
Synthesis, Characterization, and Properties of High-Energy Fillers Derived from Nitroisobutylglycerol

 , , , , , ,
, , , , , ,
Abstract
1. Introduction
2. Results and Discussion
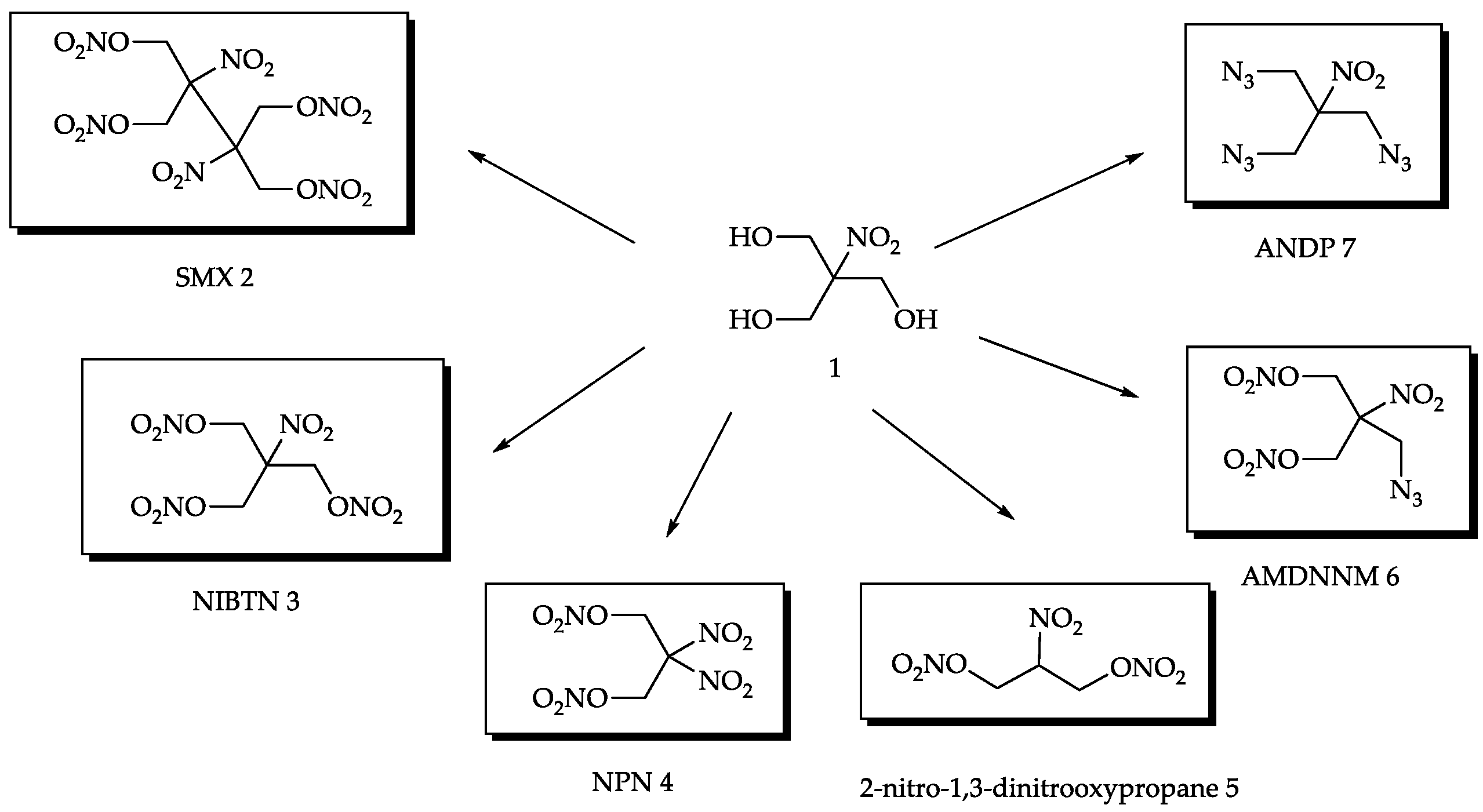
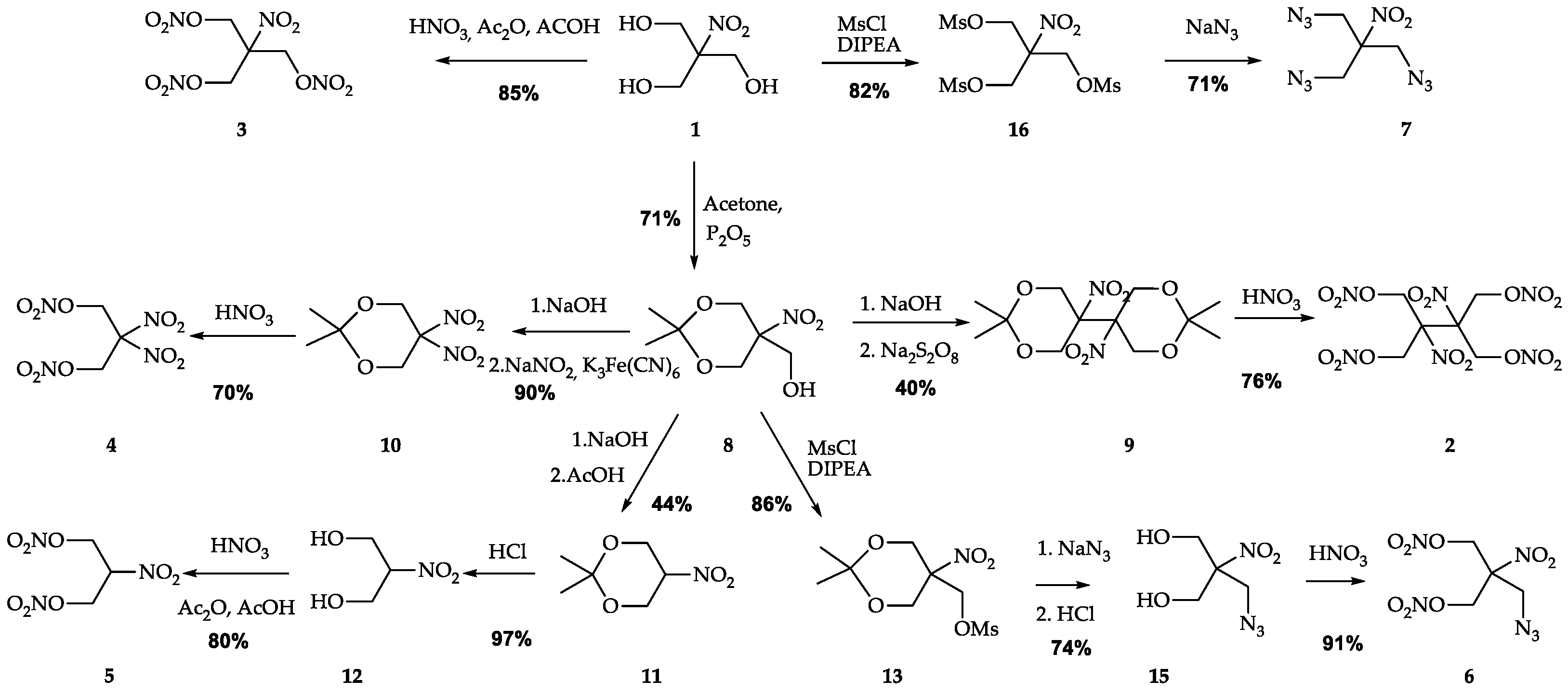
2.1. Synthesis
2.2. Physico-Chemical Properties
2.2.1. Crystal Structure Analysis
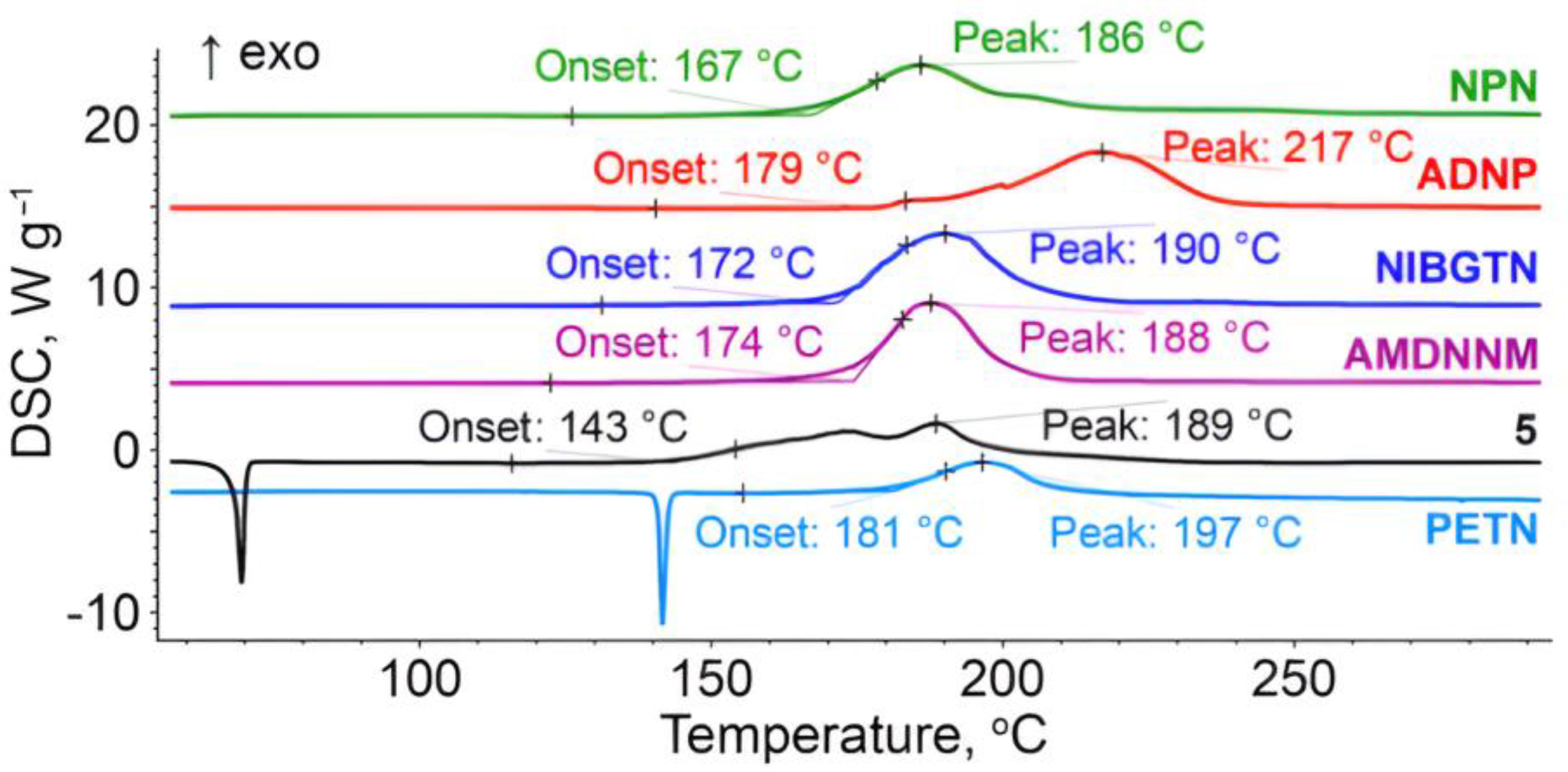
2.2.2. Thermal Analysis
2.2.3. Detonation Parameters
3. Materials and Methods
3.1. Materials
3.2. Synthetic Procedures
3.3. Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shukla, M.; Boddu, V.M.; Steevens, J.A.; Damavarapu, R.; Leszczynski, J. Energetic Materials. From Cradle to Grave, 1st ed.; Springer: Cham, Switzerland, 2017; p. 482. [Google Scholar]
- Charles, E.; Carraher, J. Seymour/Carraher’s Polymer Chemistry, 5th ed.; Marcel Dekker, Inc.: New York, NY, USA; Basel, Switzerland, 2000; p. 741. [Google Scholar]
- Wingborg, N.; Eldsäter, C. 2,2-Dinitro-1,3-Bis-Nitrooxy-Propane (NPN): A New Energetic Plasticizer. Propellants Explos. Pyrotech. 2002, 27, 314–319. [Google Scholar] [CrossRef]
- Akhavan, J. The Chemistry of Explosives; The Royal Society of Chemistry: London, UK, 2004. [Google Scholar]
- Badgujar, D. Review on Promising Insensitive Energetic Materials. Cent. Eur. J. Energetic Mater. 2017, 14, 821–843. [Google Scholar] [CrossRef] [PubMed]
- Zeman, S.; Jungová, M. Sensitivity and Performance of Energetic Materials. Propellants Explos. Pyrotech. 2016, 41, 426–451. [Google Scholar] [CrossRef]
- Kumari, D.; Balakshe, R.; Banerjee, S.; Singh, H. Energetic plasticizers for gun & rocket propellants. Rev. J. Chem. 2012, 2, 240–262. [Google Scholar] [CrossRef]
- Vinogradov, D.B.; Bulatov, P.V.; Petrov, E.Y.; Gribov, P.S.; Kondakova, N.N.; Il’icheva, N.N.; Stepanova, E.R.; Denisyuk, A.P.; Sizov, V.A.; Sinditskii, V.P.; et al. Promising Oxygen- and Nitrogen-Rich Azidonitramino Ether Plasticizers for Energetic Materials. Molecules 2022, 27, 7749. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, J.; Wang, B.; Qiu, L.; Xu, R.; Sheremetev, A.B. Recent synthetic efforts towards high energy density materials: How to design high-performance energetic structures? FirePhysChem 2022, 2, 83–139. [Google Scholar] [CrossRef]
- Kuchurov, I.V.; Arabadzhi, S.S.; Zharkov, M.N.; Fershtat, L.L.; Zlotin, S.G. Sustainable Synthesis of Polynitroesters in the Freon Medium and their in Vitro Evaluation as Potential Nitric Oxide Donors. ACS Sustain. Chem. Eng. 2018, 6, 2535–2540. [Google Scholar] [CrossRef]
- Fedorov, B.S.; Fadeev, M.A.; Eremenko, L.T. Tris(nitroxymethyl)methylamine and tris(nitroxymethyl)nitrosomethane. Mendeleev Commun. 1997, 7, 40. [Google Scholar] [CrossRef]
- Banert, K. Product Class 2: Nitrosoalkanes and Nitroso Acetals (N,N-Dialkoxyamines). In Category 5, Compounds with One Saturated Carbon Heteroatom Bond; Science of Synthesis; Banert, K., Ed.; Georg Thieme Verlag KG: Stuttgart, Germany, 2010; Volume 41. [Google Scholar]
- Urbanski, T. Chemistry and Technology of Explosives; Laverton, S., Ed.; Pergamon Press: Warszawa, Poland, 1967; Volume 3, p. 717. [Google Scholar]
- Legocki, H., Jr.; Hackel, J. Nitroalkyl esters of α,β-unsaturated acids. II. Synthesis of 2-nitro-2,2-bis(nitromethyl)ethanol. Przem. Chem. 1966, 45, 321–324. [Google Scholar]
- Sizov, V.; Pleshakov, D.; Andrey, A.; Topchiy, M.; Nechaev, M. Synthesis and Study of the Thermal and Ballistic Properties of SMX. Cent. Eur. J. Energetic Mater. 2018, 15, 30–46. [Google Scholar] [CrossRef]
- Oxley, J.C.; Smith, J.L.; Brady Iv, J.E.; Brown, A.C. Characterization and Analysis of Tetranitrate Esters. Propellants Explos. Pyrotech. 2012, 37, 24–39. [Google Scholar] [CrossRef]
- Chavez, D.E.; Hiskey, M.A.; Naud, D.L.; Parrish, D. Synthesis of an Energetic Nitrate Ester. Angew. Chem. Int. Ed. 2008, 47, 8307–8309. [Google Scholar] [CrossRef]
- Gankin, J.; Rjabikova, N.; Ilin, V.; Smirnov, S. Nitroisobytilglycerol Nitrate Preparation Method. RU2316538C1 RUS, 17 July 2006. [Google Scholar]
- Hofwimmer, F. Zeitschrift fur das Gesamte Schiess und Sprengstoffwesen (Journal for the Field of Gunpowder and Explosives); J. F. Lehmanns Verlag: Munchen, Germany, 1912; Volume VII, p. 43. [Google Scholar]
- Khmelnitsky, L.I. Handbook of Explosives: A Manual—Part II; F. E. Dzerzhinsky Military Engineering Academy: Moscow, Russia, 1962. [Google Scholar]
- Rudolf Meyer, J.K.; Axel Homburg, N. Explosives; Wiley-VCH Verlag GmbH & Co., KGaA: Weinheim, Germany, 2007; p. 230. [Google Scholar]
- Herman, P. 2,2-Dinitropropane-1,3-dinitrate. U.S. Patent US2978484A, 1 June 1951. [Google Scholar]
- Feuer, H.; Bachman, G.B.; Kispersky, J.P. A New Preparation of Potassium Dinitromethane and its Conversion to 2,2-Dinitro-1,3-propanediol1,2. J. Am. Chem. Soc. 1951, 73, 1360. [Google Scholar] [CrossRef]
- Breiner, M.M.; Chavez, D.E.; Parrish, D.A. 2-Nitro-1,3-dinitro-oxypropane. Acta Crystallogr. Sect. E Struct. Rep. Online 2013, 69, 384. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Su, S.; He, G.; Zhang, L.; Wang, F. A new group of energetic materials. Azidonitrates. In Proceedings of the Internationals Pyrotechnics Seminar, Internationals Pyrotechnics Seminar, Seattle, WA, USA, 8–11 October 1997; Volume 23, pp. 187–192. [Google Scholar]
- Schulze, M.C.; Chavez, D.E. Synthesis and Characterization of Energetic Plasticizer AMDNNM. J. Energetic Mater. 2016, 34, 129–137. [Google Scholar] [CrossRef]
- Ou, Y.; Chen, B.; Yan, H.; Jia, H.; Li, J.; Dong, S. Development of energetic additives for propellants in China. J. Propuls. Power 1995, 11, 838–847. [Google Scholar] [CrossRef]
- Bestuzheva, V.V.; Dushenok, S.A.; Zheltikov, F.A.; Lysov, A.N. Plasticized polyurethanes. Russ. J. Appl. Chem. 2008, 81, 1019–1022. [Google Scholar] [CrossRef]
- Dave, P.R.; Duddu, R.G.; Gelber, N.; Yang, K.; Surapaneni, C.R.; Damavarapu, R. Polyazido Compounds. U.S. Patent US6965042B1, 15 November 2003. [Google Scholar]
- Agrawal, J.P. Recent trends in high-energy materials. Prog. Energy Combust. Sci. 1998, 24, 1–30. [Google Scholar] [CrossRef]
- Talawar, M.B.; Sivabalan, R.; Anniyappan, M.; Gore, G.M.; Asthana, S.N.; Gandhe, B.R. Emerging trends in advanced high energy materials. Combust. Explos. Shock Waves 2007, 43, 62–72. [Google Scholar] [CrossRef]
- Muravyev, N.V.; Monogarov, K.A.; Melnikov, I.N.; Pivkina, A.N.; Kiselev, V.G. Learning to fly: Thermochemistry of energetic materials by modified thermogravimetric analysis and highly accurate quantum chemical calculations. Phys. Chem. Chem. Phys. 2021, 23, 15522–15542. [Google Scholar] [CrossRef]
- Romer, F. Uber Abkommlinge des Nitromethans. Angewante Chem. 1955, 67, 157. [Google Scholar] [CrossRef]
- Khisamutdinov, G.K.; Karpychev, Y.V.; Zhbanova, Y.S.; Kondyukov, I.Z.; Kashaev, V.A.; Il’in, V.P. Development of new methods for the synthesis of 2,3-bis(nitroxymethyl)-2,3-dinitrobutane-1,4-diol dinitrate and its intermediates. Russ. Chem. Bull. 2015, 64, 1967–1970. [Google Scholar] [CrossRef]
- Katorov, D.V.; Rudakov, G.F.; Zhilin, V.F. Synthesis of heterocyclic geminal nitro azides. Russ. Chem. Bull. 2009, 58, 2311–2317. [Google Scholar] [CrossRef]
- Kissinger, L.W.; Benziger, T.M.; Ungnade, H.E.; Rohwer, R.K. gem-Dinitro Esters. IV. Pyridine-Catalyzed Esterification of β-Dinitro Alcohols1. J. Org. Chem. 1963, 28, 2491–2494. [Google Scholar] [CrossRef]
- Latour, S.; Wuest, J.D. Simple Syntheses of 2-Hydroxymethyl-1,3-propanediol and Related Compounds. Synthesis 1987, 1987, 742–745. [Google Scholar] [CrossRef]
- Zhang, C.; Tang, G. Preparation of Triazo Alkane. CN1600778A CH, 30 March 2005. [Google Scholar]
- Lenz, T.; Klapötke, T.M.; Mühlemann, M.; Stierstorfer, J. About the Azido Derivatives of Pentaerythritol Tetranitrate. Propellants Explos. Pyrotech. 2021, 46, 723–731. [Google Scholar] [CrossRef]
- Manelis, G.B.; Nazin, G.M.; Rubtsov Yu, I.; Strunin, V.A. Thermal Decomposition and Combustion of Expolsives and and Propellants; Taylor & Francis: London, UK, 2003. [Google Scholar]
- Yan, Q.-L.; Künzel, M.; Zeman, S.; Svoboda, R.; Bartošková, M. The effect of molecular structure on thermal stability, decomposition kinetics and reaction models of nitric esters. Thermochim. Acta 2013, 566, 137–148. [Google Scholar] [CrossRef]
- Kanno, H. A simple derivation of the empirical rule TGTM = 23. J. Non-Cryst. Solids 1981, 44, 409–413. [Google Scholar] [CrossRef]
- Karton, A.; Schreiner, P.R.; Martin, J.M.L. Heats of formation of platonic hydrocarbon cages by means of high-level thermochemical procedures. J. Comput. Chem. 2016, 37, 49–58. [Google Scholar] [CrossRef]
- Liu, J. Multivariate Nitrates. In Nitrate Esters Chemistry and Technology; Liu, J., Ed.; Springer: Singapore, 2019; pp. 377–443. [Google Scholar]
- Song, X.; Wang, Y.; An, C. Thermochemical properties of nanometer CL-20 and PETN fabricated using a mechanical milling method. AIP Adv. 2018, 8, 065009. [Google Scholar] [CrossRef]
- Klapötke, T.M.; Lemarchand, G.; Lenz, T.; Mühlemann, M.; Stierstorfer, J.; Weber, R. Impact and Friction Sensitivities of PETN: I. Sensitivities of the Pure and Wetted Material. Propellants Explos. Pyrotech. 2022, 47, e202200150. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. Gaussian 09, version D. 01; Gaussian Wallingford: Wallingford, CT, USA, 2009. [Google Scholar]
- Werner, H.-J.; Knowles, P.J.; Knizia, G.; Manby, F.R.; Schütz, M. Molpro: A general-purpose quantum chemistry program package. WIREs Comput. Mol. Sci. 2012, 2, 242–253. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Karton, A.; Martin, J.M.L. Explicitly correlated Wn theory: W1-F12 and W2-F12. J. Chem. Phys. 2012, 136, 124114. [Google Scholar] [CrossRef]
- Curtiss, L.A.; Raghavachari, K.; Redfern, P.C.; Pople, J.A. Assessment of Gaussian-2 and density functional theories for the computation of enthalpies of formation. J. Chem. Phys. 1997, 106, 1063–1079. [Google Scholar] [CrossRef]
- Kiselev, V.G.; Gritsan, N.P. Theoretical Study of the Nitroalkane Thermolysis. 1. Computation of the Formation Enthalpy of the Nitroalkanes, Their Isomers and Radical Products. J. Phys. Chem. A 2008, 112, 4458–4464. [Google Scholar] [CrossRef]
- Kiselev, V.G.; Goldsmith, C.F. Accurate Thermochemistry of Novel Energetic Fused Tricyclic 1,2,3,4-Tetrazine Nitro Derivatives from Local Coupled Cluster Methods. J. Phys. Chem. A 2019, 123, 9818–9827. [Google Scholar] [CrossRef]
- Kong, L.; Bischoff, F.A.; Valeev, E.F. Explicitly Correlated R12/F12 Methods for Electronic Structure. Chem. Rev. 2012, 112, 75–107. [Google Scholar] [CrossRef]
- Liakos, D.G.; Sparta, M.; Kesharwani, M.K.; Martin, J.M.L.; Neese, F. Exploring the Accuracy Limits of Local Pair Natural Orbital Coupled-Cluster Theory. J. Chem. Theory Comput. 2015, 11, 1525–1539. [Google Scholar] [CrossRef]
- Lee, T.J.; Taylor, P.R. A diagnostic for determining the quality of single-reference electron correlation methods. Int. J. Quantum Chem. 1989, 36, 199–207. [Google Scholar] [CrossRef]
- Chase, M.W., Jr. NIST-JANAF Thermochemical Tables, Monograph 9, 4th ed.; American Chemical Society: Washington, DC, USA, 1998; Volume 9, pp. 1–1951. [Google Scholar]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Kossmann, S.; Neese, F. Comparison of two efficient approximate Hartee–Fock approaches. Chem. Phys. Lett. 2009, 481, 240–243. [Google Scholar] [CrossRef]
- UN Recommendations on Transport of Dangerous Goods. Environ. Conserv. 2009, 22, 370–371. [CrossRef]
- Muravyev, N.V.; Wozniak, D.R.; Piercey, D.G. Progress and performance of energetic materials: Open dataset, tool, and implications for synthesis. J. Mater. Chem. A 2022, 10, 11054–11073. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | ||||||
 |  |  |  |  |  | |
| ANDP 7 | SMX 2 | AMDNNM 6 | NIBTN 3 | NPN 4 | 2-Nitro-1,3-dinitrooxypropane 5 | |
| Physicochemical properties | ||||||
| Formula | C4H6N10O2 | C6H8N6O16 | C4H6N6O8 | C4H6N4O11 | C3H4N4O10 | C3H5N3O8 |
| M [g mol−1] [a] | 226.12 | 420 | 266.09 | 286.07 | 256.1 | 211.05 |
| N [%] [b] | 61.95 | 20.00 | 31.58 | 19.59 | 21.88 | 19.91 |
| Ω [%] [c] | −63.7 | 0 | −18.0 | 0 | −12.5 | −3.8 |
| ρ, g cm−3 [d] | 1.35 [25] (1.367) [26] | 1.973 [43] (1.917) [17,44] | 1.55 [26] | 1.617 [44] | 1.66 [3] | 1.760 [24] |
| [kJ mol−1] [e] | 816 | −542 [32] | 5 | −402 | −280 | −348 |
| Tm [°C] [f] or Tg [°C] [g] | 12 (m) | 86 (m) [32] | −67.4 (g) | −58.1 (g) | −81.2 (g) | 68 (m) [24] |
| Tdec. [°C] [h] | 179 | 168 [32] | 174 | 172 | 167 | 143 |
| IS [J] [i] | 0.3 | 3 [32] | 0.3 | 0.4 | 0.4 | 4 |
| FS [N] [j] | - | 50 [32] | - | - | - | 120 |
| Detonation parameters | ||||||
| D [m s−1] [k] | 7.2 | 9.2 | 7.8 | 8.1 | 7.8 | 8.4 |
| P [GPa] [l] C-J | 20 | 37 | 25 | 26 | 24 | 29 |
| Heat of explosion, J/g [m] | 4657 | 6260 | 5460 | 6160 | 5310 | 5840 |
| H [n] | 0.74 | 1.03 | 0.84 | 0.89 | 0.86 | 0.93 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rzhevskiy, S.A.; Minaeva, L.I.; Topchiy, M.A.; Melnikov, I.N.; Kiselev, V.G.; Pivkina, A.N.; Fomenkov, I.V.; Asachenko, A.F. Synthesis, Characterization, and Properties of High-Energy Fillers Derived from Nitroisobutylglycerol. Int. J. Mol. Sci. 2023, 24, 8541. https://doi.org/10.3390/ijms24108541
Rzhevskiy SA, Minaeva LI, Topchiy MA, Melnikov IN, Kiselev VG, Pivkina AN, Fomenkov IV, Asachenko AF. Synthesis, Characterization, and Properties of High-Energy Fillers Derived from Nitroisobutylglycerol. International Journal of Molecular Sciences. 2023; 24(10):8541. https://doi.org/10.3390/ijms24108541
Chicago/Turabian StyleRzhevskiy, Sergey A., Lidiya I. Minaeva, Maxim A. Topchiy, Igor N. Melnikov, Vitaly G. Kiselev, Alla N. Pivkina, Igor V. Fomenkov, and Andrey F. Asachenko. 2023. "Synthesis, Characterization, and Properties of High-Energy Fillers Derived from Nitroisobutylglycerol" International Journal of Molecular Sciences 24, no. 10: 8541. https://doi.org/10.3390/ijms24108541
APA StyleRzhevskiy, S. A., Minaeva, L. I., Topchiy, M. A., Melnikov, I. N., Kiselev, V. G., Pivkina, A. N., Fomenkov, I. V., & Asachenko, A. F. (2023). Synthesis, Characterization, and Properties of High-Energy Fillers Derived from Nitroisobutylglycerol. International Journal of Molecular Sciences, 24(10), 8541. https://doi.org/10.3390/ijms24108541