraro, E.; Mongini, T.; D′Amico, A.; Bianco, F.; Lombardo, M.E.; et al. Respiratory and cardiac function in congenital muscular dystrophies with alpha dystroglycan deficiency. Neuromuscul. Disord. NMD 2012, 22, 685–689. [Google Scholar] [CrossRef]
- Hollingsworth, K.G.; Willis, T.A.; Bates, M.G.; Dixon, B.J.; Lochmüller, H.; Bushby, K.; Bourke, J.; MacGowan, G.A.; Straub, V. Subepicardial dysfunction leads to global left ventricular systolic impairment in patients with limb girdle muscular dystrophy 2I. Eur. J. Heart Fail. 2013, 15, 986–994. [Google Scholar] [CrossRef]
- Liang, W.C.; Hayashi, Y.K.; Ogawa, M.; Wang, C.H.; Huang, W.T.; Nishino, I.; Jong, Y.J. Limb-girdle muscular dystrophy type 2I is not rare in Taiwan. Neuromuscul. Disord. NMD 2013, 23, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Schottlaender, L.V.; Petzold, A.; Wood, N.; Houlden, H. Diagnostic clues and manifesting carriers in fukutin-related protein (FKRP) limb-girdle muscular dystrophy. J. Neurol. Sci. 2015, 348, 266–268. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.N.; Wang, Z.Q.; Chen, Y.Q.; Xu, G.R.; Lin, M.T.; Wang, N. Limb-girdle muscular dystrophy type 2I: Two Chinese families and a review in Asian patients. Int. J. Neurosci. 2018, 128, 199–207. [Google Scholar] [CrossRef]
- El-Battrawy, I.; Zhao, Z.; Lan, H.; Li, X.; Yücel, G.; Lang, S.; Sattler, K.; Schünemann, J.D.; Zimmermann, W.H.; Cyganek, L.; et al. Ion Channel Dysfunctions in Dilated Cardiomyopathy in Limb-Girdle Muscular Dystrophy. Circ. Genom. Precis. Med. 2018, 11, e001893. [Google Scholar] [CrossRef]
- Ten Dam, L.; Frankhuizen, W.S.; Linssen, W.; Straathof, C.S.; Niks, E.H.; Faber, K.; Fock, A.; Kuks, J.B.; Brusse, E.; de Coo, R.; et al. Autosomal recessive limb-girdle and Miyoshi muscular dystrophies in the Netherlands: The clinical and molecular spectrum of 244 patients. Clin. Genet. 2019, 96, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Murphy, L.B.; Schreiber-Katz, O.; Rafferty, K.; Robertson, A.; Topf, A.; Willis, T.A.; Heidemann, M.; Thiele, S.; Bindoff, L.; Laurent, J.P.; et al. Global FKRP Registry: Observations in more than 300 patients with Limb Girdle Muscular Dystrophy R9. Ann. Clin. Transl. Neurol. 2020, 7, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Libell, E.M.; Richardson, J.A.; Lutz, K.L.; Ng, B.Y.; Mockler, S.R.H.; Laubscher, K.M.; Stephan, C.M.; Zimmerman, B.M.; Edens, E.R.; Reinking, B.E.; et al. Cardiomyopathy in limb girdle muscular dystrophy R9, FKRP related. Muscle Nerve 2020, 62, 626–632. [Google Scholar] [CrossRef]
- Nakanishi, T.; Sakauchi, M.; Kaneda, Y.; Tomimatsu, H.; Saito, K.; Nakazawa, M.; Osawa, M. Cardiac involvement in Fukuyama-type congenital muscular dystrophy. Pediatrics 2006, 117, e1187–e1192. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Shirasawa, H. Congenital muscular dystrophy of the Fukuyama type (FCMD) with severe myocardial fibrosis. A case report with postmortem angiography. Acta Pathol. Jpn. 1987, 37, 1823–1835. [Google Scholar] [CrossRef]
- Murakami, T.; Hayashi, Y.K.; Noguchi, S.; Ogawa, M.; Nonaka, I.; Tanabe, Y.; Ogino, M.; Takada, F.; Eriguchi, M.; Kotooka, N.; et al. Fukutin gene mutations cause dilated cardiomyopathy with minimal muscle weakness. Ann. Neurol. 2006, 60, 597–602. [Google Scholar] [CrossRef]
- Cotarelo, R.P.; Valero, M.C.; Prados, B.; Peña, A.; Rodríguez, L.; Fano, O.; Marco, J.J.; Martínez-Frías, M.L.; Cruces, J. Two new patients bearing mutations in the fukutin gene confirm the relevance of this gene in Walker-Warburg syndrome. Clin. Genet. 2008, 73, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Rosales, X.Q.; Moser, S.J.; Tran, T.; McCarthy, B.; Dunn, N.; Habib, P.; Simonetti, O.P.; Mendell, J.R.; Raman, S.V. Cardiovascular magnetic resonance of cardiomyopathy in limb girdle muscular dystrophy 2B and 2I. J. Cardiovasc. Magn. Reson. Off. J. Soc. Cardiovasc. Magn. Reson. 2011, 13, 39. [Google Scholar] [CrossRef] [PubMed]
- Fiorillo, C.; Moro, F.; Astrea, G.; Morales, M.A.; Baldacci, J.; Marchese, M.; Scapolan, S.; Bruno, C.; Battini, R.; Santorelli, F.M. Novel mutations in the fukutin gene in a boy with asymptomatic hyperCKemia. Neuromuscul. Disord. NMD 2013, 23, 1010–1015. [Google Scholar] [CrossRef]
- Amiya, E.; Morita, H.; Hatano, M.; Nitta, D.; Hosoya, Y.; Maki, H.; Motozawa, Y.; Sato, N.; Ishiura, H.; Numakura, S.; et al. Fukutin gene mutations that cause left ventricular noncompaction. Int. J. Cardiol. 2016, 222, 727–729. [Google Scholar] [CrossRef]
- Ishigaki, K.; Ihara, C.; Nakamura, H.; Mori-Yoshimura, M.; Maruo, K.; Taniguchi-Ikeda, M.; Kimura, E.; Murakami, T.; Sato, T.; Toda, T.; et al. National registry of patients with Fukuyama congenital muscular dystrophy in Japan. Neuromuscul. Disord. NMD 2018, 28, 885–893. [Google Scholar] [CrossRef]
- Carnevale, A.; Rosas-Madrigal, S.; Rosendo-Gutiérrez, R.; López-Mora, E.; Romero-Hidalgo, S.; Avila-Vazzini, N.; Jacobo-Albavera, L.; Domínguez-Pérez, M.; Vargas-Alarcón, G.; Pérez-Villatoro, F.; et al. Genomic study of dilated cardiomyopathy in a group of Mexican patients using site-directed next generation sequencing. Mol. Genet. Genom. Med. 2020, 8, e1504. [Google Scholar] [CrossRef]
- Vad, O.B.; Paludan-Müller, C.; Ahlberg, G.; Kalstø, S.M.; Ghouse, J.; Andreasen, L.; Haunsø, S.; Tveit, A.; Sajadieh, A.; Christophersen, I.E.; et al. Loss-of-Function Variants in Cytoskeletal Genes Are Associated with Early-Onset Atrial Fibrillation. J. Clin. Med. 2020, 9, 372. [Google Scholar] [CrossRef] [PubMed]
- Villarreal-Molina, M.T.; Rosas-Madrigal, S.; López-Mora, E.; Calderón-Avila, A.L.; Rodríguez-Zanella, H.; Romero-Hidalgo, S.; Rosendo-Gutierrez, R.; Carnevale, A. Homozygous Fukutin Missense Mutation in Two Mexican Siblings with Dilated Cardiomyopathy. Rev. Investig. Clin. Organo Hosp. Enferm. Nutr. 2020, 73, 132–137. [Google Scholar] [CrossRef]
- Al-Shafai, K.N.; Al-Hashemi, M.; Manickam, C.; Musa, R.; Selvaraj, S.; Syed, N.; Vempalli, F.; Ali, M.; Yacoub, M.; Estivill, X. Genetic evaluation of cardiomyopathies in Qatar identifies enrichment of pathogenic sarcomere gene variants and possible founder disease mutations in the Arabs. Mol. Genet. Genom. Med. 2021, 9, e1709. [Google Scholar] [CrossRef]
- Gaertner, A.; Burr, L.; Klauke, B.; Brodehl, A.; Laser, K.T.; Klingel, K.; Tiesmeier, J.; Schulz, U.; Knyphausen, E.Z.; Gummert, J.; et al. Compound Heterozygous FKTN Variants in a Patient with Dilated Cardiomyopathy Led to an Aberrant α-Dystroglycan Pattern. Int. J. Mol. Sci. 2022, 23, 6685. [Google Scholar] [CrossRef] [PubMed]
- Lesurf, R.; Said, A.; Akinrinade, O.; Breckpot, J.; Delfosse, K.; Liu, T.; Yao, R.; Persad, G.; McKenna, F.; Noche, R.R.; et al. Whole genome sequencing delineates regulatory, copy number, and cryptic splice variants in early onset cardiomyopathy. NPJ Genom. Med. 2022, 7, 18. [Google Scholar] [CrossRef] [PubMed]
- Willer, T.; Valero, M.C.; Tanner, W.; Cruces, J.; Strahl, S. O-mannosyl glycans: From yeast to novel associations with human disease. Curr. Opin. Struct. Biol. 2003, 13, 621–630. [Google Scholar] [CrossRef]
- Bello, L.; Melacini, P.; Pezzani, R.; D′Amico, A.; Piva, L.; Leonardi, E.; Torella, A.; Soraru, G.; Palmieri, A.; Smaniotto, G.; et al. Cardiomyopathy in patients with POMT1-related congenital and limb-girdle muscular dystrophy. Eur. J. Hum. Genet. EJHG 2012, 20, 1234–1239. [Google Scholar] [CrossRef]
- Devisme, L.; Bouchet, C.; Gonzalès, M.; Alanio, E.; Bazin, A.; Bessières, B.; Bigi, N.; Blanchet, P.; Bonneau, D.; Bonnières, M.; et al. Cobblestone lissencephaly: Neuropathological subtypes and correlations with genes of dystroglycanopathies. Brain J. Neurol. 2012, 135, 469–482. [Google Scholar] [CrossRef]
- Sparks, S.E.; Escolar, D.M. Congenital muscular dystrophies. Handb. Clin. Neurol. 2011, 101, 47–79. [Google Scholar] [CrossRef]
- Martinez, H.R.; Craigen, W.J.; Ummat, M.; Adesina, A.M.; Lotze, T.E.; Jefferies, J.L. Novel cardiovascular findings in association with a POMT2 mutation: Three siblings with α-dystroglycanopathy. Eur. J. Hum. Genet. EJHG 2014, 22, 486–491. [Google Scholar] [CrossRef][Green Version]
- Yanagisawa, A.; Bouchet, C.; Van den Bergh, P.Y.; Cuisset, J.M.; Viollet, L.; Leturcq, F.; Romero, N.B.; Quijano-Roy, S.; Fardeau, M.; Seta, N.; et al. New POMT2 mutations causing congenital muscular dystrophy: Identification of a founder mutation. Neurology 2007, 69, 1254–1260. [Google Scholar] [CrossRef] [PubMed]
- Yanagishita, M. Function of proteoglycans in the extracellular matrix. Acta Pathol. Jpn. 1993, 43, 283–293. [Google Scholar] [CrossRef]
- Wang, Q.; Chi, L. The Alterations and Roles of Glycosaminoglycans in Human Diseases. Polymers 2022, 14, 5014. [Google Scholar] [CrossRef] [PubMed]
- Doddato, G.; Fabbiani, A.; Fallerini, C.; Bruttini, M.; Hadjistilianou, T.; Landi, M.; Coradeschi, C.; Grosso, S.; Tomasini, B.; Mencarelli, M.A.; et al. Spondyloocular Syndrome: A Novel XYLT2 Variant with Description of the Neonatal Phenotype. Front. Genet. 2021, 12, 761264. [Google Scholar] [CrossRef] [PubMed]
- Munns, C.F.; Fahiminiya, S.; Poudel, N.; Munteanu, M.C.; Majewski, J.; Sillence, D.O.; Metcalf, J.P.; Biggin, A.; Glorieux, F.; Fassier, F.; et al. Homozygosity for frameshift mutations in XYLT2 result in a spondylo-ocular syndrome with bone fragility, cataracts, and hearing defects. Am. J. Hum. Genet. 2015, 96, 971–978. [Google Scholar] [CrossRef]
- Taylan, F.; Costantini, A.; Coles, N.; Pekkinen, M.; Héon, E.; Şıklar, Z.; Berberoğlu, M.; Kämpe, A.; Kıykım, E.; Grigelioniene, G.; et al. Spondyloocular Syndrome: Novel Mutations in XYLT2 Gene and Expansion of the Phenotypic Spectrum. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2016, 31, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Kranz, C.; Jungeblut, C.; Denecke, J.; Erlekotte, A.; Sohlbach, C.; Debus, V.; Kehl, H.G.; Harms, E.; Reith, A.; Reichel, S.; et al. A defect in dolichol phosphate biosynthesis causes a new inherited disorder with death in early infancy. Am. J. Hum. Genet. 2007, 80, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Lefeber, D.J.; de Brouwer, A.P.; Morava, E.; Riemersma, M.; Schuurs-Hoeijmakers, J.H.; Absmanner, B.; Verrijp, K.; van den Akker, W.M.; Huijben, K.; Steenbergen, G.; et al. Autosomal recessive dilated cardiomyopathy due to DOLK mutations results from abnormal dystroglycan O-mannosylation. PLoS Genet. 2011, 7, e1002427. [Google Scholar] [CrossRef]
- Kapusta, L.; Zucker, N.; Frenckel, G.; Medalion, B.; Ben Gal, T.; Birk, E.; Mandel, H.; Nasser, N.; Morgenstern, S.; Zuckermann, A.; et al. From discrete dilated cardiomyopathy to successful cardiac transplantation in congenital disorders of glycosylation due to dolichol kinase deficiency (DK1-CDG). Heart Fail. Rev. 2013, 18, 187–196. [Google Scholar] [CrossRef]
- Lieu, M.T.; Ng, B.G.; Rush, J.S.; Wood, T.; Basehore, M.J.; Hegde, M.; Chang, R.C.; Abdenur, J.E.; Freeze, H.H.; Wang, R.Y. Severe, fatal multisystem manifestations in a patient with dolichol kinase-congenital disorder of glycosylation. Mol. Genet. Metab. 2013, 110, 484–489. [Google Scholar] [CrossRef]
- Yu, S.; Zhang, Y.; Chen, Z.; Song, J.; Wang, C. Case Report: A Novel Compound Heterozygous Gene Mutation of Dolichol Kinase Deficiency (DOLK-CDG). Endocr. Metab. Immune Disord. Drug Targets 2022, 23, 235–241. [Google Scholar] [CrossRef]
- Komlosi, K.; Claris, O.; Collardeau-Frachon, S.; Kopp, J.; Hausser, I.; Mazereeuw-Hautier, J.; Jonca, N.; Zimmer, A.D.; Sanlaville, D.; Fischer, J. Fatal Neonatal DOLK-CDG as a Rare Form of Syndromic Ichthyosis. Front. Genet. 2021, 12, 719624. [Google Scholar] [CrossRef]
- Nagy, S.; Lau, T.; Alavi, S.; Karimiani, E.G.; Vallian, J.; Ng, B.G.; Noroozi Asl, S.; Akhondian, J.; Bahreini, A.; Yaghini, O.; et al. A recurrent homozygous missense DPM3 variant leads to muscle and brain disease. Clin. Genet. 2022, 102, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Lefeber, D.J.; Schönberger, J.; Morava, E.; Guillard, M.; Huyben, K.M.; Verrijp, K.; Grafakou, O.; Evangeliou, A.; Preijers, F.W.; Manta, P.; et al. Deficiency of Dol-P-Man synthase subunit DPM3 bridges the congenital disorders of glycosylation with the dystroglycanopathies. Am. J. Hum. Genet. 2009, 85, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Van Tol, W.; Michelakakis, H.; Georgiadou, E.; van den Bergh, P.; Moraitou, M.; Papadimas, G.K.; Papadopoulos, C.; Huijben, K.; Alsady, M.; Willemsen, M.A.; et al. Toward understanding tissue-specific symptoms in dolichol-phosphate-mannose synthesis disorders; insight from DPM3-CDG. J. Inherit. Metab. Dis. 2019, 42, 984–992. [Google Scholar] [CrossRef]
- Van Tol, W.; Ashikov, A.; Korsch, E.; Abu Bakar, N.; Willemsen, M.A.; Thiel, C.; Lefeber, D.J. A mutation in mannose-phosphate-dolichol utilization defect 1 reveals clinical symptoms of congenital disorders of glycosylation type I and dystroglycanopathy. JIMD Rep. 2019, 50, 31–39. [Google Scholar] [CrossRef]
- Thiel, C.; Wortmann, S.; Riedhammer, K.; Alhaddad, B.; Mayatepek, E.; Prokisch, H.; Distelmaier, F. Severe ichthyosis in MPDU1-CDG. J. Inherit. Metab. Dis. 2018, 41, 1293–1294. [Google Scholar] [CrossRef]
- Bastaki, F.; Bizzari, S.; Hamici, S.; Nair, P.; Mohamed, M.; Saif, F.; Malik, E.M.; Al-Ali, M.T.; Hamzeh, A.R. Single-center experience of N-linked Congenital Disorders of Glycosylation with a Summary of Molecularly Characterized Cases in Arabs. Ann. Hum. Genet. 2018, 82, 35–47. [Google Scholar] [CrossRef]
- Cantagrel, V.; Lefeber, D.J.; Ng, B.G.; Guan, Z.; Silhavy, J.L.; Bielas, S.L.; Lehle, L.; Hombauer, H.; Adamowicz, M.; Swiezewska, E.; et al. SRD5A3 is required for converting polyprenol to dolichol and is mutated in a congenital glycosylation disorder. Cell 2010, 142, 203–217. [Google Scholar] [CrossRef]
- Kasapkara, C.S.; Tümer, L.; Ezgü, F.S.; Hasanoğlu, A.; Race, V.; Matthijs, G.; Jaeken, J. SRD5A3-CDG: A patient with a novel mutation. Eur. J. Paediatr. Neurol. EJPN Off. J. Eur. Paediatr. Neurol. Soc. 2012, 16, 554–556. [Google Scholar] [CrossRef] [PubMed]
- Tuysuz, B.; Pehlivan, D.; Özkök, A.; Jhangiani, S.; Yalcinkaya, C.; Zeybek, Ç.A.; Muzny, D.M.; Lupski, J.R.; Gibbs, R.; Jaeken, J. Phenotypic Expansion of Congenital Disorder of Glycosylation Due to SRD5A3 Null Mutation. JIMD Rep. 2016, 26, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, P.G.; Ng, B.G.; Sanford, L.; Sutton, V.R.; Bartholomew, D.W.; Pastore, M.T.; Bamshad, M.J.; Kircher, M.; Buckingham, K.J.; Nickerson, D.A.; et al. SRD5A3-CDG: Expanding the phenotype of a congenital disorder of glycosylation with emphasis on adult onset features. Am. J. Med. Genet. Part A 2016, 170, 3165–3171. [Google Scholar] [CrossRef] [PubMed]
- Kamarus Jaman, N.; Rehsi, P.; Henderson, R.H.; Löbel, U.; Mankad, K.; Grunewald, S. SRD5A3-CDG: Emerging Phenotypic Features of an Ultrarare CDG Subtype. Front. Genet. 2021, 12, 737094. [Google Scholar] [CrossRef] [PubMed]
- Bayat, A.; Kløvgaard, M.; Johannesen, K.M.; Barakat, T.S.; Kievit, A.; Montomoli, M.; Parrini, E.; Pietrafusa, N.; Schelhaas, J.; van Slegtenhorst, M.; et al. Deciphering the premature mortality in PIGA-CDG–An untold story. Epilepsy Res. 2021, 170, 106530. [Google Scholar] [CrossRef]
- Fauth, C.; Steindl, K.; Toutain, A.; Farrell, S.; Witsch-Baumgartner, M.; Karall, D.; Joset, P.; Böhm, S.; Baumer, A.; Maier, O.; et al. A recurrent germline mutation in the PIGA gene causes Simpson-Golabi-Behmel syndrome type 2. Am. J. Med. Genet. Part A 2016, 170, 392–402. [Google Scholar] [CrossRef]
- Bayat, A.; Knaus, A.; Pendziwiat, M.; Afenjar, A.; Barakat, T.S.; Bosch, F.; Callewaert, B.; Calvas, P.; Ceulemans, B.; Chassaing, N.; et al. Lessons learned from 40 novel PIGA patients and a review of the literature. Epilepsia 2020, 61, 1142–1155. [Google Scholar] [CrossRef]
- Van der Crabben, S.N.; Harakalova, M.; Brilstra, E.H.; van Berkestijn, F.M.; Hofstede, F.C.; van Vught, A.J.; Cuppen, E.; Kloosterman, W.; Ploos van Amstel, H.K.; van Haaften, G.; et al. Expanding the spectrum of phenotypes associated with germline PIGA mutations: A child with developmental delay, accelerated linear growth, facial dysmorphisms, elevated alkaline phosphatase, and progressive CNS abnormalities. Am. J. Med. Genet. Part A 2014, 164, 29–35. [Google Scholar] [CrossRef]
- Swoboda, K.J.; Margraf, R.L.; Carey, J.C.; Zhou, H.; Newcomb, T.M.; Coonrod, E.; Durtschi, J.; Mallempati, K.; Kumanovics, A.; Katz, B.E.; et al. A novel germline PIGA mutation in Ferro-Cerebro-Cutaneous syndrome: A neurodegenerative X-linked epileptic encephalopathy with systemic iron-overload. Am. J. Med. Genet. Part A 2014, 164, 17–28. [Google Scholar] [CrossRef]
- Tarailo-Graovac, M.; Sinclair, G.; Stockler-Ipsiroglu, S.; Van Allen, M.; Rozmus, J.; Shyr, C.; Biancheri, R.; Oh, T.; Sayson, B.; Lafek, M.; et al. The genotypic and phenotypic spectrum of PIGA deficiency. Orphanet J. Rare Dis. 2015, 10, 23. [Google Scholar] [CrossRef]
- Shashi, V.; Zunich, J.; Kelly, T.E.; Fryburg, J.S. Neuroectodermal (CHIME) syndrome: An additional case with long term follow up of all reported cases. J. Med. Genet. 1995, 32, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Tinschert, S.; Anton-Lamprecht, I.; Albrecht-Nebe, H.; Audring, H. Zunich neuroectodermal syndrome: Migratory ichthyosiform dermatosis, colobomas, and other abnormalities. Pediatr. Dermatol. 1996, 13, 363–371. [Google Scholar] [CrossRef]
- Ng, B.G.; Hackmann, K.; Jones, M.A.; Eroshkin, A.M.; He, P.; Wiliams, R.; Bhide, S.; Cantagrel, V.; Gleeson, J.G.; Paller, A.S.; et al. Mutations in the glycosylphosphatidylinositol gene PIGL cause CHIME syndrome. Am. J. Hum. Genet. 2012, 90, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Knight Johnson, A.; Schaefer, G.B.; Lee, J.; Hu, Y.; Del Gaudio, D. Alu-mediated deletion of PIGL in a Patient with CHIME syndrome. Am. J. Med. Genet. Part A 2017, 173, 1378–1382. [Google Scholar] [CrossRef]
- Maydan, G.; Noyman, I.; Har-Zahav, A.; Neriah, Z.B.; Pasmanik-Chor, M.; Yeheskel, A.; Albin-Kaplanski, A.; Maya, I.; Magal, N.; Birk, E.; et al. Multiple congenital anomalies-hypotonia-seizures syndrome is caused by a mutation in PIGN. J. Med. Genet. 2011, 48, 383–389. [Google Scholar] [CrossRef]
- Chen, C.P.; Lin, H.M.; Leung, C.; Lin, S.P.; Su, Y.N.; Su, J.W.; Chen, Y.T.; Wang, W. Partial monosomy 9p (9p22.2→pter) and partial trisomy 18q (18q21.32→qter) in a female infant with anorectal malformations. Genet. Couns. 2012, 23, 201–206. [Google Scholar] [PubMed]
- Brady, P.D.; Moerman, P.; De Catte, L.; Deprest, J.; Devriendt, K.; Vermeesch, J.R. Exome sequencing identifies a recessive PIGN splice site mutation as a cause of syndromic congenital diaphragmatic hernia. Eur. J. Med. Genet. 2014, 57, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Couser, N.L.; Masood, M.M.; Strande, N.T.; Foreman, A.K.; Crooks, K.; Weck, K.E.; Lu, M.; Wilhelmsen, K.C.; Roche, M.; Evans, J.P.; et al. The phenotype of multiple congenital anomalies-hypotonia-seizures syndrome 1: Report and review. Am. J. Med. Genet. Part A 2015, 167, 2176–2181. [Google Scholar] [CrossRef]
- Fleming, L.; Lemmon, M.; Beck, N.; Johnson, M.; Mu, W.; Murdock, D.; Bodurtha, J.; Hoover-Fong, J.; Cohn, R.; Bosemani, T.; et al. Genotype-phenotype correlation of congenital anomalies in multiple congenital anomalies hypotonia seizures syndrome (MCAHS1)/PIGN-related epilepsy. Am. J. Med. Genet. Part A 2016, 170, 77–86. [Google Scholar] [CrossRef]
- Xiao, S.Q.; Li, M.H.; Meng, Y.L.; Li, C.; Huang, H.L.; Liu, C.X.; Lyu, Y.; Na, Q. Case Report: Compound Heterozygous Phosphatidylinositol-Glycan Biosynthesis Class N (PIGN) Mutations in a Chinese Fetus with Hypotonia-Seizures Syndrome 1. Front. Genet. 2020, 11, 594078. [Google Scholar] [CrossRef]
- Siavrienė, E.; Maldžienė, Ž.; Mikštienė, V.; Petraitytė, G.; Rančelis, T.; Dapkūnas, J.; Burnytė, B.; Benušienė, E.; Sasnauskienė, A.; Grikinienė, J.; et al. PIGN-Related Disease in Two Lithuanian Families: A Report of Two Novel Pathogenic Variants, Molecular and Clinical Characterisation. Medicina 2022, 58, 1526. [Google Scholar] [CrossRef] [PubMed]
- Kvarnung, M.; Nilsson, D.; Lindstrand, A.; Korenke, G.C.; Chiang, S.C.; Blennow, E.; Bergmann, M.; Stödberg, T.; Mäkitie, O.; Anderlid, B.M.; et al. A novel intellectual disability syndrome caused by GPI anchor deficiency due to homozygous mutations in PIGT. J. Med. Genet. 2013, 50, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Ohishi, K.; Inoue, N.; Kinoshita, T. PIG-S and PIG-T, essential for GPI anchor attachment to proteins, form a complex with GAA1 and GPI8. EMBO J. 2001, 20, 4088–4098. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.; Golas, G.A.; Davids, M.; Huizing, M.; Kane, M.S.; Krasnewich, D.M.; Malicdan, M.C.V.; Adams, D.R.; Markello, T.C.; Zein, W.M.; et al. Expanding the clinical and molecular characteristics of PIGT-CDG, a disorder of glycosylphosphatidylinositol anchors. Mol. Genet. Metab. 2015, 115, 128–140. [Google Scholar] [CrossRef]
- Bayat, A.; Knaus, A.; Juul, A.W.; Dukic, D.; Gardella, E.; Charzewska, A.; Clement, E.; Hjalgrim, H.; Hoffman-Zacharska, D.; Horn, D.; et al. PIGT-CDG, a disorder of the glycosylphosphatidylinositol anchor: Description of 13 novel patients and expansion of the clinical characteristics. Genet. Med. Off. J. Am. Coll. Med. Genet. 2019, 21, 2216–2223. [Google Scholar] [CrossRef]
- Reynolds, K.K.; Juusola, J.; Rice, G.M.; Giampietro, P.F. Prenatal presentation of Mabry syndrome with congenital diaphragmatic hernia and phenotypic overlap with Fryns syndrome. Am. J. Med. Genet. Part A 2017, 173, 2776–2781. [Google Scholar] [CrossRef]
- Morren, M.A.; Jaeken, J.; Visser, G.; Salles, I.; Van Geet, C.; Simeoni, I.; Turro, E.; Freson, K. PIGO deficiency: Palmoplantar keratoderma and novel mutations. Orphanet J. Rare Dis. 2017, 12, 101. [Google Scholar] [CrossRef]
- Krawitz, P.M.; Murakami, Y.; Hecht, J.; Krüger, U.; Holder, S.E.; Mortier, G.R.; Delle Chiaie, B.; De Baere, E.; Thompson, M.D.; Roscioli, T.; et al. Mutations in PIGO, a member of the GPI-anchor-synthesis pathway, cause hyperphosphatasia with mental retardation. Am. J. Hum. Genet. 2012, 91, 146–151. [Google Scholar] [CrossRef]
- Kuki, I.; Takahashi, Y.; Okazaki, S.; Kawawaki, H.; Ehara, E.; Inoue, N.; Kinoshita, T.; Murakami, Y. Vitamin B6-responsive epilepsy due to inherited GPI deficiency. Neurology 2013, 81, 1467–1469. [Google Scholar] [CrossRef]
- Tanigawa, J.; Mimatsu, H.; Mizuno, S.; Okamoto, N.; Fukushi, D.; Tominaga, K.; Kidokoro, H.; Muramatsu, Y.; Nishi, E.; Nakamura, S.; et al. Phenotype-genotype correlations of PIGO deficiency with variable phenotypes from infantile lethality to mild learning difficulties. Hum. Mutat. 2017, 38, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Foulquier, F.; Vasile, E.; Schollen, E.; Callewaert, N.; Raemaekers, T.; Quelhas, D.; Jaeken, J.; Mills, P.; Winchester, B.; Krieger, M.; et al. Conserved oligomeric Golgi complex subunit 1 deficiency reveals a previously uncharacterized congenital disorder of glycosylation type II. Proc. Natl. Acad. Sci. USA 2006, 103, 3764–3769. [Google Scholar] [CrossRef] [PubMed]
- Zeevaert, R.; Foulquier, F.; Dimitrov, B.; Reynders, E.; Van Damme-Lombaerts, R.; Simeonov, E.; Annaert, W.; Matthijs, G.; Jaeken, J. Cerebrocostomandibular-like syndrome and a mutation in the conserved oligomeric Golgi complex, subunit 1. Hum. Mol. Genet. 2009, 18, 517–524. [Google Scholar] [CrossRef]
- Salazar, M.; Miyake, N.; Silva, S.; Solar, B.; Papazoglu, G.M.; Asteggiano, C.G.; Matsumoto, N. COG1-congenital disorders of glycosylation: Milder presentation and review. Clin. Genet. 2021, 100, 318–323. [Google Scholar] [CrossRef]
- Quelhas, D.; Martins, E.; Azevedo, L.; Bandeira, A.; Diogo, L.; Garcia, P.; Sequeira, S.; Ferreira, A.C.; Teles, E.L.; Rodrigues, E.; et al. Congenital Disorders of Glycosylation in Portugal-Two Decades of Experience. J. Pediatr. 2021, 231, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Steet, R.A.; Bohorov, O.; Bakker, J.; Newell, J.; Krieger, M.; Spaapen, L.; Kornfeld, S.; Freeze, H.H. Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder. Nat. Med. 2004, 10, 518–523. [Google Scholar] [CrossRef] [PubMed]
- Spaapen, L.J.; Bakker, J.A.; van der Meer, S.B.; Sijstermans, H.J.; Steet, R.A.; Wevers, R.A.; Jaeken, J. Clinical and biochemical presentation of siblings with COG-7 deficiency, a lethal multiple O- and N-glycosylation disorder. J. Inherit. Metab. Dis. 2005, 28, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Morava, E.; Zeevaert, R.; Korsch, E.; Huijben, K.; Wopereis, S.; Matthijs, G.; Keymolen, K.; Lefeber, D.J.; De Meirleir, L.; Wevers, R.A. A common mutation in the COG7 gene with a consistent phenotype including microcephaly, adducted thumbs, growth retardation, VSD and episodes of hyperthermia. Eur. J. Hum. Genet. EJHG 2007, 15, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, T.; Gardeitchik, T.; Mohamed, M.; Guerrero-Castillo, S.; Freisinger, P.; Guillemyn, B.; Kariminejad, A.; Dalloyaux, D.; van Kraaij, S.; Lefeber, D.J.; et al. Mutations in ATP6V1E1 or ATP6V1A Cause Autosomal-Recessive Cutis Laxa. Am. J. Hum. Genet. 2017, 100, 216–227. [Google Scholar] [CrossRef]
- Vogt, G.; El Choubassi, N.; Herczegfalvi, Á.; Kölbel, H.; Lekaj, A.; Schara, U.; Holtgrewe, M.; Krause, S.; Horvath, R.; Schuelke, M.; et al. Expanding the clinical and molecular spectrum of ATP6V1A related metabolic cutis laxa. J. Inherit. Metab. Dis. 2021, 44, 972–986. [Google Scholar] [CrossRef]
- Alazami, A.M.; Al-Qattan, S.M.; Faqeih, E.; Alhashem, A.; Alshammari, M.; Alzahrani, F.; Al-Dosari, M.S.; Patel, N.; Alsagheir, A.; Binabbas, B.; et al. Expanding the clinical and genetic heterogeneity of hereditary disorders of connective tissue. Hum. Genet. 2016, 135, 525–540. [Google Scholar] [CrossRef]
- Winchester, B. Lysosomal metabolism of glycoproteins. Glycobiology 2005, 15, 1r–15r. [Google Scholar] [CrossRef]
- Stütz, A.E.; Wrodnigg, T.M. Carbohydrate-Processing Enzymes of the Lysosome: Diseases Caused by Misfolded Mutants and Sugar Mimetics as Correcting Pharmacological Chaperones. Adv. Carbohydr. Chem. Biochem. 2016, 73, 225–302. [Google Scholar] [CrossRef]
- Mistry, P.K.; Kishnani, P.; Wanner, C.; Dong, D.; Bender, J.; Batista, J.L.; Foster, J. Rare lysosomal disease registries: Lessons learned over three decades of real-world evidence. Orphanet J. Rare Dis. 2022, 17, 362. [Google Scholar] [CrossRef]
- Sewell, A.C.; Pontz, B.F.; Weitzel, D.; Humburg, C. Clinical heterogeneity in infantile galactosialidosis. Eur. J. Pediatr. 1987, 146, 528–531. [Google Scholar] [CrossRef]
- Strisciuglio, P.; Sly, W.S.; Dodson, W.E.; McAlister, W.H.; Martin, T.C. Combined deficiency of beta-galactosidase and neuraminidase: Natural history of the disease in the first 18 years of an American patient with late infantile onset form. Am. J. Med. Genet. 1990, 37, 573–577. [Google Scholar] [CrossRef] [PubMed]
- Say, B.; Hommes, F.A.; Malik, S.A.; Carpenter, N.J. An infant with multiple congenital abnormalities and biochemical findings suggesting a variant of galactosialidosis. J. Med. Genet. 1992, 29, 423–424. [Google Scholar] [CrossRef] [PubMed]
- Kyllerman, M.; Månsson, J.E.; Westphal, O.; Conradi, N.; Nellström, H. Infantile galactosialidosis presenting with congenital adrenal hyperplasia and renal hypertension. Pediatr. Neurol. 1993, 9, 318–322. [Google Scholar] [CrossRef]
- Senocak, F.; Sarçlar, M.; Ozkutlu, S. Echocardiographic findings in some metabolic storage diseases. Jpn. Heart J. 1994, 35, 635–643. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Haverkamp, F.; Jacobs, D.; Cantz, M.; Hansmann, M.; Fahnenstich, H.; Zerres, K. Nonimmune hydrops fetalis with galactosialidosis: Consequences for family planning. Fetal Diagn. Ther. 1996, 11, 114–119. [Google Scholar] [CrossRef]
- Claeys, M.; Van der Hoeven, M.; de Die-Smulders, C.; Bakker, J.A.; Offermans, J.P.; Forget, P.P.; Groener, J.E.; Spaapen, L.J. Early-infantile type of galactosialidosis as a cause of heart failure and neonatal ascites. J. Inherit. Metab. Dis. 1999, 22, 666–667. [Google Scholar] [CrossRef]
- Bursi, F.; Osranek, M.; Seward, J.B.; O′Leary, P.W. Mitral and aortic valve thickening associated with galactosialidosis: Echocardiographic features of a lysosomal storage disease. Echocardiography 2003, 20, 605–606. [Google Scholar] [CrossRef]
- Matsumoto, N.; Gondo, K.; Kukita, J.; Higaki, K.; Paragison, R.C.; Nanba, E. A case of galactosialidosis with a homozygous Q49R point mutation. Brain Dev. 2008, 30, 595–598. [Google Scholar] [CrossRef]
- Kartal, A.; Aydın, K. A Turkish case of galactosialidosis with a new homozygous mutation in CTSA gene. Metab. Brain Dis. 2017, 32, 973–975. [Google Scholar] [CrossRef]
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef]
- Uyama, E.; Takahashi, K.; Owada, M.; Okamura, R.; Naito, M.; Tsuji, S.; Kawasaki, S.; Araki, S. Hydrocephalus, corneal opacities, deafness, valvular heart disease, deformed toes and leptomeningeal fibrous thickening in adult siblings: A new syndrome associated with beta-glucocerebrosidase deficiency and a mosaic population of storage cells. Acta Neurol. Scand. 1992, 86, 407–420. [Google Scholar] [CrossRef]
- Mester, S.W.; Weston, M.W. Cardiac tamponade in a patient with Gaucher′s disease. Clin. Cardiol. 1992, 15, 766–767. [Google Scholar] [CrossRef] [PubMed]
- Saraçlar, M.; Atalay, S.; Koçak, N.; Ozkutlu, S. Gaucher′s disease with mitral and aortic involvement: Echocardiographic findings. Pediatr. Cardiol. 1992, 13, 56–58. [Google Scholar] [CrossRef] [PubMed]
- Sharratt, G.P.; Price, D.; Curtis, J.A.; Cornel, G. Gaucher′s disease with mitral valve calcification. Pediatr. Cardiol. 1992, 13, 127–128. [Google Scholar] [CrossRef]
- Abrahamov, A.; Elstein, D.; Gross-Tsur, V.; Farber, B.; Glaser, Y.; Hadas-Halpern, I.; Ronen, S.; Tafakjdi, M.; Horowitz, M.; Zimran, A. Gaucher′s disease variant characterised by progressive calcification of heart valves and unique genotype. Lancet 1995, 346, 1000–1003. [Google Scholar] [CrossRef]
- Beutler, E.; Kattamis, C.; Sipe, J.; Lipson, M. 1342C mutation in Gaucher′s disease. Lancet 1995, 346, 1637. [Google Scholar] [CrossRef] [PubMed]
- Chabás, A.; Cormand, B.; Grinberg, D.; Burguera, J.M.; Balcells, S.; Merino, J.L.; Mate, I.; Sobrino, J.A.; Gonzàlez-Duarte, R.; Vilageliu, L. Unusual expression of Gaucher′s disease: Cardiovascular calcifications in three sibs homozygous for the D409H mutation. J. Med. Genet. 1995, 32, 740–742. [Google Scholar] [CrossRef]
- Erduran, E.; Mocan, H.; Gedik, Y.; Kamaci, R.; Okten, A.; Değer, O. Hydrocephalus, corneal opacities, deafness, left ventricle hypertrophy, clinodactyly in an adolescent patient. A new syndrome associated with glucocerebrosidase deficiency. Genet. Couns. 1995, 6, 211–215. [Google Scholar] [PubMed]
- Hill, S.C.; Damaska, B.M.; Tsokos, M.; Kreps, C.; Brady, R.O.; Barton, N.W. Radiographic findings in type 3b Gaucher disease. Pediatr. Radiol. 1996, 26, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Hrebícek, M.; Zeman, J.; Musilová, J.; Hodanová, K.; Renkema, G.H.; Vepreková, L.; Ledvinová, J.; Hrebícek, D.; Sokolová, J.; Aerts, J.M.; et al. A case of type I Gaucher disease with cardiopulmonary amyloidosis and chitotriosidase deficiency. Virchows Arch. Int. J. Pathol. 1996, 429, 305–309. [Google Scholar] [CrossRef]
- Pasmanik-Chor, M.; Laadan, S.; Elroy-Stein, O.; Zimran, A.; Abrahamov, A.; Gatt, S.; Horowitz, M. The glucocerebrosidase D409H mutation in Gaucher disease. Biochem. Mol. Med. 1996, 59, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Uyama, E.; Uchino, M.; Ida, H.; Eto, Y.; Owada, M. D409H/D409H genotype in Gaucher-like disease. J. Med. Genet. 1997, 34, 175. [Google Scholar] [CrossRef][Green Version]
- Damiano, A.M.; Pastores, G.M.; Ware, J.E., Jr. The health-related quality of life of adults with Gaucher′s disease receiving enzyme replacement therapy: Results from a retrospective study. Qual. Life Res. Int. J. Qual. Life Asp. Treat. Care Rehabil. 1998, 7, 373–386. [Google Scholar] [CrossRef]
- Chabás, A.; Gort, L.; Montfort, M.; Castelló, F.; Domínguez, M.C.; Grinberg, D.; Vilageliu, L. Recurrence of the D409H mutation in Spanish Gaucher disease patients: Description of a new homozygous patient and haplotype analysis. J. Med. Genet. 1998, 35, 775–777. [Google Scholar] [CrossRef]
- Guemes, A.; Kosmorsky, G.S.; Moodie, D.S.; Clark, B.; Meisler, D.; Traboulsi, E.I. Corneal opacities in Gaucher disease. Am. J. Ophthalmol. 1998, 126, 833–835. [Google Scholar] [CrossRef]
- Bohlega, S.; Kambouris, M.; Shahid, M.; Al Homsi, M.; Al Sous, W. Gaucher disease with oculomotor apraxia and cardiovascular calcification (Gaucher type IIIC). Neurology 2000, 54, 261–263. [Google Scholar] [CrossRef]
- Cho, L.; Lytle, B.W.; Moodie, D.S. Type IIIC Gaucher′s disease. Circulation 2000, 102, E69–E70. [Google Scholar] [CrossRef] [PubMed]
- Stone, D.L.; Tayebi, N.; Coble, C.; Ginns, E.I.; Sidransky, E. Cardiovascular fibrosis, hydrocephalus, ophthalmoplegia, and visceral involvement in an American child with Gaucher disease. J. Med. Genet. 2000, 37, E40. [Google Scholar] [CrossRef] [PubMed]
- Inui, K.; Yanagihara, K.; Otani, K.; Suzuki, Y.; Akagi, M.; Nakayama, M.; Ida, H.; Okada, S. A new variant neuropathic type of Gaucher′s disease characterized by hydrocephalus, corneal opacities, deformed toes, and fibrous thickening of spleen and liver capsules. J. Pediatr. 2001, 138, 137–139. [Google Scholar] [CrossRef] [PubMed]
- Ono, H.; Fujiwara, M.; Ito, K.; Ueda, H.; Mizoguchi, N.; Sakura, N. Neurological features in Gaucher′s disease during enzyme replacement therapy. Acta Paediatr. 2001, 90, 229–231. [Google Scholar] [CrossRef]
- George, R.; McMahon, J.; Lytle, B.; Clark, B.; Lichtin, A. Severe valvular and aortic arch calcification in a patient with Gaucher′s disease homozygous for the D409H mutation. Clin. Genet. 2001, 59, 360–363. [Google Scholar] [CrossRef]
- Michelakakis, H.; Skardoutsou, A.; Mathioudakis, J.; Moraitou, M.; Dimitriou, E.; Voudris, C.; Karpathios, T. Early-onset severe neurological involvement and D409H homozygosity in Gaucher disease: Outcome of enzyme replacement therapy. Blood Cells Mol. Dis. 2002, 28, 1–4. [Google Scholar] [CrossRef]
- Fujiwaki, T.; Yamaguchi, S.; Tasaka, M.; Sakura, N.; Taketomi, T. Application of delayed extraction-matrix-assisted laser desorption ionization time-of-flight mass spectrometry for analysis of sphingolipids in pericardial fluid, peritoneal fluid and serum from Gaucher disease patients. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2002, 776, 115–123. [Google Scholar] [CrossRef]
- Torloni, M.R.; Franco, K.; Sass, N. Gaucher′s disease with myocardial involvement in pregnancy. Sao Paulo Med. J. Rev. Paul. De Med. 2002, 120, 90–92. [Google Scholar] [CrossRef]
- Brautbar, A.; Abrahamov, A.; Hadas-Halpern, I.; Elstein, D.; Zimran, A. Gaucher disease in Arab patients at an Israeli referral clinic. Isr. Med. Assoc. J. IMAJ 2008, 10, 600–602. [Google Scholar]
- Mireles, S.A.; Seybold, J.; Williams, G. Undiagnosed type IIIc Gaucher disease in a child with aortic and mitral valve calcification: Perioperative complications after cardiac surgery. J. Cardiothorac. Vasc. Anesth. 2010, 24, 471–474. [Google Scholar] [CrossRef]
- Cindik, N.; Ozcay, F.; Süren, D.; Akkoyun, I.; Gökdemir, M.; Varan, B.; Alehan, F.; Ozbek, N.; Tokel, K. Gaucher disease with communicating hydrocephalus and cardiac involvement. Clin. Cardiol. 2010, 33, E26–E30. [Google Scholar] [CrossRef] [PubMed]
- Perić, Z.; Kardum-Skelin, I.; Puskarić, B.J.; Letilović, T.; Vrhovac, R.; Jaksić, B. An unusual presentation of Gaucher′s disease: Aortic valve fibrosis in a patient homozygous for a rare G377S mutation. Coll. Antropol. 2010, 34, 275–278. [Google Scholar] [PubMed]
- Kozelj, M.; Zver, S.; Zadnik, V. Echocardiographic Assessment of Left Ventricular Function in Type 1 Gaucher′s Disease. Adv. Hematol. 2010, 2010, 820843. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Talluto, C.J.; Silverman, N.H. Aortic and mitral valve stenosis with regurgitation: Not due to rheumatic heart disease. Echocardiography 2011, 28, E24–E27. [Google Scholar] [CrossRef] [PubMed]
- Aksu, T.; Baysal, E.; Bıyıkoğlu, F.; Tüfekçioğlu, O. Gaucher′s disease with valvular, myocardial and aortic involvement in a patient with oculomotor apraxia. Anadolu Kardiyol. Derg. AKD Anatol. J. Cardiol. 2011, 11, E4–E5. [Google Scholar] [CrossRef]
- Altunbas, G.; Ercan, S.; Inanç, I.H.; Ozer, O.; Kervancıoğlu, S.; Davutoğlu, V. Extensive vascular and valvular involvement in Gaucher disease. Asian Cardiovasc. Thorac. Ann. 2015, 23, 446–448. [Google Scholar] [CrossRef]
- Lo Iudice, F.; Barbato, A.; Muscariello, R.; Di Nardo, C.; de Stefano, F.; Sibilio, M.; Strazzullo, P.; de Simone, G.; Galderisi, M. Left ventricular diastolic dysfunction in type I Gaucher disease: An echo Doppler study. Echocardiography 2015, 32, 890–895. [Google Scholar] [CrossRef]
- Rastogi, P.; Rao, S.; Kaur, J.; Malhotra, P.; Varma, S.; Das, R. Gaucher′s Disease with Cardiac Valve Calcification and Stenosis: A Rare Presentation due to Homozygous p.D409H Mutation in a North Indian Family. Indian J. Pediatr. 2016, 83, 877–878. [Google Scholar] [CrossRef]
- Roghi, A.; Poggiali, E.; Cassinerio, E.; Pedrotti, P.; Giuditta, M.; Milazzo, A.; Quattrocchi, G.; Cappellini, M.D. The role of cardiac magnetic resonance in assessing the cardiac involvement in Gaucher type 1 patients: Morphological and functional evaluations. J. Cardiovasc. Med. 2017, 18, 244–248. [Google Scholar] [CrossRef]
- Kör, Y.; Keskin, M.; Başpınar, O. Severe cardiac involvement in Gaucher type IIIC: A case report and review of the literature. Cardiol. Young 2017, 27, 1426–1429. [Google Scholar] [CrossRef]
- Weinreb, N.J.; Barbouth, D.S.; Lee, R.E. Causes of death in 184 patients with type 1 Gaucher disease from the United States who were never treated with enzyme replacement therapy. Blood Cells Mol. Dis. 2018, 68, 211–217. [Google Scholar] [CrossRef]
- Alzahrani, A.; Alghamdi, A.A.; Waggass, R. A Saudi Infant with Vici Syndrome: Case Report and Literature Review. Open Access Maced. J. Med. Sci. 2018, 6, 1081–1084. [Google Scholar] [CrossRef] [PubMed]
- Alsahli, S.; Bubshait, D.K.; Rahbeeni, Z.A.; Alfadhel, M. Aortic calcification in Gaucher disease: A case report. Appl. Clin. Genet. 2018, 11, 107–110. [Google Scholar] [CrossRef]
- Karakoyun, M.; Canda, E.; Kiran Tasci, E.; Dogan, E.; Coker, M.; Aydogdu, S. Two siblings with Gaucher type 3c: Different clinical presentations. J. Pediatr. Endocrinol. Metab. JPEM 2019, 32, 533–536. [Google Scholar] [CrossRef]
- Kurolap, A.; Del Toro, M.; Spiegel, R.; Gutstein, A.; Shafir, G.; Cohen, I.J.; Barrabés, J.A.; Feldman, H.B. Gaucher disease type 3c: New patients with unique presentations and review of the literature. Mol. Genet. Metab. 2019, 127, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Lazea, C.; Bucerzan, S.; Al-Khzouz, C.; Zimmermann, A.; Vesa, Ș.C.; Nașcu, I.; Creț, V.; Crișan, M.; Asăvoaie, C.; Miclea, D.; et al. Cardiac Manifestations in a Group of Romanian Patients with Gaucher Disease Type 1 (a Monocentric Study). Diagnostics 2021, 11, 989. [Google Scholar] [CrossRef] [PubMed]
- Demirci, I.; Demir, T.; Dagdelen, S.; Haymana, C.; Tasci, I.; Atmaca, A.; Ertugrul, D.; Ata, N.; Sahin, M.; Salman, S.; et al. No association of Gaucher disease with COVID-19-related outcomes: A nationwide cohort study. Intern. Med. J. 2022, 52, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, R. Fabry disease. Pharmacol. Ther. 2009, 122, 65–77. [Google Scholar] [CrossRef]
- Li, X.; Ren, X.; Zhang, Y.; Ding, L.; Huo, M.; Li, Q. Fabry disease: Mechanism and therapeutics strategies. Front. Pharmacol. 2022, 13, 1025740. [Google Scholar] [CrossRef]
- Mehta, A.; Beck, M.; Linhart, A.; Sunder-Plassmann, G.; Widmer, U. History of lysosomal storage diseases: An overview. In Fabry Disease: Perspectives from 5 Years of FOS; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; Oxford PharmaGenesis: Oxford, UK, 2006; Copyright © 2006, Oxford PharmaGenesis™. [Google Scholar]
- Saeed, S.; Imazio, M. Fabry disease: Definition, Incidence, Clinical presentations and Treatment–Focus on cardiac involvement. Pak. J. Med. Sci. 2022, 38, 2337–2344. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P.; Brand, E.; Burlina, A.; Cecchi, F.; Garman, S.C.; Kempf, J.; Laney, D.A.; Linhart, A.; Maródi, L.; Nicholls, K.; et al. Phenotypic characteristics of the p.Asn215Ser (p.N215S) GLA mutation in male and female patients with Fabry disease: A multicenter Fabry Registry study. Mol. Genet. Genom. Med. 2018, 6, 492–503. [Google Scholar] [CrossRef]
- Mehta, A.; Clarke, J.T.; Giugliani, R.; Elliott, P.; Linhart, A.; Beck, M.; Sunder-Plassmann, G. Natural course of Fabry disease: Changing pattern of causes of death in FOS–Fabry Outcome Survey. J. Med. Genet. 2009, 46, 548–552. [Google Scholar] [CrossRef]
- Caciotti, A.; Donati, M.A.; Boneh, A.; d′Azzo, A.; Federico, A.; Parini, R.; Antuzzi, D.; Bardelli, T.; Nosi, D.; Kimonis, V.; et al. Role of beta-galactosidase and elastin binding protein in lysosomal and nonlysosomal complexes of patients with GM1-gangliosidosis. Hum. Mutat. 2005, 25, 285–292. [Google Scholar] [CrossRef]
- Morrone, A.; Bardelli, T.; Donati, M.A.; Giorgi, M.; Di Rocco, M.; Gatti, R.; Parini, R.; Ricci, R.; Taddeucci, G.; D′Azzo, A.; et al. beta-galactosidase gene mutations affecting the lysosomal enzyme and the elastin-binding protein in GM1-gangliosidosis patients with cardiac involvement. Hum. Mutat. 2000, 15, 354–366. [Google Scholar] [CrossRef]
- Hadley, R.N.; Hagstrom, J.W. Cardiac lesions in a patient with familial neurovisceral lipidosis (generalized gangliosidosis). Am. J. Clin. Pathol. 1971, 55, 237–240. [Google Scholar] [CrossRef][Green Version]
- Gilbert, E.F.; Varakis, J.; Opitz, J.M.; ZuRhein, G.M.; Ware, R.; Viseskul, C.; Kaveggia, E.G.; Hartmann, H.A. Generalized gangliosidosis type II (juvenile GM1 gangliosidosis). A pathological, histochemical and ultrastructural study. Z. Kinderheilkd. 1975, 120, 151–180. [Google Scholar] [CrossRef]
- Benson, P.F.; Barbarik, A.; Brown, S.P.; Mann, T.P. GM1-generalized gangliosidosis variant with cardiomegaly. Postgrad. Med. J. 1976, 52, 159–165. [Google Scholar] [CrossRef][Green Version]
- Kohlschütter, A.; Sieg, K.; Schulte, F.J.; Hayek, H.W.; Goebel, H.H. Infantile cardiomyopathy and neuromyopathy with beta-galactosidase deficiency. Eur. J. Pediatr. 1982, 139, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, H.; Frewen, T.C.; Li, M.D.; Gordon, B.L.; Jung, J.H.; Finlay, J.P.; Roy, P.L.; Grover, D.; Spence, M. Cardiac involvement in diseases characterized by beta-galactosidase deficiency. J. Pediatr. 1985, 106, 78–80. [Google Scholar] [CrossRef]
- Miall-Allen, V.M.; Morgan, B.; Cooper, P.; Shinebourne, E.A. Peripheral arteriovenous fistula as a cause of neonatal cardiac failure. Int. J. Cardiol. 1986, 10, 177–179. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.F.; Wu, J.Y.; Tsai, F.J. Three novel beta-galactosidase gene mutations in Han Chinese patients with GM1 gangliosidosis are correlated with disease severity. J. Biomed. Sci. 2010, 17, 79. [Google Scholar] [CrossRef]
- Brunetti-Pierri, N.; Scaglia, F. GM1 gangliosidosis: Review of clinical, molecular, and therapeutic aspects. Mol. Genet. Metab. 2008, 94, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Caciotti, A.; Cellai, L.; Tonin, R.; Mei, D.; Procopio, E.; Di Rocco, M.; Andaloro, A.; Antuzzi, D.; Rampazzo, A.; Rigoldi, M.; et al. Morquio B disease: From pathophysiology towards diagnosis. Mol. Genet. Metab. 2021, 132, 180–188. [Google Scholar] [CrossRef]
- Barry, M.O.; Beardslee, M.A.; Braverman, A.C. Morquio′s syndrome: Severe aortic regurgitation and late pulmonary autograft failure. J. Heart Valve Dis. 2006, 15, 839–842. [Google Scholar]
- Dostalova, G.; Hlubocka, Z.; Lindner, J.; Hulkova, H.; Poupetova, H.; Vlaskova, H.; Sikora, J.; Linhart, A.; Zeman, J.; Magner, M. Late diagnosis of mucopolysaccharidosis type IVB and successful aortic valve replacement in a 60-year-old female patient. Cardiovasc. Pathol. Off. J. Soc. Cardiovasc. Pathol. 2018, 35, 52–56. [Google Scholar] [CrossRef]
- Venugopalan, P.; Joshi, S.N. Cardiac involvement in infantile Sandhoff disease. J. Paediatr. Child Health 2002, 38, 98–100. [Google Scholar] [CrossRef] [PubMed]
- Villamizar-Schiller, I.T.; Pabón, L.A.; Hufnagel, S.B.; Serrano, N.C.; Karl, G.; Jefferies, J.L.; Hopkin, R.J.; Prada, C.E. Neurological and cardiac responses after treatment with miglustat and a ketogenic diet in a patient with Sandhoff disease. Eur. J. Med. Genet. 2015, 58, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.F.; Chi, C.S.; Tsai, C.R. Early cardiac involvement in an infantile Sandhoff disease case with novel mutations. Brain Dev. 2017, 39, 171–176. [Google Scholar] [CrossRef]
- Sakpichaisakul, K.; Taeranawich, P.; Nitiapinyasakul, A.; Sirisopikun, T. Identification of Sandhoff disease in a Thai family: Clinical and biochemical characterization. J. Med. Assoc. Thail. Chotmaihet Thangphaet 2010, 93, 1088–1092. [Google Scholar]
- Tim-Aroon, T.; Wichajarn, K.; Katanyuwong, K.; Tanpaiboon, P.; Vatanavicharn, N.; Sakpichaisakul, K.; Kongkrapan, A.; Eu-Ahsunthornwattana, J.; Thongpradit, S.; Moolsuwan, K.; et al. Infantile onset Sandhoff disease: Clinical manifestation and a novel common mutation in Thai patients. BMC Pediatr. 2021, 21, 22. [Google Scholar] [CrossRef] [PubMed]
- Ozaal, S.; Jayasena, S.; Jayakody, S.; Schröder, S.; Jayawardana, A.; Jasinge, E. Clinical Presentation and Genetic Heterogeneity Including Two Novel Variants in Sri Lankan Patients with Infantile Sandhoff Disease. Child Neurol. Open 2022, 9, 2329048x221139495. [Google Scholar] [CrossRef] [PubMed]
- Sahyouni, J.K.; Odeh, L.B.M.; Mulla, F.; Junaid, S.; Kar, S.S.; Al Boot Almarri, N.M.J. Infantile Sandhoff disease with ventricular septal defect: A case report. J. Med. Case Rep. 2022, 16, 317. [Google Scholar] [CrossRef]
- Hampe, C.S.; Eisengart, J.B.; Lund, T.C.; Orchard, P.J.; Swietlicka, M.; Wesley, J.; McIvor, R.S. Mucopolysaccharidosis Type I: A Review of the Natural History and Molecular Pathology. Cells 2020, 9, 1838. [Google Scholar] [CrossRef]
- Butman, S.M.; Karl, L.; Copeland, J.G. Combined aortic and mitral valve replacement in an adult with Scheie′s disease. Chest 1989, 96, 209–210. [Google Scholar] [CrossRef]
- Keith, O.; Scully, C.; Weidmann, G.M. Orofacial features of Scheie (Hurler-Scheie) syndrome (alpha-L-iduronidase deficiency). Oral Surg. Oral Med. Oral Pathol. 1990, 70, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Demirsoy, S.; Gücüyener, K.; Olguntürk, R.; Tunaoğlu, S.; Oğuz, D. A case of mucopolysaccharidoses type I with heart involvement during infancy. Turk. J. Pediatr. 1990, 32, 49–52. [Google Scholar] [PubMed]
- Nicolson, S.C.; Black, A.E.; Kraras, C.M. Management of a difficult airway in a patient with Hurler-Scheie syndrome during cardiac surgery. Anesth. Analg. 1992, 75, 830–832. [Google Scholar] [CrossRef] [PubMed]
- Du Cret, R.P.; Weinberg, E.J.; Jackson, C.A.; Braunlin, E.A.; Boudreau, R.J.; Kuni, C.C.; Carpenter, B.M.; Hunter, D.W.; Krivit, W.; Bodeau, G. Resting Tl-201 scintigraphy in the evaluation of coronary artery disease in children with Hurler syndrome. Clin. Nucl. Med. 1994, 19, 975–978. [Google Scholar] [CrossRef]
- Imaizumi, M.; Gushi, K.; Kurobane, I.; Inoue, S.; Suzuki, J.; Koizumi, Y.; Suzuki, H.; Sato, A.; Gotoh, Y.; Haginoya, K.; et al. Long-term effects of bone marrow transplantation for inborn errors of metabolism: A study of four patients with lysosomal storage diseases. Acta Paediatr. Jpn. Overseas Ed. 1994, 36, 30–36. [Google Scholar] [CrossRef]
- Minakata, K.; Konishi, Y.; Matsumoto, M.; Miwa, S. Surgical treatment for Scheie′s syndrome (mucopolysaccharidosis type I-S): Report of two cases. Jpn. Circ. J. 1998, 62, 700–703. [Google Scholar] [CrossRef]
- Kakkis, E.D.; Muenzer, J.; Tiller, G.E.; Waber, L.; Belmont, J.; Passage, M.; Izykowski, B.; Phillips, J.; Doroshow, R.; Walot, I.; et al. Enzyme-replacement therapy in mucopolysaccharidosis I. N. Engl. J. Med. 2001, 344, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Braunlin, E.A.; Stauffer, N.R.; Peters, C.H.; Bass, J.L.; Berry, J.M.; Hopwood, J.J.; Krivit, W. Usefulness of bone marrow transplantation in the Hurler syndrome. Am. J. Cardiol. 2003, 92, 882–886. [Google Scholar] [CrossRef]
- Vijay, S.; Wraith, J.E. Clinical presentation and follow-up of patients with the attenuated phenotype of mucopolysaccharidosis type I. Acta Paediatr. 2005, 94, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Lin, S.P.; Chuang, C.K.; Chen, M.R.; Chen, B.F.; Wraith, J.E. Mucopolysaccharidosis I under enzyme replacement therapy with laronidase—A mortality case with autopsy report. J. Inherit. Metab. Dis. 2005, 28, 1146–1148. [Google Scholar] [CrossRef] [PubMed]
- Hingston, E.J.; Hunter, M.L.; Hunter, B.; Drage, N. Hurler′s syndrome: Dental findings in a case treated with bone marrow transplantation in infancy. Int. J. Paediatr. Dent. 2006, 16, 207–212. [Google Scholar] [CrossRef]
- Braunlin, E.A.; Berry, J.M.; Whitley, C.B. Cardiac findings after enzyme replacement therapy for mucopolysaccharidosis type I. Am. J. Cardiol. 2006, 98, 416–418. [Google Scholar] [CrossRef]
- Sifuentes, M.; Doroshow, R.; Hoft, R.; Mason, G.; Walot, I.; Diament, M.; Okazaki, S.; Huff, K.; Cox, G.F.; Swiedler, S.J.; et al. A follow-up study of MPS I patients treated with laronidase enzyme replacement therapy for 6 years. Mol. Genet. Metab. 2007, 90, 171–180. [Google Scholar] [CrossRef]
- Tokic, V.; Barisic, I.; Huzjak, N.; Petkovic, G.; Fumic, K.; Paschke, E. Enzyme replacement therapy in two patients with an advanced severe (Hurler) phenotype of mucopolysaccharidosis I. Eur. J. Pediatr. 2007, 166, 727–732. [Google Scholar] [CrossRef]
- Soliman, O.I.; Timmermans, R.G.; Nemes, A.; Vletter, W.B.; Wilson, J.H.; ten Cate, F.J.; Geleijnse, M.L. Cardiac abnormalities in adults with the attenuated form of mucopolysaccharidosis type I. J. Inherit. Metab. Dis. 2007, 30, 750–757. [Google Scholar] [CrossRef]
- Hirth, A.; Berg, A.; Greve, G. Successful treatment of severe heart failure in an infant with Hurler syndrome. J. Inherit. Metab. Dis. 2007, 30, 820. [Google Scholar] [CrossRef]
- Hansen, M.D.; Filipovich, A.H.; Davies, S.M.; Mehta, P.; Bleesing, J.; Jodele, S.; Hayashi, R.; Barnes, Y.; Shenoy, S. Allogeneic hematopoietic cell transplantation (HCT) in Hurler′s syndrome using a reduced intensity preparative regimen. Bone Marrow Transplant. 2008, 41, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Nemes, A.; Timmermans, R.G.; Wilson, J.H.; Soliman, O.I.; Krenning, B.J.; ten Cate, F.J.; Geleijnse, M.L. The mild form of mucopolysaccharidosis type I (Scheie syndrome) is associated with increased ascending aortic stiffness. Heart Vessel. 2008, 23, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Khedhiri, S.; Chkioua, L.; Bouzidi, H.; Dandana, A.; Ben Turkia, H.; Miled, A.; Laradi, S. Mucopolysaccharidoses type I and IVA: Clinical features and consanguinity in Tunisia. Pathol.-Biol. 2009, 57, 392–397. [Google Scholar] [CrossRef]
- Fesslová, V.; Corti, P.; Sersale, G.; Rovelli, A.; Russo, P.; Mannarino, S.; Butera, G.; Parini, R. The natural course and the impact of therapies of cardiac involvement in the mucopolysaccharidoses. Cardiol. Young 2009, 19, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Moseley, K.; Pavlova, Z. Postmortem studies on a patient with mucopolysaccharidosis type I: Histopathological findings after one year of enzyme replacement therapy. J. Inherit. Metab. Dis. 2009, 32 (Suppl. S1), 53–57. [Google Scholar] [CrossRef]
- Goksel, O.S.; El, H.; Tireli, E.; Dayioglu, E. Combined aortic and mitral valve replacement in a child with mucopolysaccharidosis type I: A case report. J. Heart Valve Dis. 2009, 18, 214–216. [Google Scholar]
- Mercimek-Mahmutoglu, S.; Reilly, C.; Human, D.; Waters, P.J.; Stoeckler-Ipsiroglu, S. Progression of organ manifestations upon enzyme replacement therapy in a patient with mucopolysaccharidosis type I/Hurler. World J. Pediatr. WJP 2009, 5, 319–321. [Google Scholar] [CrossRef]
- Bahadir, C.; Kurtulus, D.; Cihandide, E. Mucopolysaccharidosis type-IS presenting with onset of carpal tunnel syndrome at adolescence. J. Clin. Rheumatol. Pract. Rep. Rheum. Musculoskelet. Dis. 2009, 15, 402–404. [Google Scholar] [CrossRef]
- Gabrielli, O.; Clarke, L.A.; Bruni, S.; Coppa, G.V. Enzyme-replacement therapy in a 5-month-old boy with attenuated presymptomatic MPS I: 5-year follow-up. Pediatrics 2010, 125, e183–e187. [Google Scholar] [CrossRef]
- Thomas, J.A.; Beck, M.; Clarke, J.T.; Cox, G.F. Childhood onset of Scheie syndrome, the attenuated form of mucopolysaccharidosis I. J. Inherit. Metab. Dis. 2010, 33, 421–427. [Google Scholar] [CrossRef]
- Furukawa, Y.; Hamaguchi, A.; Nozaki, I.; Iizuka, T.; Sasagawa, T.; Shima, Y.; Demura, S.; Murakami, H.; Kawahara, N.; Okuyama, T.; et al. Cervical pachymeningeal hypertrophy as the initial and cardinal manifestation of mucopolysaccharidosis type I in monozygotic twins with a novel mutation in the alpha-L-iduronidase gene. J. Neurol. Sci. 2011, 302, 121–125. [Google Scholar] [CrossRef]
- Watanabe, N.; Anagnostopoulos, P.V.; Azakie, A. Aortic stenosis in a patient with Hurler′s syndrome after bone marrow transplantation. Cardiol. Young 2011, 21, 349–350. [Google Scholar] [CrossRef]
- Van den Broek, L.; Backx, A.P.; Coolen, H.; Wijburg, F.A.; Wevers, R.; Morava, E.; Neeleman, C. Fatal coronary artery disease in an infant with severe mucopolysaccharidosis type I. Pediatrics 2011, 127, e1343–e1346. [Google Scholar] [CrossRef] [PubMed]
- Harada, H.; Uchiwa, H.; Nakamura, M.; Ohno, S.; Morita, H.; Katoh, A.; Yoshino, M.; Ikeda, H. Laronidase replacement therapy improves myocardial function in mucopolysaccharidosis I. Mol. Genet. Metab. 2011, 103, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Rojas, M.V.; Bay, L.; Sanchez, L.; van Kuijck, M.; Ospina, S.; Cabello, J.F.; Martins, A.M. Clinical manifestations and treatment of mucopolysaccharidosis type I patients in Latin America as compared with the rest of the world. J. Inherit. Metab. Dis. 2011, 34, 1029–1037. [Google Scholar] [CrossRef]
- Grigull, L.; Sykora, K.W.; Tenger, A.; Bertram, H.; Meyer-Marcotty, M.; Hartmann, H.; Bültmann, E.; Beilken, A.; Zivicnjak, M.; Mynarek, M.; et al. Variable disease progression after successful stem cell transplantation: Prospective follow-up investigations in eight patients with Hurler syndrome. Pediatr. Transplant. 2011, 15, 861–869. [Google Scholar] [CrossRef]
- Yosunkaya, E.; Karaca, E.; Yilmaz, S.B.; Gezdirici, A.; Guven, G.; Seven, M.; Yuksel, A. Sudden vision loss in a mucopolysaccharidosis I patient receiving enzyme replacement therapy. Genet. Couns. 2011, 22, 371–376. [Google Scholar]
- Rocha, R.V.; Alvarez, R.J.; Bermudez, C.A. Valve surgery in a mucopolysaccharidosis type I patient: Early prosthetic valve endocarditis. Eur. J. Cardio-Thorac. Surg. Off. J. Eur. Assoc. Cardio-Thorac. Surg. 2012, 41, 448–449. [Google Scholar] [CrossRef]
- Arn, P.; Whitley, C.; Wraith, J.E.; Webb, H.W.; Underhill, L.; Rangachari, L.; Cox, G.F. High rate of postoperative mortality in patients with mucopolysaccharidosis I: Findings from the MPS I Registry. J. Pediatr. Surg. 2012, 47, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, G.H.; Fernández, I.; Dominguez, M.; Clarke, L.A. Left ventricular aneurysm in an adult patient with mucopolysaccharidosis type I: Comment on pathogenesis of a novel complication. Mol. Genet. Metab. 2012, 106, 470–473. [Google Scholar] [CrossRef]
- Jurecka, A.; Marucha, J.; Jurkiewicz, E.; Różdżyńska-Świątkowska, A.; Tylki-Szymańska, A. Enzyme replacement therapy in an attenuated case of mucopolysaccharidosis type I (Scheie syndrome): A 6.5-year detailed follow-up. Pediatr. Neurol. 2012, 47, 461–465. [Google Scholar] [CrossRef]
- Wiseman, D.H.; Mercer, J.; Tylee, K.; Malaiya, N.; Bonney, D.K.; Jones, S.A.; Wraith, J.E.; Wynn, R.F. Management of mucopolysaccharidosis type IH (Hurler′s syndrome) presenting in infancy with severe dilated cardiomyopathy: A single institution′s experience. J. Inherit. Metab. Dis. 2013, 36, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Abelin Genevois, K.; Garin, C.; Solla, F.; Guffon, N.; Kohler, R. Surgical management of thoracolumbar kyphosis in mucopolysaccharidosis type 1 in a reference center. J. Inherit. Metab. Dis. 2014, 37, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Braunlin, E.; Orchard, P.J.; Whitley, C.B.; Schroeder, L.; Reed, R.C.; Manivel, J.C. Unexpected coronary artery findings in mucopolysaccharidosis. Report of four cases and literature review. Cardiovasc. Pathol. Off. J. Soc. Cardiovasc. Pathol. 2014, 23, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Aldenhoven, M.; Wynn, R.F.; Orchard, P.J.; O′Meara, A.; Veys, P.; Fischer, A.; Valayannopoulos, V.; Neven, B.; Rovelli, A.; Prasad, V.K.; et al. Long-term outcome of Hurler syndrome patients after hematopoietic cell transplantation: An international multicenter study. Blood 2015, 125, 2164–2172. [Google Scholar] [CrossRef]
- Al-Sannaa, N.A.; Bay, L.; Barbouth, D.S.; Benhayoun, Y.; Goizet, C.; Guelbert, N.; Jones, S.A.; Kyosen, S.O.; Martins, A.M.; Phornphutkul, C.; et al. Early treatment with laronidase improves clinical outcomes in patients with attenuated MPS I: A retrospective case series analysis of nine sibships. Orphanet J. Rare Dis. 2015, 10, 131. [Google Scholar] [CrossRef]
- Brazier, A.; Hasan, R.; Jenkins, P.; Hoschtitzky, A. Urgent resection of a giant left atrial appendage aneurysm and mitral valve replacement in a complex case of Hurler-Scheie syndrome. BMJ Case Rep. 2015, 2015, bcr2015211551. [Google Scholar] [CrossRef]
- Ghosh, A.; Miller, W.; Orchard, P.J.; Jones, S.A.; Mercer, J.; Church, H.J.; Tylee, K.; Lund, T.; Bigger, B.W.; Tolar, J.; et al. Enzyme replacement therapy prior to haematopoietic stem cell transplantation in Mucopolysaccharidosis Type I: 10 year combined experience of 2 centres. Mol. Genet. Metab. 2016, 117, 373–377. [Google Scholar] [CrossRef]
- Gabrielli, O.; Clarke, L.A.; Ficcadenti, A.; Santoro, L.; Zampini, L.; Volpi, N.; Coppa, G.V. 12 year follow up of enzyme-replacement therapy in two siblings with attenuated mucopolysaccharidosis I: The important role of early treatment. BMC Med. Genet. 2016, 17, 19. [Google Scholar] [CrossRef]
- Horovitz, D.D.; Acosta, A.X.; Giugliani, R.; Hlavatá, A.; Hlavatá, K.; Tchan, M.C.; Lopes Barth, A.; Cardoso, L., Jr.; Embiruçu de Araújo Leão, E.K.; Esposito, A.C.; et al. Alternative laronidase dose regimen for patients with mucopolysaccharidosis I: A multinational, retrospective, chart review case series. Orphanet J. Rare Dis. 2016, 11, 51. [Google Scholar] [CrossRef][Green Version]
- Laraway, S.; Mercer, J.; Jameson, E.; Ashworth, J.; Hensman, P.; Jones, S.A. Outcomes of Long-Term Treatment with Laronidase in Patients with Mucopolysaccharidosis Type I. J. Pediatr. 2016, 178, 219–226.e1. [Google Scholar] [CrossRef]
- Bolourchi, M.; Renella, P.; Wang, R.Y. Aortic Root Dilatation in Mucopolysaccharidosis I-VII. Int. J. Mol. Sci. 2016, 17, 2004. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.R.; Roberts, W.C. Outcome of Combined Mitral and Aortic Valve Replacement in Adults with Mucopolysaccharidosis (the Hurler Syndrome). Am. J. Cardiol. 2017, 120, 2113–2118. [Google Scholar] [CrossRef] [PubMed]
- Kiely, B.T.; Kohler, J.L.; Coletti, H.Y.; Poe, M.D.; Escolar, M.L. Early disease progression of Hurler syndrome. Orphanet J. Rare Dis. 2017, 12, 32. [Google Scholar] [CrossRef]
- Lum, S.H.; Stepien, K.M.; Ghosh, A.; Broomfield, A.; Church, H.; Mercer, J.; Jones, S.; Wynn, R. Long term survival and cardiopulmonary outcome in children with Hurler syndrome after haematopoietic stem cell transplantation. J. Inherit. Metab. Dis. 2017, 40, 455–460. [Google Scholar] [CrossRef]
- Andrade, M.F.A.; Guimarães, I.C.B.; Acosta, A.X.; Leão, E.; Moreira, M.I.G.; Mendes, C.M.C. Left ventricular assessment in patients with mucopolysaccharidosis using conventional echocardiography and myocardial deformation by two-dimensional speckle-tracking method. J. Pediatr. 2019, 95, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Braunlin, E.; Miettunen, K.; Lund, T.; Luquette, M.; Orchard, P. Hematopoietic cell transplantation for severe MPS I in the first six months of life: The heart of the matter. Mol. Genet. Metab. 2019, 126, 117–120. [Google Scholar] [CrossRef]
- Yamazaki, N.; Kosuga, M.; Kida, K.; Takei, G.; Fukuhara, Y.; Matsumoto, H.; Senda, M.; Honda, A.; Ishiguro, A.; Koike, T.; et al. Early enzyme replacement therapy enables a successful hematopoietic stem cell transplantation in mucopolysaccharidosis type IH: Divergent clinical outcomes in two Japanese siblings. Brain Dev. 2019, 41, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Moghadam, S.H.; Ghahvechi, M.; Mozafari, F.; Sayarifard, F.; Mousavi, M.S.; Rostami, R.; Ziaee, V. Mucopolysaccharidosis Type I in Children, a Forgotten Diagnosis Responsible for Undiagnosed Musculoskeletal Complaints: Report of Two Cases. Acta Med. 2019, 62, 161–165. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Encarnacion, C.O.; Hang, D.; Earing, M.; Mitchell, M.E. Mucopolysaccharidoses Causing Valvular Heart Disease: Report and Review of Surgical Management. World J. Pediatr. Congenit. Heart Surg. 2020, 11, Np22–Np24. [Google Scholar] [CrossRef]
- Zhou, Y.A.; Li, P.; Zhang, Y.; Xiong, Q.; Li, C.; Zhao, Z.; Wang, Y.; Xiao, H. Identification of a novel compound heterozygous IDUA mutation underlies Mucopolysaccharidoses type I in a Chinese pedigree. Mol. Genet. Genom. Med. 2020, 8, e1058. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, K.; Kubo, T.; Ochi, Y.; Baba, Y.; Hirota, T.; Yamasaki, N.; Kitaoka, H. Cardiac manifestations and effects of enzyme replacement therapy for over 10 years in adults with the attenuated form of mucopolysaccharidosis type I. Mol. Genet. Metab. Rep. 2020, 25, 100662. [Google Scholar] [CrossRef] [PubMed]
- Sadeghian, H.; Sadeghian, A.; Eslami, B.; Abbasi, S.H.; Lotfi-Tokaldany, M. Combined Aortic and Mitral Valve Stenosis in Mucopolysaccharidosis Syndrome Type I-S: A Report of a Rare Case. J. Tehran Heart Cent. 2021, 16, 31–33. [Google Scholar] [CrossRef]
- Wraith, J.E.; Scarpa, M.; Beck, M.; Bodamer, O.A.; De Meirleir, L.; Guffon, N.; Meldgaard Lund, A.; Malm, G.; Van der Ploeg, A.T.; Zeman, J. Mucopolysaccharidosis type II (Hunter syndrome): A clinical review and recommendations for treatment in the era of enzyme replacement therapy. Eur. J. Pediatr. 2008, 167, 267–277. [Google Scholar] [CrossRef]
- Lin, H.Y.; Chuang, C.K.; Huang, Y.H.; Tu, R.Y.; Lin, F.J.; Lin, S.J.; Chiu, P.C.; Niu, D.M.; Tsai, F.J.; Hwu, W.L.; et al. Causes of death and clinical characteristics of 34 patients with Mucopolysaccharidosis II in Taiwan from 1995–2012. Orphanet J. Rare Dis. 2016, 11, 85. [Google Scholar] [CrossRef]
- Kurihara, M.; Kumagai, K.; Goto, K.; Imai, M.; Yagishita, S. Severe type Hunter′s syndrome. Polysomnographic and neuropathological study. Neuropediatrics 1992, 23, 248–256. [Google Scholar] [CrossRef]
- Kettles, D.I.; Sheppard, M.; Liebmann, R.D.; Davidson, C. Left ventricular aneurysm, aortic valve disease and coronary narrowing in a patient with Hunter′s syndrome. Cardiovasc. Pathol. Off. J. Soc. Cardiovasc. Pathol. 2002, 11, 94–96. [Google Scholar] [CrossRef]
- Mohan, U.R.; Hay, A.A.; Cleary, M.A.; Wraith, J.E.; Patel, R.G. Cardiovascular changes in children with mucopolysaccharide disorders. Acta Paediatr. 2002, 91, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, K.; Gibson, S.C.; Pathi, V.L. Mitral valve replacement for mitral stenosis secondary to Hunter′s syndrome. Ann. Thorac. Surg. 2005, 80, 1911–1912. [Google Scholar] [CrossRef]
- Pinto, L.L.; Schwartz, I.V.; Puga, A.C.; Vieira, T.A.; Munoz, M.V.; Giugliani, R. Prospective study of 11 Brazilian patients with mucopolysaccharidosis II. J. Pediatr. 2006, 82, 273–278. [Google Scholar] [CrossRef][Green Version]
- Neely, J.; Carpenter, J.; Hsu, W.; Jordan, L.; Restrepo, L. Cerebral infarction in Hunter syndrome. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2006, 13, 1054–1057. [Google Scholar] [CrossRef]
- Guffon, N.; Bertrand, Y.; Forest, I.; Fouilhoux, A.; Froissart, R. Bone marrow transplantation in children with Hunter syndrome: Outcome after 7 to 17 years. J. Pediatr. 2009, 154, 733–737. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, T.; Kirvassilis, G.; Tsourelis, L.; Ieromonachos, C.; Zarkalis, D.; Alivizatos, P. Mitral valve replacement and Hunter syndrome: Case report. Heart Surg. Forum 2009, 12, E54–E56. [Google Scholar] [CrossRef]
- Okuyama, T.; Tanaka, A.; Suzuki, Y.; Ida, H.; Tanaka, T.; Cox, G.F.; Eto, Y.; Orii, T. Japan Elaprase Treatment (JET) study: Idursulfase enzyme replacement therapy in adult patients with attenuated Hunter syndrome (Mucopolysaccharidosis II, MPS II). Mol. Genet. Metab. 2010, 99, 18–25. [Google Scholar] [CrossRef]
- Kampmann, C.; Beck, M.; Morin, I.; Loehr, J.P. Prevalence and characterization of cardiac involvement in Hunter syndrome. J. Pediatr. 2011, 159, 327–331.e322. [Google Scholar] [CrossRef]
- Wang, R.Y.; Covault, K.K.; Halcrow, E.M.; Gardner, A.J.; Cao, X.; Newcomb, R.L.; Dauben, R.D.; Chang, A.C. Carotid intima-media thickness is increased in patients with mucopolysaccharidoses. Mol. Genet. Metab. 2011, 104, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Sohn, Y.B.; Choi, E.W.; Kim, S.J.; Park, S.W.; Kim, S.H.; Cho, S.Y.; Jeong, S.I.; Huh, J.; Kang, I.S.; Lee, H.J.; et al. Retrospective analysis of the clinical manifestations and survival of Korean patients with mucopolysaccharidosis type II: Emphasis on the cardiovascular complication and mortality cases. Am. J. Med. Genet. Part A 2012, 158, 90–96. [Google Scholar] [CrossRef]
- Grinberg, H.; Quaio, C.R.; Avila, M.S.; Ferreira, S.M.; Vieira, M.L.; Benvenuti, L.A.; Kim, C.A.; Bocchi, E.A. The first cardiac transplant experience in a patient with mucopolysaccharidosis. Cardiovasc. Pathol. Off. J. Soc. Cardiovasc. Pathol. 2012, 21, 358–360. [Google Scholar] [CrossRef]
- Uluganyan, M.; Velibey, Y.; Karaca, G.; Güngör, B. Echocardiographic demonstration of isolated mitral valve involvement in a patient with mucopolysaccharidosis. Turk Kardiyol. Dern. Ars. Turk Kardiyol. Dern. Yayin. Organidir 2012, 40, 199. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tanaka, A.; Okuyama, T.; Suzuki, Y.; Sakai, N.; Takakura, H.; Sawada, T.; Tanaka, T.; Otomo, T.; Ohashi, T.; Ishige-Wada, M.; et al. Long-term efficacy of hematopoietic stem cell transplantation on brain involvement in patients with mucopolysaccharidosis type II: A nationwide survey in Japan. Mol. Genet. Metab. 2012, 107, 513–520. [Google Scholar] [CrossRef]
- Quaio, C.R.; Grinberg, H.; Vieira, M.L.; Paula, A.C.; Leal, G.N.; Gomy, I.; Leistner-Segal, S.; Giugliani, R.; Bertola, D.R.; Kim, C.A. Report of a Large Brazilian Family with a Very Attenuated Form of Hunter Syndrome (MPS II). JIMD Rep. 2012, 4, 125–128. [Google Scholar] [CrossRef] [PubMed]
- Brands, M.M.; Frohn-Mulder, I.M.; Hagemans, M.L.; Hop, W.C.; Oussoren, E.; Helbing, W.A.; van der Ploeg, A.T. Mucopolysaccharidosis: Cardiologic features and effects of enzyme-replacement therapy in 24 children with MPS I, II and VI. J. Inherit. Metab. Dis. 2013, 36, 227–234. [Google Scholar] [CrossRef]
- Tajima, G.; Sakura, N.; Kosuga, M.; Okuyama, T.; Kobayashi, M. Effects of idursulfase enzyme replacement therapy for Mucopolysaccharidosis type II when started in early infancy: Comparison in two siblings. Mol. Genet. Metab. 2013, 108, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Fujiwara, M.; Kobayashi, H.; Ida, H. Massive accumulation of glycosaminoglycans in the aortic valve of a patient with Hunter syndrome during enzyme replacement therapy. Pediatr. Cardiol. 2013, 34, 2077–2079. [Google Scholar] [CrossRef] [PubMed]
- Lampe, C.; Bosserhoff, A.K.; Burton, B.K.; Giugliani, R.; de Souza, C.F.; Bittar, C.; Muschol, N.; Olson, R.; Mendelsohn, N.J. Long-term experience with enzyme replacement therapy (ERT) in MPS II patients with a severe phenotype: An international case series. J. Inherit. Metab. Dis. 2014, 37, 823–829. [Google Scholar] [CrossRef]
- Lin, S.M.; Lin, H.Y.; Chuang, C.K.; Lin, S.P.; Chen, M.R. Cardiovascular abnormalities in Taiwanese patients with mucopolysaccharidosis. Mol. Genet. Metab. 2014, 111, 493–498. [Google Scholar] [CrossRef]
- Tomanin, R.; Zanetti, A.; D′Avanzo, F.; Rampazzo, A.; Gasparotto, N.; Parini, R.; Pascarella, A.; Concolino, D.; Procopio, E.; Fiumara, A.; et al. Clinical efficacy of enzyme replacement therapy in paediatric Hunter patients, an independent study of 3.5 years. Orphanet J. Rare Dis. 2014, 9, 129. [Google Scholar] [CrossRef]
- Madireddi, J.; Sarada, P.; Shetty, R.K.; Prabhu, M.; Girish, K.M. Hunter syndrome with its typical heart: A close mimic to rheumatic heart. BMJ Case Rep. 2015, 2015, bcr2015209359. [Google Scholar] [CrossRef]
- Dalmau Serra, J.; Vitoria Miñana, I.; Calderón Fernández, R.; Cortell Aznar, I. Clinical response to long term enzyme replacement treatment in children, adolescent and adult patients with Hunter syndrome. Med. Clin. 2015, 145, 392–398. [Google Scholar] [CrossRef]
- Parini, R.; Rigoldi, M.; Tedesco, L.; Boffi, L.; Brambilla, A.; Bertoletti, S.; Boncimino, A.; Del Longo, A.; De Lorenzo, P.; Gaini, R.; et al. Enzymatic replacement therapy for Hunter disease: Up to 9 years experience with 17 patients. Mol. Genet. Metab. Rep. 2015, 3, 65–74. [Google Scholar] [CrossRef]
- Lin, H.Y.; Chuang, C.K.; Chen, M.R.; Lin, S.M.; Hung, C.L.; Chang, C.Y.; Chiu, P.C.; Tsai, W.H.; Niu, D.M.; Tsai, F.J.; et al. Cardiac structure and function and effects of enzyme replacement therapy in patients with mucopolysaccharidoses I, II, IVA and VI. Mol. Genet. Metab. 2016, 117, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Alkhzouz, C.; Lazea, C.; Bucerzan, S.; Nascu, I.; Kiss, E.; Denes, C.L.; Grigorescu-Sido, P. Clinical and Genetic Characteristics of Romanian Patients with Mucopolysaccharidosis Type II. JIMD Rep. 2017, 33, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Bounds, R.L.; Kuebler, J.; Cholette, J.M.; Alfieris, G.M.; Emani, S.M.; Wittlieb-Weber, C.A. Left Main Coronary Artery Atresia in an Infant with Inclusion-Cell Disease. World J. Pediatr. Congenit. Heart Surg. 2018, 9, 246–250. [Google Scholar] [CrossRef]
- Chlebowski, M.M.; Heese, B.A.; Malloy-Walton, L.E. Early childhood onset of high-grade atrioventricular block in Hunter syndrome. Cardiol. Young 2018, 28, 786–787. [Google Scholar] [CrossRef]
- Suzuki, K.; Sakai, H.; Takahashi, K. Perioperative airway management for aortic valve replacement in an adult with mucopolysaccharidosis type II (Hunter syndrome). JA Clin. Rep. 2018, 4, 24. [Google Scholar] [CrossRef]
- Selvanathan, A.; Ellaway, C.; Wilson, C.; Owens, P.; Shaw, P.J.; Bhattacharya, K. Effectiveness of Early Hematopoietic Stem Cell Transplantation in Preventing Neurocognitive Decline in Mucopolysaccharidosis Type II: A Case Series. JIMD Rep. 2018, 41, 81–89. [Google Scholar] [CrossRef]
- Curran, L.; Davison, J.; Shaughnessy, L.; Shore, D.; Franklin, R.C. Visual Loss Post Ross Procedure in an Adolescent with Newly Diagnosed Mucopolysaccharidosis Type II. Ann. Thorac. Surg. 2019, 108, e297–e299. [Google Scholar] [CrossRef]
- Broomfield, A.; Davison, J.; Roberts, J.; Stewart, C.; Hensman, P.; Beesley, C.; Tylee, K.; Rust, S.; Schwahn, B.; Jameson, E.; et al. Ten years of enzyme replacement therapy in paediatric onset mucopolysaccharidosis II in England. Mol. Genet. Metab. 2020, 129, 98–105. [Google Scholar] [CrossRef]
- Do Valle, D.A.; Cirino, R.H.D.; Santos, M.; Pellissari, E.C.; Scola, R.H. Enzyme Replacement Therapy Decreases Left Ventricular Mass Index in Patients with Hunter Syndrome? Pediatr. Cardiol. 2020, 41, 361–365. [Google Scholar] [CrossRef]
- Suzuki, Y.; Taylor, M.; Orii, K.; Fukao, T.; Orii, T.; Tomatsu, S. Assessment of Activity of Daily Life in Mucopolysaccharidosis Type II Patients with Hematopoietic Stem Cell Transplantation. Diagnostics 2020, 10, 46. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Chuang, C.K.; Lee, C.L.; Chen, M.R.; Sung, K.T.; Lin, S.M.; Hou, C.J.; Niu, D.M.; Chang, T.M.; Hung, C.L.; et al. Cardiac Evaluation using Two-Dimensional Speckle-Tracking Echocardiography and Conventional Echocardiography in Taiwanese Patients with Mucopolysaccharidoses. Diagnostics 2020, 10, 62. [Google Scholar] [CrossRef] [PubMed]
- Ayuna, A.; Stepien, K.M.; Hendriksz, C.J.; Balerdi, M.; Garg, A.; Woolfson, P. Cardiac rhythm abnormalities–An underestimated cardiovascular risk in adult patients with Mucopolysaccharidoses. Mol. Genet. Metab. 2020, 130, 133–139. [Google Scholar] [CrossRef]
- Lin, H.Y.; Chen, M.R.; Lee, C.L.; Lin, S.M.; Hung, C.L.; Niu, D.M.; Chang, T.M.; Chuang, C.K.; Lin, S.P. Aortic Root Dilatation in Taiwanese Patients with Mucopolysaccharidoses and the Long-Term Effects of Enzyme Replacement Therapy. Diagnostics 2020, 11, 16. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Chen, M.R.; Lee, C.L.; Lin, S.M.; Hung, C.L.; Niu, D.M.; Chang, T.M.; Chuang, C.K.; Lin, S.P. Natural progression of cardiac features and long-term effects of enzyme replacement therapy in Taiwanese patients with mucopolysaccharidosis II. Orphanet J. Rare Dis. 2021, 16, 99. [Google Scholar] [CrossRef] [PubMed]
- Poitier, B.; Amrane, M.; Bruneval, P.; Achouh, P. Surgical management of an aortic root dilatation in a patient suffering from Hunter syndrome. Interact. Cardiovasc. Thorac. Surg. 2021, 33, 819–821. [Google Scholar] [CrossRef] [PubMed]
- Racoma, M.J.C.; Calibag, M.; Cordero, C.P.; Abacan, M.A.R.; Chiong, M.A.D. A review of the clinical outcomes in idursulfase-treated and untreated Filipino patients with mucopolysaccharidosis type II: Data from the local lysosomal storage disease registry. Orphanet J. Rare Dis. 2021, 16, 323. [Google Scholar] [CrossRef]
- Mori, N.; Kitahara, H.; Muramatsu, T.; Matsuura, K.; Nakayama, T.; Matsumiya, G.; Kobayashi, Y. Transcatheter aortic valve implantation for severe aortic stenosis in a patient with mucopolysaccharidosis type II (Hunter syndrome) accompanied by severe airway obstruction. J. Cardiol. Cases 2022, 25, 49–51. [Google Scholar] [CrossRef]
- Sestito, S.; Rinninella, G.; Rampazzo, A.; D′Avanzo, F.; Zampini, L.; Santoro, L.; Gabrielli, O.; Fiumara, A.; Barone, R.; Volpi, N.; et al. Cardiac involvement in MPS patients: Incidence and response to therapy in an Italian multicentre study. Orphanet J. Rare Dis. 2022, 17, 251. [Google Scholar] [CrossRef]
- Stephan, B.O.; Quaio, C.R.; Spolador, G.M.; de Paula, A.C.; Curiati, M.A.; Martins, A.M.; Leal, G.N.; Tenorio, A.; Finzi, S.; Chimelo, F.T.; et al. Impact of ERT and follow-up of 17 patients from the same family with a mild form of MPS II. Clinics 2022, 77, 100082. [Google Scholar] [CrossRef]
- Zhang, Z.; Ma, M.; Zhang, W.; Zhou, Y.; Yao, F.; Zhu, L.; Wei, M.; Qiu, Z. Phenotypic and genetic characteristics of 130 patients with mucopolysaccharidosis type II: A single-center retrospective study in China. Front. Genet. 2023, 14, 1103620. [Google Scholar] [CrossRef]
- Stapleton, M.; Kubaski, F.; Mason, R.W.; Yabe, H.; Suzuki, Y.; Orii, K.E.; Orii, T.; Tomatsu, S. Presentation and Treatments for Mucopolysaccharidosis Type II (MPS II.; Hunter Syndrome). Expert Opin. Orphan Drugs 2017, 5, 295–307. [Google Scholar] [CrossRef]
- Cohen, M.A.; Stuart, G.M. Delivery of anesthesia for children with Mucopolysaccharidosis Type III (Sanfilippo syndrome): A review of 86 anesthetics. Paediatr. Anaesth. 2017, 27, 363–369. [Google Scholar] [CrossRef]
- Zelei, T.; Csetneki, K.; Vokó, Z.; Siffel, C. Epidemiology of Sanfilippo syndrome: Results of a systematic literature review. Orphanet J. Rare Dis. 2018, 13, 53. [Google Scholar] [CrossRef]
- Seker Yilmaz, B.; Davison, J.; Jones, S.A.; Baruteau, J. Novel therapies for mucopolysaccharidosis type III. J. Inherit. Metab. Dis. 2021, 44, 129–147. [Google Scholar] [CrossRef]
- Lavery, C.; Hendriksz, C.J.; Jones, S.A. Mortality in patients with Sanfilippo syndrome. Orphanet J. Rare Dis. 2017, 12, 168. [Google Scholar] [CrossRef]
- Mueller, P.; Attenhofer Jost, C.H.; Rohrbach, M.; Valsangiacomo Buechel, E.R.; Seifert, B.; Balmer, C.; Kretschmar, O.; Baumgartner, M.R.; Weber, R. Cardiac disease in children and young adults with various lysosomal storage diseases: Comparison of echocardiographic and ECG changes among clinical groups. Int. J. Cardiol. Heart Vessel. 2013, 2, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, C.M.; Truxal, K.V.; McBride, K.L.; Kovalchin, J.P.; Flanigan, K.M. Natural history of echocardiographic abnormalities in mucopolysaccharidosis III. Mol. Genet. Metab. 2018, 124, 131–134. [Google Scholar] [CrossRef]
- Nijmeijer, S.C.M.; de Bruin-Bon, R.; Wijburg, F.A.; Kuipers, I.M. Cardiac disease in mucopolysaccharidosis type III. J. Inherit. Metab. Dis. 2019, 42, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Chen, M.R.; Lin, S.M.; Hung, C.L.; Niu, D.M.; Chang, T.M.; Chuang, C.K.; Lin, S.P. Cardiac characteristics and natural progression in Taiwanese patients with mucopolysaccharidosis III. Orphanet J. Rare Dis. 2019, 14, 140. [Google Scholar] [CrossRef] [PubMed]
- Cirino Ballantyne, M.; Chiu, B.; Sergi, C.M. Sanfilippo syndrome type A: Early cardiac involvement of two patients with cardiac manifestations. Cardiovasc. Pathol. Off. J. Soc. Cardiovasc. Pathol. 2022, 60, 107430. [Google Scholar] [CrossRef] [PubMed]
- Altin, H.; Dorum, S.; Ture, E. Tissue doppler echocardiographic evaluation of cardiac functions in children with mucopolysaccharidosis type III disease. Niger. J. Clin. Pract. 2022, 25, 1717–1724. [Google Scholar]
- Shawky, R.M.; Abd el-Monim, M.T.; el-Sebai, A.A.; el-Sayed, S.M. Cardiac and ocular manifestations in Egyptian patients with mucopolysaccharidoses. East. Mediterr. Health J. Rev. Sante Mediterr. Orient. Al-Majallah Al-Sihhiyah Li-Sharq Al-Mutawassit 2001, 7, 981–991. [Google Scholar] [CrossRef]
- Zhao, H.G.; Li, H.H.; Bach, G.; Schmidtchen, A.; Neufeld, E.F. The molecular basis of Sanfilippo syndrome type B. Proc. Natl. Acad. Sci. USA 1996, 93, 6101–6105. [Google Scholar] [CrossRef] [PubMed]
- Muenzer, J.; Beekman, R.H.; Profera, L.M.; Bove, E.L. Severe mitral insufficiency in mucopolysaccharidosis type III-B (Sanfilippo syndrome). Pediatr. Cardiol. 1993, 14, 130–132. [Google Scholar] [CrossRef] [PubMed]
- Kourouklis, S.; Chatzis, D.; Skafida, M.; Liagkas, K.; Paradellis, G.; Kyriakides, Z. Outlet type of interventricular septal defect in SanFilippo type-B syndrome. Int. J. Cardiol. 2007, 122, e4–e5. [Google Scholar] [CrossRef]
- Moog, U.; van Mierlo, I.; van Schrojenstein Lantman-de Valk, H.M.; Spaapen, L.; Maaskant, M.A.; Curfs, L.M. Is Sanfilippo type B in your mind when you see adults with mental retardation and behavioral problems? Am. J. Med. Genet. Part C Semin. Med. Genet. 2007, 145, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Ruijter, G.J.; Valstar, M.J.; van de Kamp, J.M.; van der Helm, R.M.; Durand, S.; van Diggelen, O.P.; Wevers, R.A.; Poorthuis, B.J.; Pshezhetsky, A.V.; Wijburg, F.A. Clinical and genetic spectrum of Sanfilippo type C (MPS IIIC) disease in The Netherlands. Mol. Genet. Metab. 2008, 93, 104–111. [Google Scholar] [CrossRef]
- Fedele, A.O. Sanfilippo syndrome: Causes, consequences, and treatments. Appl. Clin. Genet. 2015, 8, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, M.; Kumagai, K.; Yagishita, S. Sanfilippo syndrome type C: A clinicopathological autopsy study of a long-term survivor. Pediatr. Neurol. 1996, 14, 317–321. [Google Scholar] [CrossRef]
- Kampmann, C.; Abu-Tair, T.; Gökce, S.; Lampe, C.; Reinke, J.; Mengel, E.; Hennermann, J.B.; Wiethoff, C.M. Heart and Cardiovascular Involvement in Patients with Mucopolysaccharidosis Type IVA (Morquio-A Syndrome). PLoS ONE 2016, 11, e0162612. [Google Scholar] [CrossRef]
- Ireland, M.A.; Rowlands, D.B. Mucopolysaccharidosis type IV as a cause of mitral stenosis in an adult. Br. Heart J. 1981, 46, 113–115. [Google Scholar] [CrossRef]
- John, R.M.; Hunter, D.; Swanton, R.H. Echocardiographic abnormalities in type IV mucopolysaccharidosis. Arch. Dis. Child. 1990, 65, 746–749. [Google Scholar] [CrossRef]
- Nicolini, F.; Corradi, D.; Bosio, S.; Gherli, T. Aortic valve replacement in a patient with morquio syndrome. Heart Surg. Forum 2008, 11, E96–E98. [Google Scholar] [CrossRef]
- Pagel, P.S.; Almassi, G.H. Perioperative implications of Morquio syndrome in a 31-year-old woman undergoing aortic valve replacement. J. Cardiothorac. Vasc. Anesth. 2009, 23, 855–857. [Google Scholar] [CrossRef]
- Politei, J.; Porras-Hurtado, G.L.; Guelbert, N.; Fainboim, A.; Horovitz, D.D.G.; Satizábal, J.M. Enzyme replacement therapy interruption in mucopolysaccharidosis type IVA patients and its impact in different clinical outcomes. JIMD Rep. 2021, 58, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Golda, A.; Jurecka, A.; Tylki-Szymanska, A. Cardiovascular manifestations of mucopolysaccharidosis type VI (Maroteaux-Lamy syndrome). Int. J. Cardiol. 2012, 158, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Aminzadeh, M.; Malekpour, N.; Ghandil, P. Identification of arylsulfatase B gene mutations and clinical presentations of Iranian patients with Mucopolysaccharidosis VI. Gene 2019, 706, 1–5. [Google Scholar] [CrossRef]
- Jurecka, A.; Zakharova, E.; Cimbalistiene, L.; Gusina, N.; Kulpanovich, A.; Golda, A.; Opoka-Winiarska, V.; Piotrowska, E.; Voskoboeva, E.; Tylki-Szymańska, A. Mucopolysaccharidosis type VI: A predominantly cardiac phenotype associated with homozygosity for p.R152W mutation in the ARSB gene. Am. J. Med. Genet. Part A 2013, 161, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Marwick, T.H.; Bastian, B.; Hughes, C.F.; Bailey, B.P. Mitral stenosis in the Maroteaux-Lamy syndrome: A treatable cause of dyspnoea. Postgrad. Med. J. 1992, 68, 287–288. [Google Scholar] [CrossRef][Green Version]
- Tan, C.T.; Schaff, H.V.; Miller, F.A., Jr.; Edwards, W.D.; Karnes, P.S. Valvular heart disease in four patients with Maroteaux-Lamy syndrome. Circulation 1992, 85, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Herskhovitz, E.; Young, E.; Rainer, J.; Hall, C.M.; Lidchi, V.; Chong, K.; Vellodi, A. Bone marrow transplantation for Maroteaux-Lamy syndrome (MPS VI): Long-term follow-up. J. Inherit. Metab. Dis. 1999, 22, 50–62. [Google Scholar] [CrossRef]
- Dilber, E.; Celiker, A.; Karagöz, T.; Kalkanoğlu, H.S. Permanent transfemoral pacemaker implantation in a child with Maroteaux Lamy syndrome. Pacing Clin. Electrophysiol. PACE 2002, 25, 1784–1785. [Google Scholar] [CrossRef] [PubMed]
- Oudit, G.Y.; Butany, J.; Williams, W.G.; Siu, S.C.; Clarke, J.T.; Iwanochko, R.M. Left ventricular aneurysm in a patient with mucopolysaccharidosis type VI (Maroteaux-Lamy syndrome): Clinical and pathological correlation. Cardiovasc. Pathol. Off. J. Soc. Cardiovasc. Pathol. 2007, 16, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, M.; Barone, R.; Fiumara, A.; Astarita, L.; Parenti, G.; Rampazzo, A.; Sala, S.; Sorge, G.; Parini, R. Mucopolysaccharidosis VI: The Italian experience. Eur. J. Pediatr. 2009, 168, 1203–1206. [Google Scholar] [CrossRef]
- Scarpa, M.; Buffone, E.; Marca, P.L.; Campello, M.; Rampazzo, A. Difficulties in diagnosing slowly progressive mucopolysaccharidosis VI: A case series. J. Pediatr. Rehabil. Med. 2010, 3, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Jurecka, A.; Golda, A.; Opoka-Winiarska, V.; Piotrowska, E.; Tylki-Szymańska, A. Mucopolysaccharidosis type VI (Maroteaux-Lamy syndrome) with a predominantly cardiac phenotype. Mol. Genet. Metab. 2011, 104, 695–699. [Google Scholar] [CrossRef]
- Furujo, M.; Kubo, T.; Kosuga, M.; Okuyama, T. Enzyme replacement therapy attenuates disease progression in two Japanese siblings with mucopolysaccharidosis type VI. Mol. Genet. Metab. 2011, 104, 597–602. [Google Scholar] [CrossRef]
- Hendriksz, C.J.; Giugliani, R.; Harmatz, P.; Lampe, C.; Martins, A.M.; Pastores, G.M.; Steiner, R.D.; Leão Teles, E.; Valayannopoulos, V. Design, baseline characteristics, and early findings of the MPS VI (mucopolysaccharidosis VI) Clinical Surveillance Program (CSP). J. Inherit. Metab. Dis. 2013, 36, 373–384. [Google Scholar] [CrossRef]
- Braunlin, E.; Rosenfeld, H.; Kampmann, C.; Johnson, J.; Beck, M.; Giugliani, R.; Guffon, N.; Ketteridge, D.; CM, S.M.; Scarpa, M.; et al. Enzyme replacement therapy for mucopolysaccharidosis VI: Long-term cardiac effects of galsulfase (Naglazyme®) therapy. J. Inherit. Metab. Dis. 2013, 36, 385–394. [Google Scholar] [CrossRef]
- Yano, S.; Li, C.; Pavlova, Z. The transforming growth factor-Beta signaling pathway involvement in cardiovascular lesions in mucopolysaccharidosis-I. JIMD Rep. 2013, 7, 55–58. [Google Scholar] [CrossRef]
- Brands, M.M.; Oussoren, E.; Ruijter, G.J.; Vollebregt, A.A.; van den Hout, H.M.; Joosten, K.F.; Hop, W.C.; Plug, I.; van der Ploeg, A.T. Up to five years experience with 11 mucopolysaccharidosis type VI patients. Mol. Genet. Metab. 2013, 109, 70–76. [Google Scholar] [CrossRef]
- Horovitz, D.D.; Magalhães, T.S.; Acosta, A.; Ribeiro, E.M.; Giuliani, L.R.; Palhares, D.B.; Kim, C.A.; de Paula, A.C.; Kerstenestzy, M.; Pianovski, M.A.; et al. Enzyme replacement therapy with galsulfase in 34 children younger than five years of age with MPS VI. Mol. Genet. Metab. 2013, 109, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Leal, G.N.; de Paula, A.C.; Morhy, S.S.; Andrade, J.L.; Kim, C.A. Advantages of early replacement therapy for mucopolysaccharidosis type VI: Echocardiographic follow-up of siblings. Cardiol. Young 2014, 24, 229–235. [Google Scholar] [CrossRef]
- Kampmann, C.; Lampe, C.; Whybra-Trümpler, C.; Wiethoff, C.M.; Mengel, E.; Arash, L.; Beck, M.; Miebach, E. Mucopolysaccharidosis VI: Cardiac involvement and the impact of enzyme replacement therapy. J. Inherit. Metab. Dis. 2014, 37, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Giugliani, R.; Lampe, C.; Guffon, N.; Ketteridge, D.; Leão-Teles, E.; Wraith, J.E.; Jones, S.A.; Piscia-Nichols, C.; Lin, P.; Quartel, A.; et al. Natural history and galsulfase treatment in mucopolysaccharidosis VI (MPS VI, Maroteaux-Lamy syndrome)—10-year follow-up of patients who previously participated in an MPS VI Survey Study. Am. J. Med. Genet. Part A 2014, 164, 1953–1964. [Google Scholar] [CrossRef]
- Choy, Y.S.; Bhattacharya, K.; Balasubramaniam, S.; Fietz, M.; Fu, A.; Inwood, A.; Jin, D.K.; Kim, O.H.; Kosuga, M.; Kwun, Y.H.; et al. Identifying the need for a multidisciplinary approach for early recognition of mucopolysaccharidosis VI (MPS VI). Mol. Genet. Metab. 2015, 115, 41–47. [Google Scholar] [CrossRef]
- Franco, J.F.; Soares, D.C.; Torres, L.C.; Leal, G.N.; Cunha, M.T.; Honjo, R.S.; Bertola, D.R.; Kim, C.A. Short Communication Impact of early enzyme-replacement therapy for mucopolysaccharidosis VI: Results of a long-term follow-up of Brazilian siblings. Genet. Mol. Res. GMR 2016, 15. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Chuang, C.K.; Wang, C.H.; Chien, Y.H.; Wang, Y.M.; Tsai, F.J.; Chou, Y.Y.; Lin, S.J.; Pan, H.P.; Niu, D.M.; et al. Long-term galsulfase enzyme replacement therapy in Taiwanese mucopolysaccharidosis VI patients: A case series. Mol. Genet. Metab. Rep. 2016, 7, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Torre, S.; Scarpelli, M.; Salviati, A.; Buffone, E.; Faggian, G.; Luciani, G.B. Aortic and Mitral Valve Involvement in Maroteaux-Lamy Syndrome VI: Surgical Implications in the Enzyme Replacement Therapy Era. Ann. Thorac. Surg. 2016, 102, e23–e25. [Google Scholar] [CrossRef][Green Version]
- Kılıç, M.; Dursun, A.; Coşkun, T.; Tokatlı, A.; Özgül, R.K.; Yücel-Yılmaz, D.; Karaca, M.; Doğru, D.; Alehan, D.; Kadayıfçılar, S.; et al. Genotypic-phenotypic features and enzyme replacement therapy outcome in patients with mucopolysaccharidosis VI from Turkey. Am. J. Med. Genet. Part A 2017, 173, 2954–2967. [Google Scholar] [CrossRef]
- Furujo, M.; Kosuga, M.; Okuyama, T. Enzyme replacement therapy attenuates disease progression in two Japanese siblings with mucopolysaccharidosis type VI: 10-Year follow up. Mol. Genet. Metab. Rep. 2017, 13, 69–75. [Google Scholar] [CrossRef]
- Bell, D.J.; He, C.; Pauli, J.L.; Naidoo, R. Maroteaux-Lamy syndrome: A rare and challenging case of mitral valve replacement. Asian Cardiovasc. Thorac. Ann. 2018, 26, 560–562. [Google Scholar] [CrossRef]
- Lin, H.Y.; Lee, C.L.; Lo, Y.T.; Wang, T.J.; Huang, S.F.; Chen, T.L.; Wang, Y.S.; Niu, D.M.; Chuang, C.K.; Lin, S.P. The relationships between urinary glycosaminoglycan levels and phenotypes of mucopolysaccharidoses. Mol. Genet. Genom. Med. 2018, 6, 982–992. [Google Scholar] [CrossRef] [PubMed]
- Harmatz, P.R.; Lampe, C.; Parini, R.; Sharma, R.; Teles, E.L.; Johnson, J.; Sivam, D.; Sisic, Z. Enzyme replacement therapy outcomes across the disease spectrum: Findings from the mucopolysaccharidosis VI Clinical Surveillance Program. J. Inherit. Metab. Dis. 2019, 42, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Lampe, C.; Harmatz, P.R.; Parini, R.; Sharma, R.; Teles, E.L.; Johnson, J.; Sivam, D.; Sisic, Z. Enzyme replacement therapy initiated in adulthood: Findings from the mucopolysaccharidosis VI Clinical Surveillance Program. Mol. Genet. Metab. 2019, 127, 355–360. [Google Scholar] [CrossRef]
- Honjo, R.S.; Vaca, E.C.N.; Leal, G.N.; Abellan, D.M.; Ikari, N.M.; Jatene, M.B.; Martins, A.M.; Kim, C.A. Mucopolysaccharidosis type VI: Case report with first neonatal presentation with ascites fetalis and rapidly progressive cardiac manifestation. BMC Med. Genet. 2020, 21, 37. [Google Scholar] [CrossRef]
- Horovitz, D.D.G.; Leão, E.; Ribeiro, E.M.; Martins, A.M.; Barth, A.L.; Neri, J.; Kerstenetzky, M.; Siqueira, A.C.M.; Ribeiro, B.F.R.; Kim, C.A.; et al. Long-term impact of early initiation of enzyme replacement therapy in 34 MPS VI patients: A resurvey study. Mol. Genet. Metab. 2021, 133, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Garcia, P.; Phillips, D.; Johnson, J.; Martin, K.; Randolph, L.M.; Rosenfeld, H.; Harmatz, P. Long-term outcomes of patients with mucopolysaccharidosis VI treated with galsulfase enzyme replacement therapy since infancy. Mol. Genet. Metab. 2021, 133, 100–108. [Google Scholar] [CrossRef]
- Zauk, J.; Wyatt, K. Transversus thoracis muscle plane blocks for a patient with Maroteaux-Lamy syndrome undergoing mitral valve replacement. J. Clin. Anesth. 2021, 72, 110269. [Google Scholar] [CrossRef]
- Salik, I.; Rodhouse, H.B.; Mehta, B.; Villion, A. Preoperative cardiac POCUS for urgent surgery in a patient with Maroteaux-Lamy syndrome. J. Clin. Anesth. 2021, 72, 110296. [Google Scholar] [CrossRef]
- Demis, A.A.; Oikonomidou, S.; Daglis, F.; Polymenakos, S.; Panagiotou, M. Double valve replacement in a patient with Maroteaux–Lamy syndrome as an ultimate team challenge. J. Cardiothorac. Surg. 2021, 16, 141. [Google Scholar] [CrossRef] [PubMed]
- İnci, A.; Okur, İ.; Tümer, L.; Biberoğlu, G.; Öktem, M.; Ezgü, F. Clinical and event-based outcomes of patients with mucopolysaccharidosis VI receiving enzyme replacement therapy in Turkey: A case series. Orphanet J. Rare Dis. 2021, 16, 438. [Google Scholar] [CrossRef]
- Guffon, N.; Chowdary, P.; Teles, E.L.; Hughes, D.; Hennermann, J.B.; Huot-Marchand, P.; Faudot-Vernier, E.; Lacombe, O.; Fiquet, A.; Richard, M.P.; et al. Oral treatment for mucopolysaccharidosis VI: Outcomes of the first phase IIa study with odiparcil. J. Inherit. Metab. Dis. 2022, 45, 340–352. [Google Scholar] [CrossRef]
- Andrade, I.; Ribeiro, R.; Carneiro, Z.A.; Giugliani, R.; Pereira, C.; Cozma, C.; Grinberg, D.; Vilageliu, L.; Lourenco, C.M. Fifteen years of enzyme replacement therapy for mucopolysaccharidosis type VI (Maroteaux-Lamy syndrome): A case report. J. Med. Case Rep. 2022, 16, 46. [Google Scholar] [CrossRef]
- Haleem, A.A. Clinical, Endocrine and Genetic spectrums of Mucopolysaccharidoses type VI in Duhok city, Kurdistan Region, Iraq. Cell. Mol. Biol. 2022, 68, 63–69. [Google Scholar] [CrossRef]
- Parini, R.; Deodato, F. Intravenous Enzyme Replacement Therapy in Mucopolysaccharidoses: Clinical Effectiveness and Limitations. Int. J. Mol. Sci. 2020, 21, 2975. [Google Scholar] [CrossRef]
- Sands, M.S. Mucopolysaccharidosis type VII: A powerful experimental system and therapeutic challenge. Pediatr. Endocrinol. Rev. PER 2014, 12 (Suppl. S1), 159–165. [Google Scholar] [PubMed]
- Vogler, C.; Levy, B.; Kyle, J.W.; Sly, W.S.; Williamson, J.; Whyte, M.P. Mucopolysaccharidosis VII: Postmortem biochemical and pathological findings in a young adult with beta-glucuronidase deficiency. Mod. Pathol. Off. J. United States Can. Acad. Pathol. Inc 1994, 7, 132–137. [Google Scholar]
- Toda, Y.; Takeuchi, M.; Morita, K.; Iwasaki, T.; Oe, K.; Yokoyama, M.; Hirakawa, M. Complete heart block during anesthetic management in a patient with mucopolysaccharidosis type VII. Anesthesiology 2001, 95, 1035–1037. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.E.; Volpe, L.; Bullaro, J.; Kakkis, E.D.; Sly, W.S. First human treatment with investigational rhGUS enzyme replacement therapy in an advanced stage MPS VII patient. Mol. Genet. Metab. 2015, 114, 203–208. [Google Scholar] [CrossRef]
- Gniadek, T.J.; Singer, N.; Barker, N.J.; Spevak, P.J.; Crain, B.J.; Valle, D.; Halushka, M.K. Cardiovascular pathologies in mucopolysaccharidosis type VII (Sly Syndrome). Cardiovasc. Pathol. Off. J. Soc. Cardiovasc. Pathol. 2015, 24, 322–326. [Google Scholar] [CrossRef]
- Montaño, A.M.; Lock-Hock, N.; Steiner, R.D.; Graham, B.H.; Szlago, M.; Greenstein, R.; Pineda, M.; Gonzalez-Meneses, A.; Çoker, M.; Bartholomew, D.; et al. Clinical course of sly syndrome (mucopolysaccharidosis type VII). J. Med. Genet. 2016, 53, 403–418. [Google Scholar] [CrossRef] [PubMed]
- Lew, V.; Pena, L.; Edwards, R.; Wang, R.Y. Cardiovascular Histopathology of a 11-Year Old with Mucopolysaccharidosis VII Demonstrates Fibrosis, Macrophage Infiltration, and Arterial Luminal Stenosis. JIMD Rep. 2018, 39, 31–37. [Google Scholar] [CrossRef]
- Kantaputra, P.N.; Smith, L.J.; Casal, M.L.; Kuptanon, C.; Chang, Y.C.; Nampoothiri, S.; Paiyarom, A.; Veerasakulwong, T.; Trachoo, O.; Ketudat Cairns, J.R.; et al. Oral manifestations in patients and dogs with mucopolysaccharidosis Type VII. Am. J. Med. Genet. Part A 2019, 179, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Bilginer Gurbuz, B.; Aypar, E.; Coskun, T.; Alehan, D.; Dursun, A.; Tokatli, A.; Sivri, H.S. The effectiveness of enzyme replacement therapy on cardiac findings in patients with mucopolysaccharidosis. J. Pediatr. Endocrinol. Metab. JPEM 2019, 32, 1049–1053. [Google Scholar] [CrossRef]
- Marek, J.; Kuchynka, P.; Mikulenka, V.; Palecek, T.; Sikora, J.; Hulkova, H.; Lambert, L.; Linkova, H.; Zemanek, D.; Tesarova, M.; et al. Combined valve replacement and aortocoronary bypass in an adult mucopolysaccharidosis type VII patient. Cardiovasc. Pathol. Off. J. Soc. Cardiovasc. Pathol. 2021, 50, 107297. [Google Scholar] [CrossRef]
- Oldham, A.; Oxborrow, N.J.; Woolfson, P.; Jenkins, P.; Gadepalli, C.; Ashworth, J.; Saxena, A.; Rothera, M.; Hendriksz, C.J.; Tol, G.; et al. MPS VII–Extending the classical phenotype. Mol. Genet. Metab. Rep. 2022, 33, 100922. [Google Scholar] [CrossRef]
- Poswar, F.O.; Henriques Nehm, J.; Kubaski, F.; Poletto, E.; Giugliani, R. Diagnosis and Emerging Treatment Strategies for Mucopolysaccharidosis VII (Sly Syndrome). Ther. Clin. Risk Manag. 2022, 18, 1143–1155. [Google Scholar] [CrossRef] [PubMed]
- Trabszo, C.; Ramms, B.; Chopra, P.; Lüllmann-Rauch, R.; Stroobants, S.; Sproß, J.; Jeschke, A.; Schinke, T.; Boons, G.J.; Esko, J.D.; et al. Arylsulfatase K inactivation causes mucopolysaccharidosis due to deficient glucuronate desulfation of heparan and chondroitin sulfate. Biochem. J. 2020, 477, 3433–3451. [Google Scholar] [CrossRef]
- Verheyen, S.; Blatterer, J.; Speicher, M.R.; Bhavani, G.S.; Boons, G.J.; Ilse, M.B.; Andrae, D.; Sproß, J.; Vaz, F.M.; Kircher, S.G.; et al. Novel subtype of mucopolysaccharidosis caused by arylsulfatase K (ARSK) deficiency. J. Med. Genet. 2022, 59, 957–964. [Google Scholar] [CrossRef]
- Depre, C.; Vanoverschelde, J.L.; Taegtmeyer, H. Glucose for the heart. Circulation 1999, 99, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Kolwicz, S.C., Jr.; Tian, R. Glucose metabolism and cardiac hypertrophy. Cardiovasc. Res. 2011, 90, 194–201. [Google Scholar] [CrossRef] [PubMed]


| Arrhythmogenic Disorders | Cardiomyopathies | Cardiac and Valvular Defects | |||
|---|---|---|---|---|---|
| AT | Arrhythmia | ASH | Atrial septal hypertrophy | AD | Aortic dilation |
| AF | Atrial fibrillation | BVD | Biventricular dilation | AC | Aortic coarctation |
| BR | Bradycardia | BVH | Biventricular hypertrophy | AF | Atrial fibrosis |
| HF | Heart failure | CD | Cardiac dilation | AI | Aortic insufficiency |
| LQTS | Long-QT syndromes | CH | Cardiac hypertrophy | ARe | Aortic regurgitation |
| TC | Tachycardia | DI | Diastolic impairment (or dysfunction) | ASD | Atrial septal defects |
| VF | Ventricular fibrillation | DCM | Dilated cardiomyopathy | AVB | Atrioventricular block |
| HCM | Hypertrophic cardiomyopathy | AVD | Aortic valve dysplasia | ||
| HOCM | Hypertrophic obstructive cardiomyopathy | AVP | Atrial valve prolapse | ||
| HoRV | Hypoplastic right ventricle | AVS | Aortic valve stenosis | ||
| HoLV | Hypoplastic left ventricle | BAV | Bicuspid aortic valve | ||
| IC | Ischemic cardiomyopathy | BPV | Bicuscpid pulmonary valve | ||
| IOC | Iron overload cardiomyopathy | CMG | Cardiomegaly | ||
| LCDM | Dilated ventricular cardiomyopathy | CVD | Cardiac valve defects | ||
| LHCM | Left HCM | DAR | Dilated aortic root | ||
| LVHCM | Left ventricular HCM | DC | Dextrocardia | ||
| LVCO | Left ventricular cavity obliteration | DOV | Double outlet ventricle | ||
| LVD | Left ventricular dilation | EA | Ebstein anomaly | ||
| LVH | Left ventricular hypertrophy | ECD | Endocardial cushion defect | ||
| LVRWMA | Left ventricular regional wall motion abnormality * | HSS | Hypertrophic subaortic stenosis | ||
| NCM | Non-compaction cardiomyopathy | LAE | Left atrial enlargement | ||
| RAD | Right atrial dilation | LBBB | Left bundle branch block | ||
| RCM | Restrictive cardiomyopathy | LVCR | Left ventricular concentric remodeling | ||
| RHCM | Right HCM | MF | Myocardial fibrosis | ||
| RVD | Right ventricular dilation | MI | Mitral insuffiency | ||
| RVDe | Right ventricular defect | MVP | Mitral valve prolapse | ||
| RVH | Right ventricular hypertrophy | MVR | Mitral valve regurgitation | ||
| RVHCM | Right ventricular HCM | MVS | Mitral valve stenosis | ||
| SH | Septal hypertrophy | OA | Overriding aorta | ||
| SI | Systolic impairment (or dysfunction) | PDA | Patent (persistent) ductus arteriosus | ||
| VD | Ventricular dysfunction | PFO | Patent (persistent) foramen ovale | ||
| VEFR | Ventricular ejection fraction reduced | PMV | Parachute mitral valve | ||
| VH | Ventricular hypertrophy | PPS | Peripheral pulmonary stenosis | ||
| PVSD | Perimembranous ventricular septal defect | ||||
| RAE | Right atrial enlargement | ||||
| RBBB | Right bundle branch block (associated with structure) | ||||
| RDA | Right-descending aorta | ||||
| RHHS | Right hypoplastic heart syndrome | ||||
| SAI | Small aortic isthmus | ||||
| SD | Septal defect | ||||
| SCA | Single coronary artery | ||||
| TGA | Transposition of the great arteries | ||||
| TI | Tricuspid insufficiency | ||||
| ToF | Tetralogy of Fallot (VSD, aortic dextroposition, PPS, RVH) | ||||
| TVR | Tricuspid valve regurgitation | ||||
| TVS | Tricuspid valve stenosis | ||||
| VC | Valvular calcification | ||||
| VF | Ventricular fibrosis | ||||
| VSD | Ventricular septal defect | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conte, F.; Sam, J.-E.; Lefeber, D.J.; Passier, R. Metabolic Cardiomyopathies and Cardiac Defects in Inherited Disorders of Carbohydrate Metabolism: A Systematic Review. Int. J. Mol. Sci. 2023, 24, 8632. https://doi.org/10.3390/ijms24108632
Conte F, Sam J-E, Lefeber DJ, Passier R. Metabolic Cardiomyopathies and Cardiac Defects in Inherited Disorders of Carbohydrate Metabolism: A Systematic Review. International Journal of Molecular Sciences. 2023; 24(10):8632. https://doi.org/10.3390/ijms24108632
Chicago/Turabian StyleConte, Federica, Juda-El Sam, Dirk J. Lefeber, and Robert Passier. 2023. "Metabolic Cardiomyopathies and Cardiac Defects in Inherited Disorders of Carbohydrate Metabolism: A Systematic Review" International Journal of Molecular Sciences 24, no. 10: 8632. https://doi.org/10.3390/ijms24108632
APA StyleConte, F., Sam, J.-E., Lefeber, D. J., & Passier, R. (2023). Metabolic Cardiomyopathies and Cardiac Defects in Inherited Disorders of Carbohydrate Metabolism: A Systematic Review. International Journal of Molecular Sciences, 24(10), 8632. https://doi.org/10.3390/ijms24108632