
Quantitative Aspects of the Human Cell Proteome


Abstract
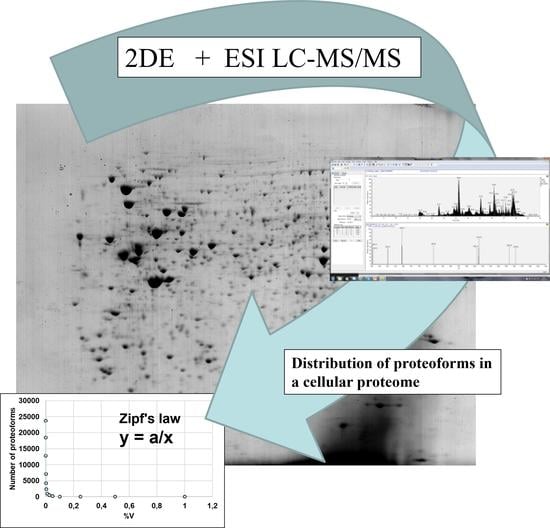
1. Introduction
2. Quantification
2.1. Panoramic Quantification
2.2. Aspects of Cell Size
2.3. Aspects of Proteome Variation
2.4. Aspects of Sensitivity
2.5. Aspects of Cancer
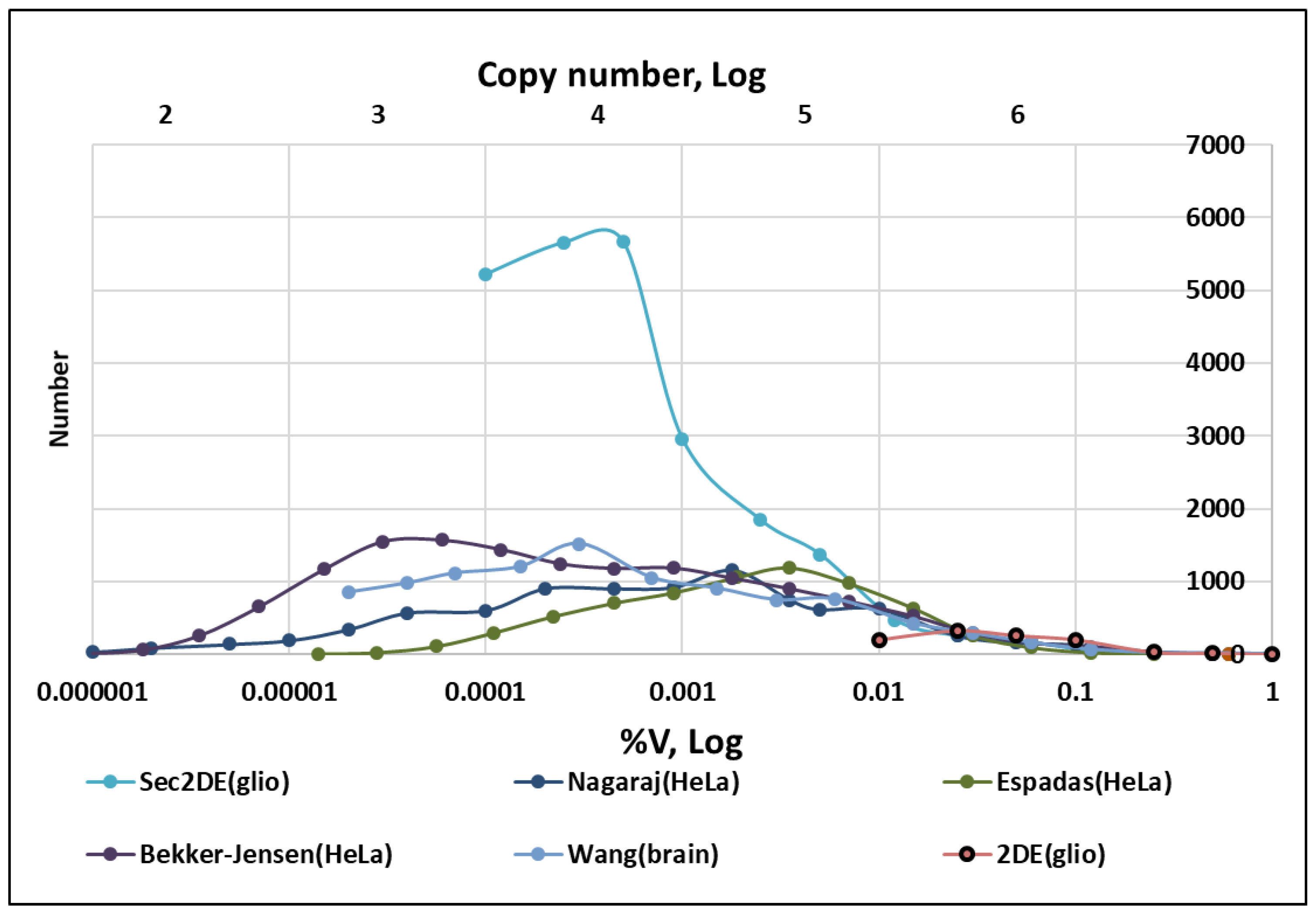
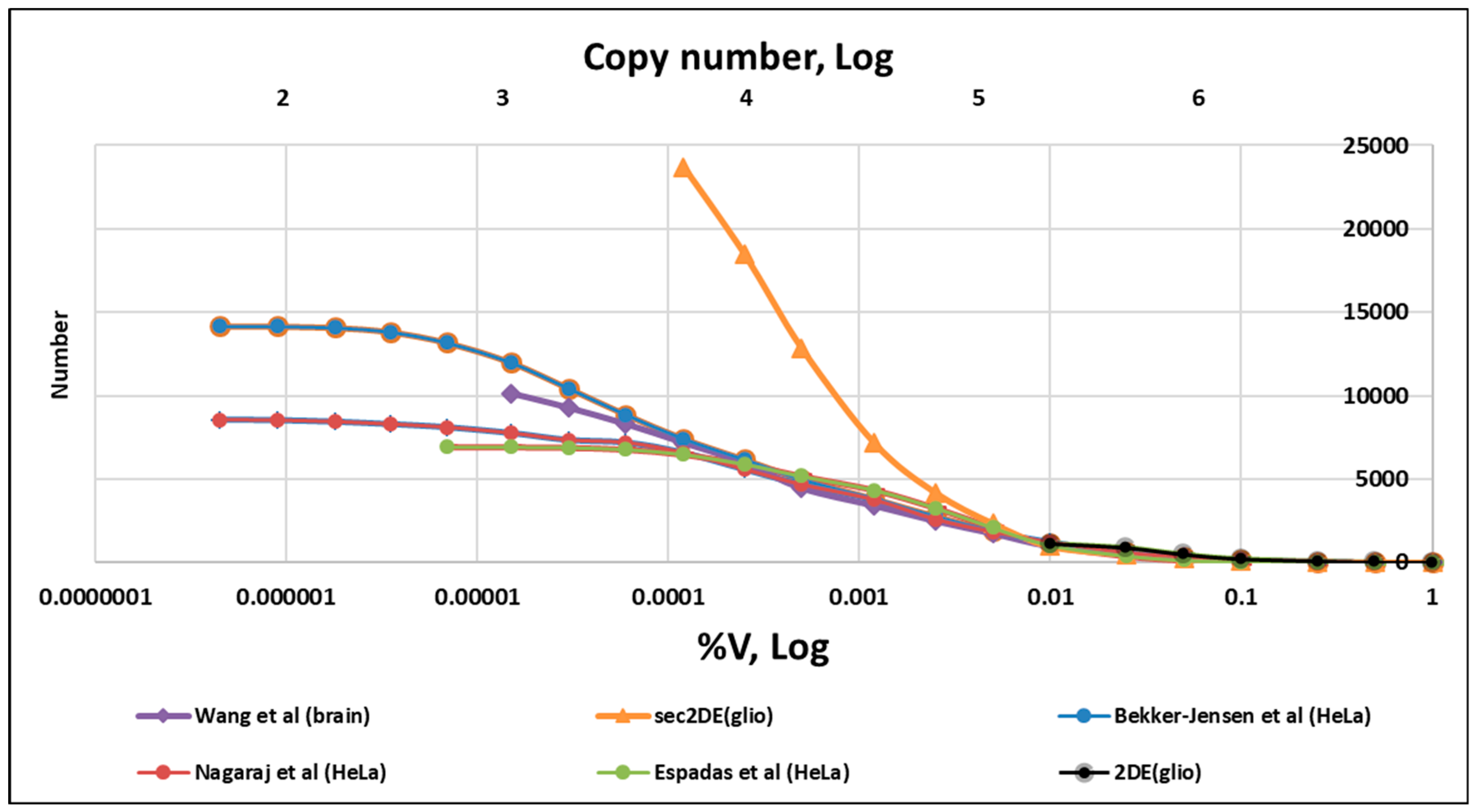
2.6. Analysis of Proteomics Datasets
3. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MS | Mass spectrometry |
| ESI LC-MS/MS | Liquid chromatography–electrospray ionization tandem mass spectrometry |
| 2DE | Two-dimensional gel electrophoresis |
| Sec2DE | Sectional two-dimensional gel electrophoresis |
| emPAI | Exponentially modified Protein Abundance Index |
| iBAQ | intensity Based Absolute Quantification |
| SNP | Single-Nucleotide Polymorphism |
| TMT | Tandem Mass Tag |
| TS | Tissue Specificity |
References
- Smith, L.M.; Agar, J.N.; Chamot-Rooke, J.; Danis, P.O.; Ge, Y.; Loo, J.A.; Paša-Tolić, L.; Tsybin, Y.O.; Kelleher, N.L. The Consortium for Top-Down Proteomics The Human Proteoform Project: Defining the human proteome. Sci. Adv. 2021, 7, eabk0734. [Google Scholar] [CrossRef]
- Smith, L.M.; Kelleher, N.L. Proteoform: A single term describing protein complexity. Nat. Methods 2013, 10, 186–187. [Google Scholar] [CrossRef]
- Schlüter, H.; Apweiler, R.; Holzhütter, H.-G.; Jungblut, P.R. Finding one’s way in proteomics: A protein species nomenclature. Chem. Cent. J. 2009, 3, 11. [Google Scholar] [CrossRef]
- Toby, T.K.; Fornelli, L.; Kelleher, N.L. Progress in Top-Down Proteomics and the Analysis of Proteoforms. Annu. Rev. Anal. Chem. 2016, 9, 499–519. [Google Scholar] [CrossRef]
- Ponomarenko, E.A.; Poverennaya, E.V.; Ilgisonis, E.V.; Pyatnitskiy, M.A.; Kopylov, A.T.; Zgoda, V.G.; Lisitsa, A.V.; Archakov, A.I. The Size of the Human Proteome: The Width and Depth. Int. J. Anal. Chem. 2016, 2016, 7436849. [Google Scholar] [CrossRef]
- Naryzhny, S.N.; Lisitsa, A.V.; Zgoda, V.G.; Ponomarenko, E.A.; Archakov, A.I. 2DE-based approach for estimation of number of protein species in a cell. Electrophoresis 2013, 35, 895–900. [Google Scholar] [CrossRef]
- Ramsköld, D.; Wang, E.T.; Burge, C.B.; Sandberg, R. An Abundance of Ubiquitously Expressed Genes Revealed by Tissue Transcriptome Sequence Data. PLoS Comput. Biol. 2009, 5, e1000598. [Google Scholar] [CrossRef]
- Zahn, M.; Michel, P.-A.; Gateau, A.; Nikitin, F.; Schaeffer, M.; Audot, E.; Gaudet, P.; Duek, P.D.; Teixeira, D.; de Laval, V.R.; et al. The neXtProt knowledgebase in 2020: Data, tools and usability improvements. Nucleic Acids Res. 2019, 48, D328–D334. [Google Scholar] [CrossRef]
- Aebersold, R.; Agar, J.N.; Amster, I.J.; Baker, M.S.; Bertozzi, C.R.; Boja, E.S.; Costello, C.E.; Cravatt, B.F.; Fenselau, C.; Garcia, B.A.; et al. How many human proteoforms are there? Nat. Chem. Biol. 2018, 14, 206–214. [Google Scholar] [CrossRef]
- Dehart, C.J.; Fornelli, L.; Anderson, L.C.; Fellers, R.T.; Lu, D.; Hendrickson, C.L.; Lahav, G.; Gunawardena, J.; Kelleher, N.L. A multi-modal proteomics strategy for characterizing posttranslational modifications of tumor suppressor p53 reveals many sites but few modified forms. bioRxiv 2018, bioRxiv:455527. [Google Scholar] [CrossRef]
- Nakamura, T.; Oda, Y. Mass spectrometry-based quantitative proteomics. Biotechnol. Genet. Eng. Rev. 2007, 24, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Ueberheide, B.M.; Waldemarson, S.; Myung, S.; Molloy, K.; Eriksson, J.; Chait, B.T.; Neubert, T.A.; Fenyö, D. Protein Quantitation Using Mass Spectrometry. Comput. Biol. 2010, 673, 211–222. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Hein, M.Y.; Cox, J.; Mann, M. A “Proteomic Ruler” for Protein Copy Number and Concentration Estimation without Spike-in Standards. Mol. Cell. Proteom. 2014, 13, 3497–3506. [Google Scholar] [CrossRef] [PubMed]
- Millán-Oropeza, A.; Blein-Nicolas, M.; Monnet, V.; Zivy, M.; Henry, C. Comparison of Different Label-Free Techniques for the Semi-Absolute Quantification of Protein Abundance. Proteomes 2022, 10, 2. [Google Scholar] [CrossRef]
- DeSouza, L.V.; Siu, K.M. Mass spectrometry-based quantification. Clin. Biochem. 2013, 46, 421–431. [Google Scholar] [CrossRef]
- Dai, Y.; Buxton, K.E.; Schaffer, L.V.; Miller, R.M.; Millikin, R.J.; Scalf, M.; Frey, B.L.; Shortreed, M.R.; Smith, L.M. Constructing Human Proteoform Families Using Intact-Mass and Top-Down Proteomics with a Multi-Protease Global Post-Translational Modification Discovery Database. J. Proteome Res. 2019, 18, 3671–3680. [Google Scholar] [CrossRef] [PubMed]
- Tran, J.C.; Zamdborg, L.; Ahlf, D.R.; Lee, J.E.; Catherman, A.D.; Durbin, K.R.; Tipton, J.D.; Vellaichamy, A.; Kellie, J.F.; Li, M.; et al. Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature 2011, 480, 254–258. [Google Scholar] [CrossRef]
- Catherman, A.D.; Durbin, K.R.; Ahlf, D.R.; Early, B.P.; Fellers, R.T.; Tran, J.; Thomas, P.; Kelleher, N.L. Large-scale Top-down Proteomics of the Human Proteome: Membrane Proteins, Mitochondria, and Senescence. Mol. Cell Proteom. 2013, 12, 3465–3473. [Google Scholar] [CrossRef]
- Anderson, L.C.; DeHart, C.J.; Kaiser, N.K.; Fellers, R.T.; Smith, D.F.; Greer, J.B.; LeDuc, R.D.; Blakney, G.T.; Thomas, P.M.; Kelleher, N.L.; et al. Identification and Characterization of Human Proteoforms by Top-Down LC-21 Tesla FT-ICR Mass Spectrometry. J. Proteome Res. 2016, 16, 1087–1096. [Google Scholar] [CrossRef]
- Schaffer, L.V.; Millikin, R.J.; Miller, R.M.; Anderson, L.C.; Fellers, R.T.; Ge, Y.; Kelleher, N.L.; LeDuc, R.; Liu, X.; Payne, S.H.; et al. Identification and Quantification of Proteoforms by Mass Spectrometry. Proteomics 2019, 19, e1800361. [Google Scholar] [CrossRef]
- McWhite, C.D.; Sae-Lee, W.; Yuan, Y.; Mallam, A.L.; Gort-Freitas, N.A.; Ramundo, S.; Onishi, M.; Marcotte, E.M. Alternative proteoforms and proteoform-dependent assemblies in humans and plants. bioRxiv 2022. [Google Scholar] [CrossRef]
- Naryzhny, S.; Zgoda, V.; Kopylov, A.; Petrenko, E.; Kleist, O.; Archakov, A. Variety and Dynamics of Proteoforms in the Human Proteome: Aspects of Markers for Hepatocellular Carcinoma. Proteomes 2017, 5, 33. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Burgess-Cassler, A.; Johansen, J.J.; Santek, D.A.; Ide, J.R.; Kendrick, N.C. Computerized quantitative analysis of coomassie-blue-stained serum proteins separated by two-dimensional electrophoresis. Clin. Chem. 1989, 35, 2297–2304. [Google Scholar] [CrossRef]
- Luo, S.; Wehr, N.B.; Levine, R.L. Quantitation of protein on gels and blots by infrared fluorescence of Coomassie blue and Fast Green. Anal. Biochem. 2006, 350, 233–238. [Google Scholar] [CrossRef]
- Thiede, B.; Koehler, C.J.; Strozynski, M.; Treumann, A.; Stein, R.; Zimny-Arndt, U.; Schmid, M.; Jungblut, P.R. High Resolution Quantitative Proteomics of HeLa Cells Protein Species Using Stable Isotope Labeling with Amino Acids in Cell Culture(SILAC), Two-Dimensional Gel Electrophoresis(2DE) and Nano-Liquid Chromatograpohy Coupled to an LTQ-OrbitrapMass Spectrometer. Mol. Cell. Proteom. 2013, 12, 529–538. [Google Scholar] [CrossRef]
- Naryzhny, S.N.; Zgoda, V.G.; Maynskova, M.A.; Novikova, S.E.; Ronzhina, N.L.; Vakhrushev, I.V.; Khryapova, E.V.; Lisitsa, A.V.; Tikhonova, O.V.; Ponomarenko, E.A.; et al. Combination of virtual and experimental 2DE together with ESI LC-MS/MS gives a clearer view about proteomes of human cells and plasma. Electrophoresis 2015, 37, 302–309. [Google Scholar] [CrossRef]
- Naryzhny, S.N.; Maynskova, M.A.; Zgoda, V.G.; Ronzhina, N.L.; Kleyst, O.A.; Vakhrushev, I.V.; Archakov, A.I. Virtual-Experimental 2DE Approach in Chromosome-Centric Human Proteome Project. J. Proteome Res. 2015, 15, 525–530. [Google Scholar] [CrossRef]
- Bianconi, E.; Piovesan, A.; Facchin, F.; Beraudi, A.; Casadei, R.; Frabetti, F.; Vitale, L.; Pelleri, M.C.; Tassani, S.; Piva, F.; et al. An estimation of the number of cells in the human body. Ann. Hum. Biol. 2013, 40, 463–471. [Google Scholar] [CrossRef]
- Milo, R.; Jorgensen, P.; Moran, U.; Weber, G.; Springer, M. BioNumbers—The database of key numbers in molecular and cell biology. Nucleic Acids Res. 2009, 38, D750–D753. [Google Scholar] [CrossRef]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [PubMed]
- Lodish, H.; Berk, A.; Zipursky, L.; Matsudaira, P.; Baltimore, D.; Darnell, J. Molecular Cell Biology, 4th ed.; W. H. Freeman: New York, NY, USA, 2000. [Google Scholar]
- Giles, C. The Platelet Count and Mean Platelet Volume. Br. J. Haematol. 2008, 48, 31–37. [Google Scholar] [CrossRef]
- Lundberg, E.; Gry, M.; Oksvold, P.; Kononen, J.; Andersson-Svahn, H.; Pontén, F.; Uhlén, M.; Asplund, A. The correlation between cellular size and protein expression levels—Normalization for global protein profiling. J. Proteom. 2008, 71, 448–460. [Google Scholar] [CrossRef]
- Milo, R. What is the total number of protein molecules per cell volume? A call to rethink some published values. Bioessays 2013, 35, 1050–1055. [Google Scholar] [CrossRef]
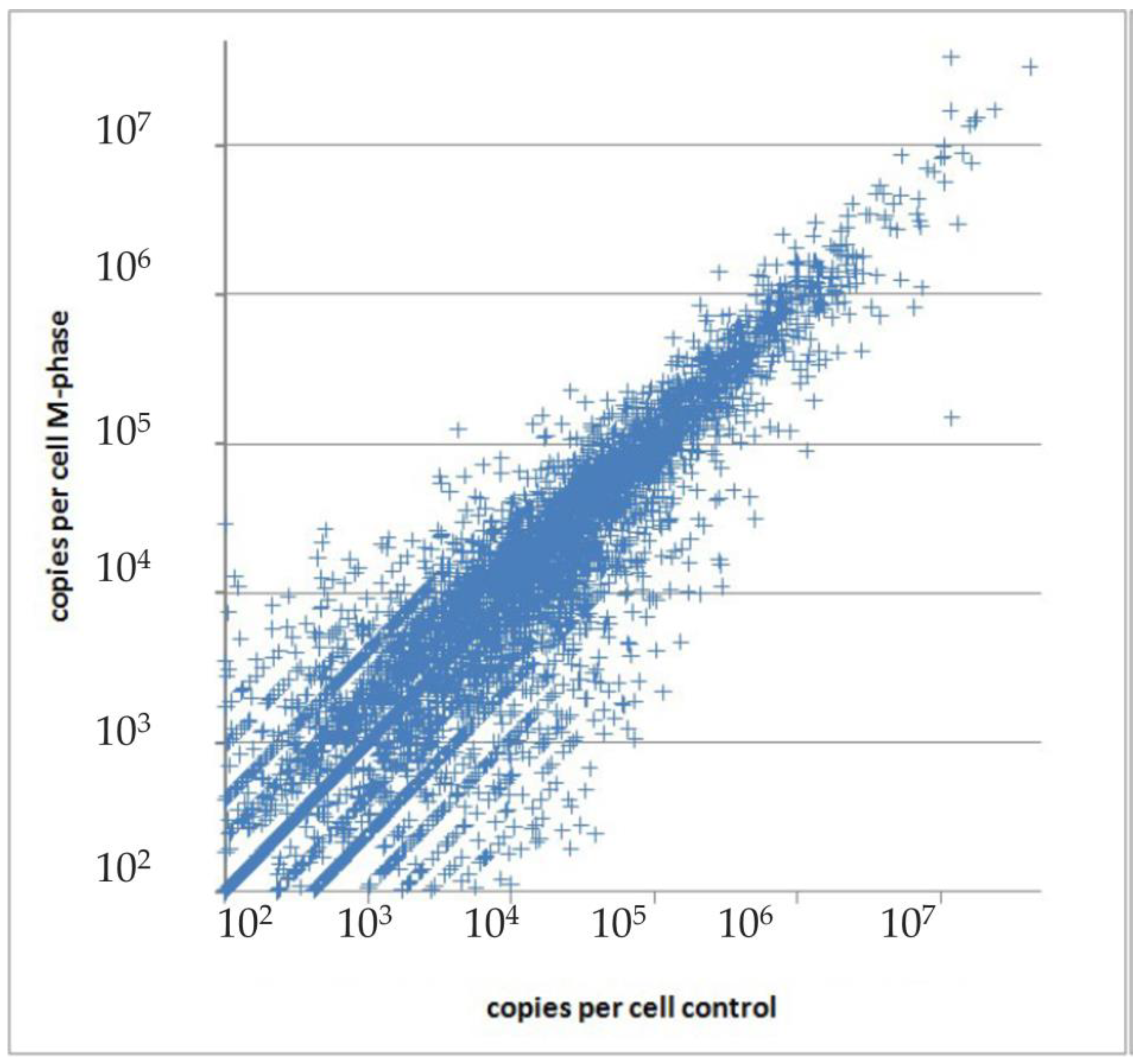
- Cohen, A.A.; Geva-Zatorsky, N.; Eden, E.; Frenkel-Morgenstern, M.; Issaeva, I.; Sigal, A.; Milo, R.; Cohen-Saidon, C.; Liron, Y.; Kam, Z.; et al. Dynamic Proteomics of Individual Cancer Cells in Response to a Drug. Science 2008, 322, 1511–1516. [Google Scholar] [CrossRef]
- Farkash-Amar, S.; Eden, E.; Cohen, A.; Geva-Zatorsky, N.; Cohen, L.; Milo, R.; Sigal, A.; Danon, T.; Alon, U. Dynamic Proteomics of Human Protein Level and Localization across the Cell Cycle. PLoS ONE 2012, 7, e48722. [Google Scholar] [CrossRef]
- Beck, M.; Schmidt, A.; Malmstroem, J.; Claassen, M.; Ori, A.; Szymborska, A.; Herzog, F.; Rinner, O.; Ellenberg, J.; Aebersold, R. The quantitative proteome of a human cell line. Mol. Syst. Biol. 2011, 7, 549. [Google Scholar] [CrossRef]
- Dissmeyer, N.; Coux, O.; Rodriguez, M.S.; Barrio, R. PROTEOSTASIS: A European Network to Break Barriers and Integrate Science on Protein Homeostasis. Trends Biochem. Sci. 2019, 44, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Slavov, N. Scaling Up Single-Cell Proteomics. Mol. Cell. Proteom. 2021, 21, 100179. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.T. Single-cell Proteomics: Progress and Prospects. Mol. Cell. Proteom. 2020, 19, 1739–1748. [Google Scholar] [CrossRef]
- Budnik, B.; Levy, E.; Harmange, G.; Slavov, N. SCoPE-MS: Mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biol. 2018, 19, 161. [Google Scholar] [CrossRef] [PubMed]
- Gebreyesus, S.T.; Siyal, A.A.; Kitata, R.B.; Chen, E.S.-W.; Enkhbayar, B.; Angata, T.; Lin, K.-I.; Chen, Y.-J.; Tu, H.-L. Streamlined single-cell proteomics by an integrated microfluidic chip and data-independent acquisition mass spectrometry. Nat. Commun. 2022, 13, 37. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Wang, M.; Lin, S.; Jian, R.; Li, X.; Chan, J.; Dong, G.; Fang, H.; Robinson, A.E.; Snyder, M.P.; et al. A Quantitative Proteome Map of the Human Body. Cell 2020, 183, 269–283.e19. [Google Scholar] [CrossRef]
- Wang, D.; Eraslan, B.; Wieland, T.; Hallström, B.; Hopf, T.; Zolg, D.P.; Zecha, J.; Asplund, A.; Li, L.; Meng, C.; et al. A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Biol. 2019, 15, e8503. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-S.; Pinto, S.M.; Getnet, D.; Nirujogi, R.S.; Manda, S.S.; Chaerkady, R.; Madugundu, A.K.; Kelkar, D.S.; Isserlin, R.; Jain, S.; et al. A draft map of the human proteome. Nature 2014, 509, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, M.; Schlegl, J.; Hahne, H.; Gholami, A.M.; Lieberenz, M.; Savitski, M.M.; Ziegler, E.; Butzmann, L.; Gessulat, S.; Marx, H.; et al. Mass-spectrometry-based draft of the human proteome. Nature 2014, 509, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Orsburn, B.C. Evaluation of the Sensitivity of Proteomics Methods Using the Absolute Copy Number of Proteins in a Single Cell as a Metric. Proteomes 2021, 9, 34. [Google Scholar] [CrossRef]
- Espadas, G.; Borràs, E.; Chiva, C.; Sabidó, E. Evaluation of different peptide fragmentation types and mass analyzers in data-dependent methods using an Orbitrap Fusion Lumos Tribrid mass spectrometer. Proteomics 2017, 17, 1600416. [Google Scholar] [CrossRef]
- Nagaraj, N.; Wisniewski, J.R.; Geiger, T.; Cox, J.; Kircher, M.; Kelso, J.; Pääbo, S.; Mann, M. Deep proteome and transcriptome mapping of a human cancer cell line. Mol. Syst. Biol. 2011, 7, 548. [Google Scholar] [CrossRef]
- Bekker-Jensen, D.B.; Kelstrup, C.D.; Batth, T.S.; Larsen, S.C.; Haldrup, C.; Bramsen, J.B.; Sørensen, K.D.; Høyer, S.; Ørntoft, T.F.; Andersen, C.L.; et al. An Optimized Shotgun Strategy for the Rapid Generation of Comprehensive Human Proteomes. Cell Syst. 2017, 4, 587–599.e4. [Google Scholar] [CrossRef]
- Naryzhny, S.N.; Maynskova, M.A.; Zgoda, V.G.; Ronzhina, N.L.; Novikova, S.E.; Belyakova, N.V.; Kleyst, O.A.; Legina, O.K.; Pantina, R.A. Proteomic profiling of high-grade glioblastoma using virtual-experimental 2DE. J. Proteom. Bioinform. 2016, 9, 158–165. [Google Scholar] [CrossRef]
- Burkhart, J.M.; Vaudel, M.; Gambaryan, S.; Radau, S.; Walter, U.; Martens, L.; Geiger, J.; Sickmann, A.; Zahedi, R.P. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood 2012, 120, e73–e82. [Google Scholar] [CrossRef]
- Stewart, P.A.; Welsh, E.A.; Slebos, R.J.C.; Fang, B.; Izumi, V.; Chambers, M.; Zhang, G.; Cen, L.; Pettersson, F.; Zhang, Y.; et al. Proteogenomic landscape of squamous cell lung cancer. Nat. Commun. 2019, 10, 3578. [Google Scholar] [CrossRef]
- Zhuo, H.; Zhao, Y.; Cheng, X.; Xu, M.; Wang, L.; Lin, L.; Lyu, Z.; Hong, X.; Cai, J. Tumor endothelial cell-derived cadherin-2 promotes angiogenesis and has prognostic significance for lung adenocarcinoma. Mol. Cancer 2019, 18, 34. [Google Scholar] [CrossRef] [PubMed]
- Edwards, N.J.; Oberti, M.; Thangudu, R.R.; Cai, S.; McGarvey, P.B.; Jacob, S.; Madhavan, S.; Ketchum, K.A. The CPTAC Data Portal: A Resource for Cancer Proteomics Research. J. Proteome Res. 2015, 14, 2707–2713. [Google Scholar] [CrossRef]
- Chen, F.; Chandrashekar, D.S.; Varambally, S.; Creighton, C.J. Pan-cancer molecular subtypes revealed by mass-spectrometry-based proteomic characterization of more than 500 human cancers. Nat. Commun. 2019, 10, 5679. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.N.; Friedrich, B.; Schnaubelt, M.; Chan, D.W.; Zhang, H.; Aebersold, R. Orthogonal Proteomic Platforms and Their Implications for the Stable Classification of High-Grade Serous Ovarian Cancer Subtypes. iScience 2020, 23, 101079. [Google Scholar] [CrossRef]
- Naryzhny, S.; Maynskova, M.; Zgoda, V.; Archakov, A. Zipf’s Law in Proteomics. J. Proteom. Bioinform. 2017, 10, 79–84. [Google Scholar] [CrossRef]
- Naryzhny, S.; Maynskova, M.; Zgoda, V.; Archakov, A. Dataset of protein species from human liver. Data Brief 2017, 12, 584–588. [Google Scholar] [CrossRef]
- Doll, S.; Dreßen, M.; Geyer, P.E.; Itzhak, D.N.; Braun, C.; Doppler, S.A.; Meier, F.; Deutsch, M.-A.; Lahm, H.; Lange, R.; et al. Region and cell-type resolved quantitative proteomic map of the human heart. Nat. Commun. 2017, 8, 1469. [Google Scholar] [CrossRef]
- Moreno-Sánchez, I.; Font-Clos, F.; Corral, Á. Large-Scale Analysis of Zipf’s Law in English Texts. PLoS ONE 2016, 11, e0147073. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, C.; Kaneko, K. Zipf’s Law in Gene Expression. Phys. Rev. Lett. 2003, 90, 088102. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, V.A.; Knott, G.D.; Bonner, R.F. General Statistics of Stochastic Process of Gene Expression in Eukaryotic Cells. Genetics 2002, 161, 1321–1332. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, K.; Hatano, N. Universality of Zipf’s Law. J. Phys. Soc. Jpn. 2002, 71, 1211–1213. [Google Scholar] [CrossRef]
- Corominas-Murtra, B.; Solé, R.V. Universality of Zipf’s law. Phys. Rev. E 2010, 82, 011102. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Average Volume (μm3) | BNID [30] ¹ |
|---|---|---|
| Platelet | 10 | [33] |
| Sperm cell | 30 | 109,891 |
| Red blood cell | 100 | 107,600 |
| Lymphocyte | 130 | 111,439 |
| Neutrophil | 300 | 108,241 |
| Beta cell | 1000 | 109,227 |
| Enterocyte | 1400 | 111,216 |
| Fibroblast | 2000 | 108,244 |
| HeLa, cervix | 3000 | 103,725 |
| Hepatocyte | 3400 | [32] |
| Osteoblast | 4000 | 108,088 |
| Cardiomyocyte | 15,000 | 108,243 |
| Fat cell | 600,000 | 107,668 |
| Oocyte | 4,000,000 | 101,664 |
| Sample | Equation | Number | Reference |
|---|---|---|---|
| Glio (2DE spots) | y = 13.185x−1.085 R2 = 0.9261 | 1000 | [51] |
| Glio (sec2DE) | y = 13.653x−0.889 R2 = 0.9845 | 24,000 | [51] |
| HepG2 (2DE spots) | y = 17.459x−1 R2 = 0.9776 | 1300 | [27] |
| HepG2 (sec2DE) | y = 9.99x−0.984 R2 = 0.9758 | 20,000 | [27] |
| Liver (sec2DE) | y = 11.549x−0.98 R2 = 0.8952 | 15,000 | [53] |
| Liver (2DE spots) | y = 13.452x−1.16 R2 = 0.9113 | 700 | [53] |
| MCF7 (2DE spots) | y = 14.487x−1.06 R2 = 0.9792 | 700 | [59] |
| Sample | Equation | Number | Reference |
|---|---|---|---|
| Liver | y = 7.0162x−1.056 R2 = 0.9398 | 16,000 | [45] |
| Fetal liver | y = 11.951x−0.943 R2 = 0.9484 | 16,000 | [45] |
| Liver | y = 10.955x−0.958 R2 = 0.9007 | 5000 | [43] |
| Liver | y = 7.2197x−1.047 R2 = 0.8961 | 5500 | [43] |
| Adrenal | y = 7.2765x−1.003 R2 = 0.8627 | 7000 | [43] |
| Adult Adrenal | y = 6.6905x−1.051 R2 = 0.9225 | 15,000 | [45] |
| Adult Colon | y = 13.745x−0.915 R2 = 0.9766 | 15,000 | [45] |
| Colon | y = 11.996x−0.944 R2 = 0.976 | 5000 | [43] |
| Adult Esophagus | y = 15.288x−0.876 R2 = 0.9544 | 9000 | [45] |
| Frontal Cortex | y = 12.544x−0.953 R2 = 0.9297 | 16,000 | [45] |
| Adult gallbladder | y = 14.781x−0.911 R2 = 0.9727 | 10,000 | [45] |
| Adult Pancreas | y = 12.281x−0.948 R2 = 0.9537 | 17,000 | [45] |
| Pancreas | y = 12.537x−0.894 R2 = 0.8619 | 7000 | [43] |
| Prostate | y = 12.246x−0.939 R2 = 0.973 | 4000 ¹ (11,000) | [44] |
| Adult Prostate | y = 14.466x−0.916 R2 = 0.9773 | 17,000 | [45] |
| Adult Rectum | y = 11.495x−0.932 R2 = 0.9755 | 17,000 | [45] |
| Adult Retina | y = 5.8187x−1.079 R2 = 0.9095 | 19,000 | [45] |
| Spinal Cord | y = 11.821x−0.946 R2 = 0.9288 | 15,000 | [45] |
| Adult Testis | y = 8.169x−1.045 R2 = 0.8192 | 20,000 | [45] |
| Testis | y = 11.105x−0.882 R2 = 0.8996 | 9000 | [44] |
| Fetal Testis | y = 5.439x−1.092 R2 = 0.9224 | 15,000 | [45] |
| Placenta | y = 9.3737x−0.998 R2 = 0.9267 | 11,000 | [45] |
| Kidney | y = 5.9506x−1.075 R2 = 0.9228 | 12,000 | [45] |
| Heart | y = 15.719x−0.927 R2 = 0.9755 | 1500 | [44] |
| Heart | y = 9.1228x−1.019 R2 = 0.9233 | 5000 | [43] |
| Heart | y = 12.319x−0.893 R2 = 0.9824 | 13,000 | [45] |
| Aorta | y = 12.254x−1.009 R2 = 0.9655 | 1200 | [61] |
| Aortic valve | y = 17.85x−0.698 R2 = 0.8931 | 6800 | [61] |
| Stomach | y = 10.254x−1.017 R2 = 0.8661 | 5000 | [43] |
| Stomach | y = 15.361x−0.905 R2 = 0.9698 | 4000 | [44] |
| Thyroid | y = 9.698x−1.023 R2 = 0.9185 | 5000 | [43] |
| Muscle | y = 11.563x−0.974 R2 = 0.9422 | 3500 | [43] |
| Muscle | y = 13.174x−0.994 R2 = 0.9409 | 9000 | [44] |
| Brain | y = 10.672x−0.985 R2 = 0.8955 | 6000 | [43] |
| Fetal brain | y = 8.584x−0.981 R2 = 0.9314 | 15,000 | [45] |
| Lung | y = 9.2953x−1.001 R2 = 0.9583 | 12,500 | [45] |
| Lung | y = 8.5254x−1.023 R2 = 0.6913 | 6000 | [43] |
| Ovary | y = 7.3857x−1.053 R2 = 0.931 | 19,000 | [45] |
| Fetal ovary | y = 7.4986x−1.045 R2 = 0.9368 | 17,000 | [45] |
| Ovary | y = 9.8454x−0.929 R2 = 0.9009 | 6800 | [43] |
| Platelets | y = 13.949x−0.949 R2 = 0.9909 | 3600 | [52] |
| Platelets | y = 7.3257x−1.127 R2 = 0.9575 | 11,300 | [45] |
| Uterus | y = 7.7271x−1.059 R2 = 0.9477 | 6000 | [43] |
| B cells | y = 6.5677x−1.051 R2 = 0.9319 | 17,000 | [45] |
| CD4 Cells | y = 7.5448x−1.051 R2 = 0.9533 | 14,000 | [45] |
| NK Cells | y = 7.8551x−1.029 R2 = 0.9616 | 16,000 | [45] |
| HeLa | y = 6.9393x−0.963 R2 = 0.9312 | 6000 ¹ (7000) | [48] |
| HeLa | y = 13.715x−0.931 R2 = 0.9453 | 4700 ¹ (10,200) | [49] |
| HeLa | y = 12.004x−0.931 R2 = 0.9187 | 6200 ¹ (14,000) | [50] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naryzhny, S. Quantitative Aspects of the Human Cell Proteome. Int. J. Mol. Sci. 2023, 24, 8524. https://doi.org/10.3390/ijms24108524
Naryzhny S. Quantitative Aspects of the Human Cell Proteome. International Journal of Molecular Sciences. 2023; 24(10):8524. https://doi.org/10.3390/ijms24108524
Chicago/Turabian StyleNaryzhny, Stanislav. 2023. "Quantitative Aspects of the Human Cell Proteome" International Journal of Molecular Sciences 24, no. 10: 8524. https://doi.org/10.3390/ijms24108524
APA StyleNaryzhny, S. (2023). Quantitative Aspects of the Human Cell Proteome. International Journal of Molecular Sciences, 24(10), 8524. https://doi.org/10.3390/ijms24108524