
Protein–DNA Interactions Regulate Human Papillomavirus DNA Replication, Transcription, and Oncogenesis




Abstract
1. Introduction
2. The HPV Genome
3. Evolution and Diversity of HPV Types
4. Regulation of DNA Replication, Transcription, and Oncogenesis by E2 Protein
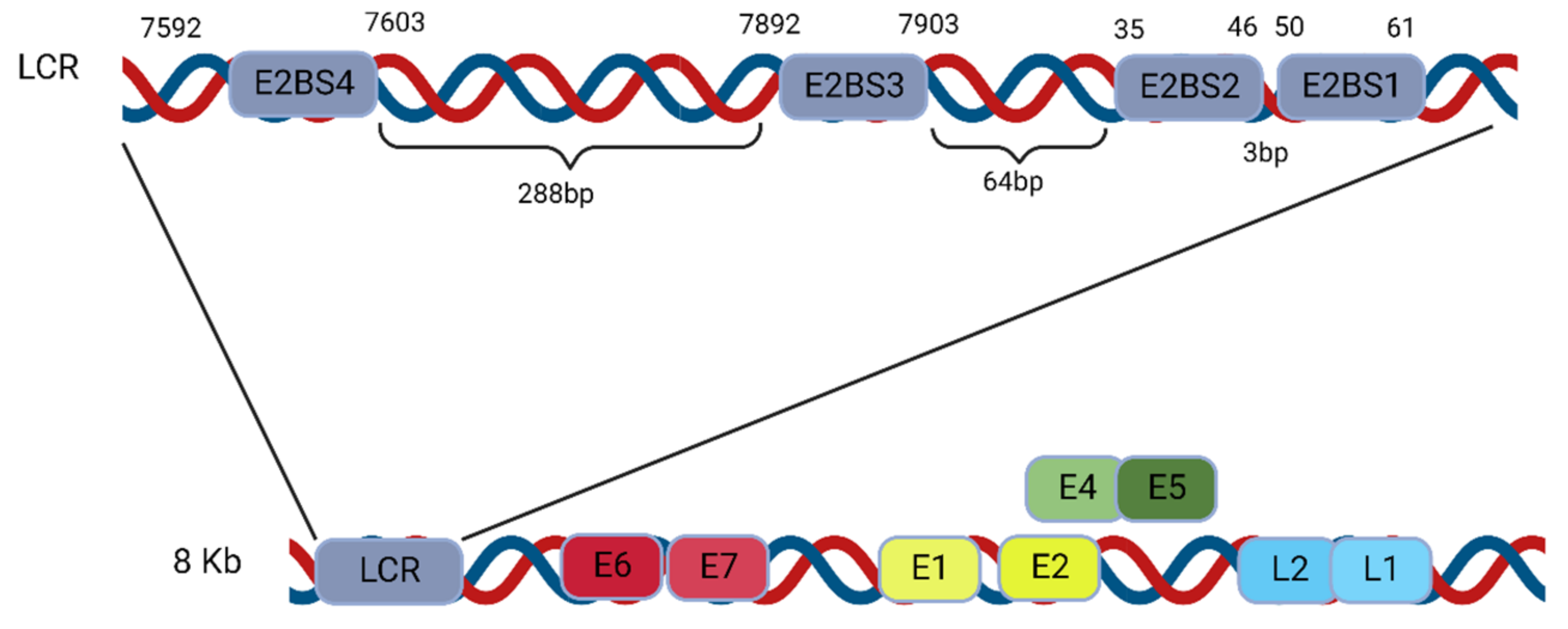
4.1. E2 Binding Sites in the HPV Genomes
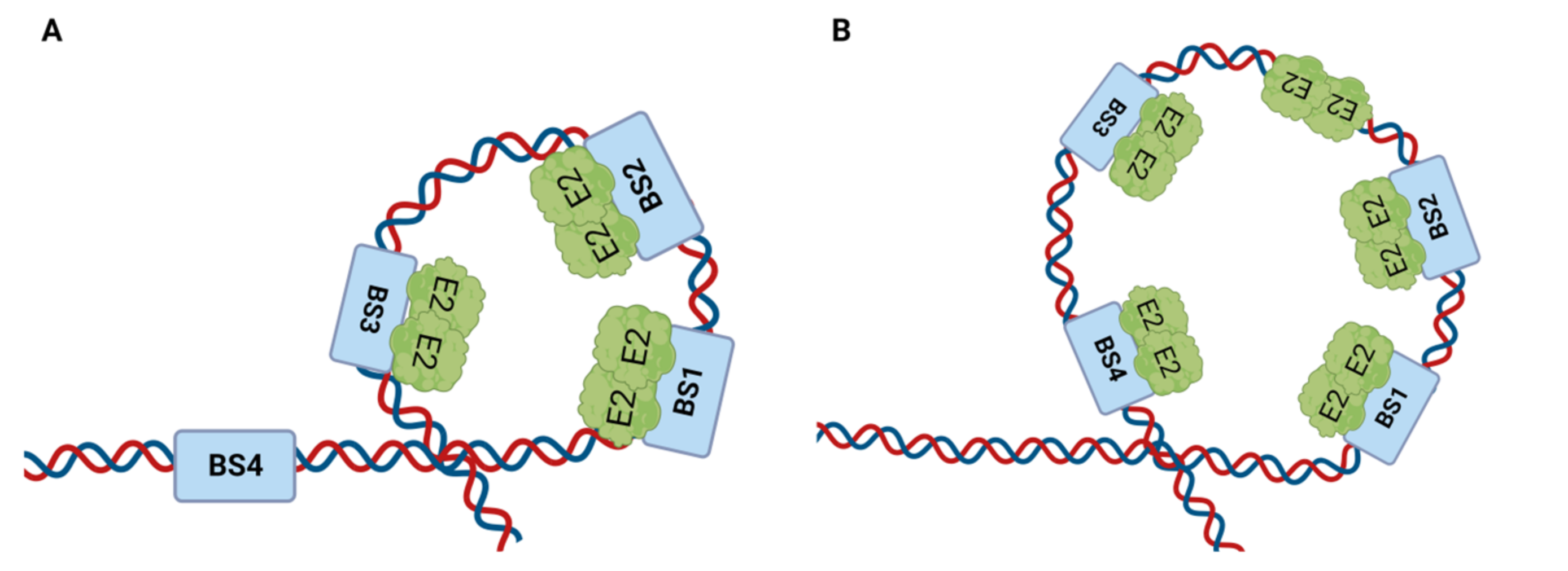
4.1.1. E2 Binding to Four Binding Sites (BS1–4) of the LCR
4.1.2. E2 Binding to Its Binding Sites at the LCR Modulates DNA Replication Initiation
4.2. HPV Life Cycle and Regulation of DNA Replication
4.3. E2 Protein-Mediated Assembly of the Replication Initiation Complex
4.4. HPV Genome Maintenance and Regulation
4.5. Transcriptional Regulation
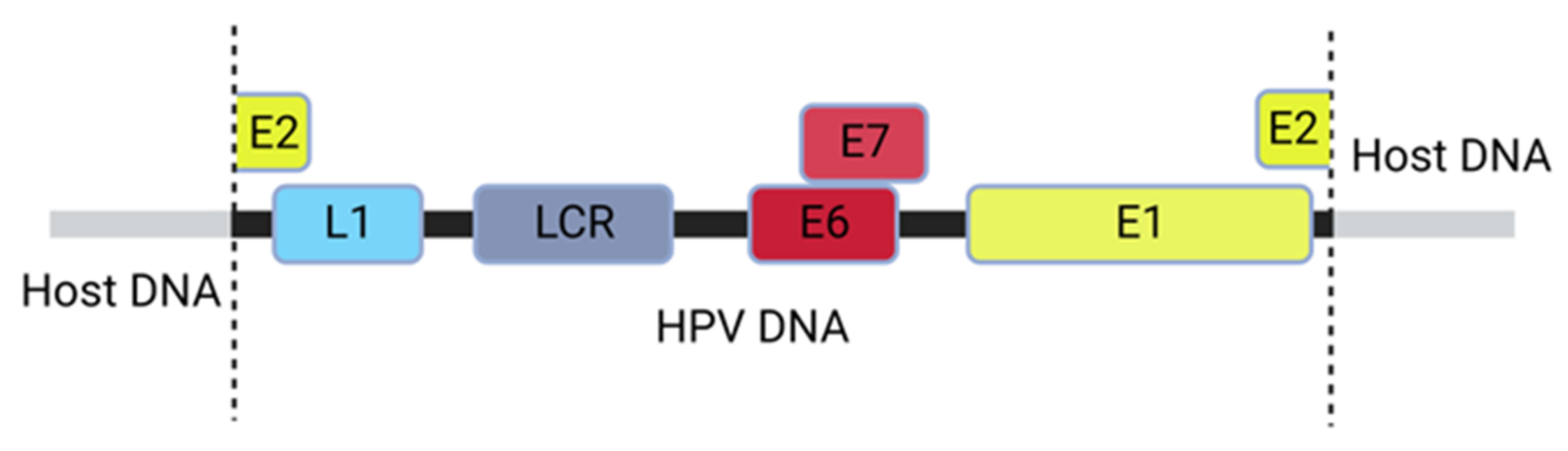
4.6. HPV Infection Leading to Oncogenesis
4.7. Roles of HPV-Encoded E5, E6, and E7 Proteins
5. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- IARC. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans: Human Papillomaviruses. In Working Group on the Evaluation of Carcinogenic Risks to Humans; World Health Organization: Geneva, Switzerland, 1995; Volume 90. [Google Scholar]
- Graham, S.V. Human Papillomavirus E2 Protein: Linking Replication, Transcription, and RNA Processing. J. Virol. 2016, 90, 8384–8388. [Google Scholar] [CrossRef]
- Muller, M.; Demeret, C. The HPV E2-Host Protein-Protein Interactions: A Complex Hijacking of the Cellular Network. Open Virol. J. 2012, 6, 173–189. [Google Scholar] [CrossRef] [PubMed]
- Kajitani, N.; Satsuka, A.; Kawate, A.; Sakai, H. Productive Lifecycle of Human Papillomaviruses that Depends Upon Squamous Epithelial Differentiation. Front. Microbiol. 2012, 3, 152. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.V. Human papillomavirus: Gene expression, regulation and prospects for novel diagnostic methods and antiviral therapies. Future Microbiol. 2010, 5, 1493–1506. [Google Scholar] [CrossRef] [PubMed]
- Graham Sheila, V. The human papillomavirus replication cycle, and its links to cancer progression: A comprehensive review. Clin. Sci. 2017, 131, 2201–2221. [Google Scholar] [CrossRef]
- D’Abramo, C.M.; Archambault, J. Small molecule inhibitors of human papillomavirus protein—Protein interactions. Open Virol. J. 2011, 5, 80–95. [Google Scholar] [CrossRef]
- Ribeiro, A.L.; Caodaglio, A.S.; Sichero, L. Regulation of HPV transcription. Clinics 2018, 73 (Suppl. S1), e486s. [Google Scholar] [CrossRef]
- Ong, C.K.; Chan, S.Y.; Campo, M.S.; Fujinaga, K.; Mavromara-Nazos, P.; Labropoulou, V.; Pfister, H.; Tay, S.K.; ter Meulen, J.; Villa, L.L.; et al. Evolution of human papillomavirus type 18: An ancient phylogenetic root in Africa and intratype diversity reflect coevolution with human ethnic groups. J. Virol. 1993, 67, 6424–6431. [Google Scholar] [CrossRef]
- Zur Hausen, H.; Gissmann, L.; Steiner, W.; Dippold, W.; Dreger, I. Human papilloma viruses and cancer. In Comparative Leukemia Research 1975; Karger Publishers: Basel, Switzerland, 1976; Volume 43l, pp. 569–571. [Google Scholar]
- Bzhalava, D.; Eklund, C.; Dillner, J. International standardization and classification of human papillomavirus types. Virology 2015, 476, 341–344. [Google Scholar] [CrossRef]
- De Koning, M.N.; Quint, K.D.; Bruggink, S.C.; Gussekloo, J.; Bouwes Bavinck, J.N.; Feltkamp, M.C.; Quint, W.G.; Eekhof, J.A. High prevalence of cutaneous warts in elementary school children and the ubiquitous presence of wart-associated human papillomavirus on clinically normal skin. Br. J. Dermatol. 2015, 172, 196–201. [Google Scholar] [CrossRef]
- Egawa, N.; Egawa, K.; Griffin, H.; Doorbar, J. Human Papillomaviruses; Epithelial Tropisms, and the Development of Neoplasia. Viruses 2015, 7, 3863–3890. [Google Scholar] [CrossRef] [PubMed]
- Agalliu, I.; Gapstur, S.; Chen, Z.; Wang, T.; Anderson, R.L.; Teras, L.; Kreimer, A.R.; Hayes, R.B.; Freedman, N.D.; Burk, R.D. Associations of Oral α-, β-, and γ-Human Papillomavirus Types With Risk of Incident Head and Neck Cancer. JAMA Oncol. 2016, 2, 599. [Google Scholar] [CrossRef] [PubMed]
- Sias, C.; Salichos, L.; Lapa, D.; Del Nonno, F.; Baiocchini, A.; Capobianchi, M.R.; Garbuglia, A.R. Alpha, Beta, gamma human PapillomaViruses (HPV) detection with a different sets of primers in oropharyngeal swabs, anal and cervical samples. Virol. J. 2019, 16, 27. [Google Scholar] [CrossRef] [PubMed]
- Braaten, K.P.; Laufer, M.R. Human Papillomavirus (HPV), HPV-Related Disease, and the HPV Vaccine. Rev. Obstet. Gynecol. 2008, 1, 2–10. [Google Scholar] [PubMed]
- Burd, E.M. Human Papillomavirus and Cervical Cancer. Clin. Microbiol. Rev. 2003, 16, 1–17. [Google Scholar] [CrossRef]
- Yilmaz, G.; Biswas-Fiss, E.E.; Biswas, S.B. Genetic variations in the DNA replication origins of human papillomavirus family correlate with their oncogenic potential. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 979–990. [Google Scholar] [CrossRef] [PubMed]
- Sakai, H.; Yasugi, T.; Benson, J.D.; Dowhanick, J.J.; Howley, P.M. Targeted mutagenesis of the human papillomavirus type 16 E2 transactivation domain reveals separable transcriptional activation and DNA replication functions. J. Virol. 1996, 70, 1602–1611. [Google Scholar] [CrossRef]
- Antson, A.A.; Burns, J.E.; Moroz, O.V.; Scott, D.J.; Sanders, C.M.; Bronstein, I.B.; Dodson, G.G.; Wilson, K.S.; Maitland, N.J. Structure of the intact transactivation domain of the human papillomavirus E2 protein. Nature 2000, 403, 805–809. [Google Scholar] [CrossRef]
- Hegde, R.S. The Papillomavirus E2 Proteins: Structure, Function, and Biology. Annu. Rev. Biophys. Biomol. Struct. 2002, 31, 343–360. [Google Scholar] [CrossRef]
- McBride, A.A. The papillomavirus E2 proteins. Virology 2013, 445, 57–79. [Google Scholar] [CrossRef]
- Lace, M.J.; Anson, J.R.; Thomas, G.S.; Turek, L.P.; Haugen, T.H. The E8^E2 gene product of human papillomavirus type 16 represses early transcription and replication but is dispensable for viral plasmid persistence in keratinocytes. J. Virol. 2008, 82, 10841–10853. [Google Scholar] [CrossRef] [PubMed]
- Hawley-Nelson, P.; Androphy, E.J.; Lowy, D.R.; Schiller, J.T. The specific DNA recognition sequence of the bovine papillomavirus E2 protein is an E2-dependent enhancer. EMBO J 1988, 7, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Bedrosian, C.L.; Bastia, D. The DNA-binding domain of HPV-16 E2 protein interaction with the viral enhancer: Protein-induced DNA bending and role of the nonconserved core sequence in binding site affinity. Virology 1990, 174, 557–575. [Google Scholar] [CrossRef] [PubMed]
- Thain, A.; Webster, K.; Emery, D.; Clarke, A.R.; Gaston, K. DNA Binding and Bending by the Human Papillomavirus Type 16 E2 Protein. J. Biol. Chem. 1997, 272, 8236–8242. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, G.; Biswas-Fiss, E.E.; Biswas, S.B. Sequence-Dependent Interaction of the Human Papillomavirus E2 Protein with the DNA Elements on Its DNA Replication Origin. Int. J. Mol. Sci. 2023, 24, 6555. [Google Scholar] [CrossRef]
- Aksoy, P.; Gottschalk, E.Y.; Meneses, P.I. HPV entry into cells. Mutat. Res./Rev. Mutat. Res. 2017, 772, 13–22. [Google Scholar] [CrossRef]
- Giroglou, T.; Florin, L.; Schäfer, F.; Streeck, R.E.; Sapp, M. Human Papillomavirus Infection Requires Cell Surface Heparan Sulfate. J. Virol. 2001, 75, 1565–1570. [Google Scholar] [CrossRef]
- Letian, T.; Tianyu, Z. Cellular receptor binding and entry of human papillomavirus. Virol. J. 2010, 7, 2. [Google Scholar] [CrossRef]
- Wang, J.W.; Roden, R.B.S. L2, the minor capsid protein of papillomavirus. Virology 2013, 445, 175–186. [Google Scholar] [CrossRef]
- Schelhaas, M.; Shah, B.; Holzer, M.; Blattmann, P.; Kühling, L.; Day, P.M.; Schiller, J.T.; Helenius, A. Entry of Human Papillomavirus Type 16 by Actin-Dependent, Clathrin- and Lipid Raft-Independent Endocytosis. PLoS Pathog. 2012, 8, e1002657. [Google Scholar] [CrossRef]
- Spoden, G.; Freitag, K.; Husmann, M.; Boller, K.; Sapp, M.; Lambert, C.; Florin, L. Clathrin- and Caveolin-Independent Entry of Human Papillomavirus Type 16—Involvement of Tetraspanin-Enriched Microdomains (TEMs). PLoS ONE 2008, 3, e3313. [Google Scholar] [CrossRef] [PubMed]
- Kuo, S.R.; Liu, J.S.; Broker, T.R.; Chow, L.T. Cell-free replication of the human papillomavirus DNA with homologous viral E1 and E2 proteins and human cell extracts. J. Biol. Chem. 1994, 269, 24058–24065. [Google Scholar] [CrossRef] [PubMed]
- Romanczuk, H.; Thierry, F.; Howley, P.M. Mutational analysis of cis elements involved in E2 modulation of human papillomavirus type 16 P97 and type 18 P105 promoters. J. Virol. 1990, 64, 2849–2859. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Haberstroh, F.S.; White, E.A.; Livingston, D.M.; DeCaprio, J.A.; Howley, P.M. SMCX and components of the TIP60 complex contribute to E2 regulation of the HPV E6/E7 promoter. Virology 2014, 468–470, 311–321. [Google Scholar] [CrossRef]
- Smith, J.A.; White, E.A.; Sowa, M.E.; Powell, M.L.C.; Ottinger, M.; Harper, J.W.; Howley, P.M. Genome-wide siRNA screen identifies SMCX, EP400, and Brd4 as E2-dependent regulators of human papillomavirus oncogene expression. Proc. Natl. Acad. Sci. USA 2010, 107, 3752–3757. [Google Scholar] [CrossRef]
- Chiang, C.M.; Ustav, M.; Stenlund, A.; Ho, T.F.; Broker, T.R.; Chow, L.T. Viral E1 and E2 proteins support replication of homologous and heterologous papillomaviral origins. Proc. Natl. Acad. Sci. USA 1992, 89, 5799–5803. [Google Scholar] [CrossRef]
- McBride, A.A. Replication and partitioning of papillomavirus genomes. Adv. Virus Res. 2008, 72, 155–205. [Google Scholar]
- Vecchio, A.M.D.; Romanczuk, H.; Howley, P.M.; Baker, C.C. Transient replication of human papillomavirus DNAs. J. Virol. 1992, 66, 5949–5958. [Google Scholar] [CrossRef]
- Bramhill, D.; Kornberg, A. A model for initiation at origins of DNA replication. Cell 1988, 54, 915–918. [Google Scholar] [CrossRef]
- Funnell, B.E.; Baker, T.A.; Kornberg, A. In vitro assembly of a prepriming complex at the origin of the Escherichia coli chromosome. J. Biol. Chem. 1987, 262, 10327–10334. [Google Scholar] [CrossRef]
- Alfano, C.; McMacken, R. Ordered Assembly of Nucleoprotein Structures at the Bacteriophage λ Replication Origin during the Initiation of DNA Replication. J. Biol. Chem. 1989, 264, 10699–10708. [Google Scholar] [CrossRef] [PubMed]
- Gaczynska, M.; Osmulski, P.A.; Jiang, Y.; Lee, J.-K.; Bermudez, V.; Hurwitz, J. Atomic force microscopic analysis of the binding of the Schizosaccharomyces pombe origin recognition complex and the spOrc4 protein with origin DNA. Proc. Natl. Acad. Sci. USA 2004, 101, 17952–17957. [Google Scholar] [CrossRef] [PubMed]
- Sim, J.; Ozgur, S.; Lin, B.Y.; Yu, J.-H.; Broker, T.R.; Chow, L.T.; Griffith, J. Remodeling of the Human Papillomavirus Type 11 Replication Origin into Discrete Nucleoprotein Particles and Looped Structures by the E2 Protein. J. Mol. Biol. 2008, 375, 1165–1177. [Google Scholar] [CrossRef]
- Berg, M.; Stenlund, A. Functional interactions between papillomavirus E1 and E2 proteins. J. Virol. 1997, 71, 3853–3863. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.A.; Florin, L.; Sapp, C.; Sapp, M. Dissection of human papillomavirus type 33 L2 domains involved in nuclear domains (ND) 10 homing and reorganization. Virology 2003, 314, 161–167. [Google Scholar] [CrossRef]
- Darshan, M.S.; Lucchi, J.; Harding, E.; Moroianu, J. The L2 Minor Capsid Protein of Human Papillomavirus Type 16 Interacts with a Network of Nuclear Import Receptors. J. Virol. 2004, 78, 12179–12188. [Google Scholar] [CrossRef]
- Florin, L.; Sapp, C.; Streeck, R.E.; Sapp, M. Assembly and Translocation of Papillomavirus Capsid Proteins. J. Virol. 2002, 76, 10009–10014. [Google Scholar] [CrossRef]
- You, J.; Croyle, J.L.; Nishimura, A.; Ozato, K.; Howley, P.M. Interaction of the bovine papillomavirus E2 protein with Brd4 tethers the viral DNA to host mitotic chromosomes. Cell 2004, 117, 349–360. [Google Scholar] [CrossRef]
- McBride, A.A.; Oliveira, J.G.; McPhillips, M.G. Partitioning viral genomes in mitosis: Same idea, different targets. Cell Cycle 2006, 5, 1499–1502. [Google Scholar] [CrossRef]
- Iftner, T.; Haedicke-Jarboui, J.; Wu, S.-Y.; Chiang, C.-M. Involvement of Brd4 in different steps of the papillomavirus life cycle. Virus Res. 2017, 231, 76–82. [Google Scholar] [CrossRef]
- Donaldson, M.M.; Boner, W.; Morgan, I.M. TopBP1 Regulates Human Papillomavirus Type 16 E2 Interaction with Chromatin. J. Virol. 2007, 81, 4338–4342. [Google Scholar] [CrossRef] [PubMed]
- Parish, J.L.; Bean, A.M.; Park, R.B.; Androphy, E.J. ChlR1 Is Required for Loading Papillomavirus E2 onto Mitotic Chromosomes and Viral Genome Maintenance. Mol. Cell 2006, 24, 867–876. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A.; Warburton, A. The role of integration in oncogenic progression of HPV-associated cancers. PLoS Pathog. 2017, 13, e1006211. [Google Scholar] [CrossRef] [PubMed]
- Bernard, B.A.; Bailly, C.; Lenoir, M.C.; Darmon, M.; Thierry, F.; Yaniv, M. The human papillomavirus type 18 (HPV18) E2 gene product is a repressor of the HPV18 regulatory region in human keratinocytes. J. Virol. 1989, 63, 4317–4324. [Google Scholar] [CrossRef]
- Dong, G.; Broker, T.R.; Chow, L.T. Human papillomavirus type 11 E2 proteins repress the homologous E6 promoter by interfering with the binding of host transcription factors to adjacent elements. J. Virol. 1994, 68, 1115–1127. [Google Scholar] [CrossRef]
- Nishimura, A.; Ono, T.; Ishimoto, A.; Dowhanick, J.J.; Frizzell, M.A.; Howley, P.M.; Sakai, H. Mechanisms of human papillomavirus E2-mediated repression of viral oncogene expression and cervical cancer cell growth inhibition. J. Virol. 2000, 74, 3752–3760. [Google Scholar] [CrossRef]
- Rank, N.M.; Lambert, P.F. Bovine papillomavirus type 1 E2 transcriptional regulators directly bind two cellular transcription factors, TFIID and TFIIB. J. Virol. 1995, 69, 6323–6334. [Google Scholar] [CrossRef]
- Jang, M.K.; Kwon, D.; McBride, A.A. Papillomavirus E2 proteins and the host BRD4 protein associate with transcriptionally active cellular chromatin. J. Virol. 2009, 83, 2592–2600. [Google Scholar] [CrossRef]
- Võsa, L.; Sudakov, A.; Remm, M.; Ustav, M.; Kurg, R. Identification and analysis of papillomavirus E2 protein binding sites in the human genome. J. Virol. 2012, 86, 348–357. [Google Scholar] [CrossRef]
- Gilbert, W. The lac repressor and the lac operator. Ciba Found. Symp. 1972, 7, 245–259. [Google Scholar]
- Lin, S.Y.; Riggs, A.D. Lac repressor binding to DNA not containing the lac operator and to synthetic poly dAT. Nature 1970, 228, 1184–1186. [Google Scholar] [CrossRef] [PubMed]
- Kovelman, R.; Bilter, G.K.; Glezer, E.; Tsou, A.Y.; Barbosa, M.S. Enhanced transcriptional activation by E2 proteins from the oncogenic human papillomaviruses. J. Virol. 1996, 70, 7549–7560. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.-H.; Gloss, B.; Bernard, H.-U. During negative regulation of the human papillomavirus-16E6 promoter, the viral E2 protein can displace Sp1 from a proximal promoter element. Nucleic Acids Res. 1992, 20, 251–256. [Google Scholar] [CrossRef]
- Van Dyne, E.A.; Henley, S.J.; Saraiya, M.; Thomas, C.C.; Markowitz, L.E.; Benard, V.B. Trends in Human Papillomavirus-Associated Cancers—United States, 1999–2015. MMWR Morb. Mortal. Wkly. Rep. 2018, 67, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Vaccarella, S.; Laversanne, M.; Ferlay, J.; Bray, F. Cervical cancer in Africa, Latin America and the Caribbean and Asia: Regional inequalities and changing trends. Int. J. Cancer 2017, 141, 1997–2001. [Google Scholar] [CrossRef]
- Okunade, K.S. Human papillomavirus and cervical cancer. J. Obstet. Gynaecol. 2020, 40, 602–608. [Google Scholar] [CrossRef]
- Oyervides-Muñoz, M.A.; Pérez-Maya, A.A.; Rodríguez-Gutiérrez, H.F.; Gómez-Macias, G.S.; Fajardo-Ramírez, O.R.; Treviño, V.; Barrera-Saldaña, H.A.; Garza-Rodríguez, M.L. Understanding the HPV integration and its progression to cervical cancer. Infect. Genet. Evol. 2018, 61, 134–144. [Google Scholar] [CrossRef]
- Williams, V.M.; Filippova, M.; Soto, U.; Duerksen-Hughes, P.J. HPV-DNA integration and carcinogenesis: Putative roles for inflammation and oxidative stress. Future Virol. 2011, 6, 45–57. [Google Scholar] [CrossRef]
- Zhou, L.; Qiu, Q.; Zhou, Q.; Li, J.; Yu, M.; Li, K.; Xu, L.; Ke, X.; Xu, H.; Lu, B.; et al. Long-read sequencing unveils high-resolution HPV integration and its oncogenic progression in cervical cancer. Nat. Commun. 2022, 13, 2563. [Google Scholar] [CrossRef]
- Yim, E.K.; Park, J.S. The role of HPV E6 and E7 oncoproteins in HPV-associated cervical carcinogenesis. Cancer Res. Treat. 2005, 37, 319–324. [Google Scholar] [CrossRef]
- Holmes, A.; Lameiras, S.; Jeannot, E.; Marie, Y.; Castera, L.; Sastre-Garau, X.; Nicolas, A. Mechanistic signatures of HPV insertions in cervical carcinomas. NPJ Genom. Med. 2016, 1, 16004. [Google Scholar] [CrossRef] [PubMed]
- Kamal, M.; Lameiras, S.; Deloger, M.; Morel, A.; Vacher, S.; Lecerf, C.; Dupain, C.; Jeannot, E.; Girard, E.; Baulande, S.; et al. Human papilloma virus (HPV) integration signature in Cervical Cancer: Identification of MACROD2 gene as HPV hot spot integration site. Br. J. Cancer 2021, 124, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Mainguené, J.; Vacher, S.; Kamal, M.; Hamza, A.; Masliah-Planchon, J.; Baulande, S.; Ibadioune, S.; Borcoman, E.; Cacheux, W.; Calugaru, V.; et al. Human papilloma virus integration sites and genomic signatures in head and neck squamous cell carcinoma. Mol. Oncol. 2022, 16, 3001–3016. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993, 75, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Kelley, M.L.; Keiger, K.E.; Lee, C.J.; Huibregtse, J.M. The global transcriptional effects of the human papillomavirus E6 protein in cervical carcinoma cell lines are mediated by the E6AP ubiquitin ligase. J. Virol. 2005, 79, 3737–3747. [Google Scholar] [CrossRef]
- Nguyen, M.; Song, S.; Liem, A.; Androphy, E.; Liu, Y.; Lambert, P.F. A mutant of human papillomavirus type 16 E6 deficient in binding alpha-helix partners displays reduced oncogenic potential in vivo. J. Virol. 2002, 76, 13039–13048. [Google Scholar] [CrossRef]
- Boyer, S.N.; Wazer, D.E.; Band, V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996, 56, 4620–4624. [Google Scholar]
- Thomas, M.; Banks, L. Inhibition of Bak-induced apoptosis by HPV-18 E6. Oncogene 1998, 17, 2943–2954. [Google Scholar] [CrossRef]
- Thomas, M.; Banks, L. Human papillomavirus (HPV) E6 interactions with Bak are conserved amongst E6 proteins from high and low risk HPV types. J. Gen. Virol. 1999, 80, 1513–1517. [Google Scholar] [CrossRef]
- Filippova, M.; Parkhurst, L.; Duerksen-Hughes, P.J. The Human Papillomavirus 16 E6 Protein Binds to Fas-associated Death Domain and Protects Cells from Fas-triggered Apoptosis. J. Biol. Chem. 2004, 279, 25729–25744. [Google Scholar] [CrossRef]
- Tungteakkhun, S.S.; Filippova, M.; Neidigh, J.W.; Fodor, N.; Duerksen-Hughes, P.J. The interaction between human papillomavirus type 16 and FADD is mediated by a novel E6 binding domain. J. Virol. 2008, 82, 9600–9614. [Google Scholar] [CrossRef]
- Filippova, M.; Song, H.; Connolly, J.L.; Dermody, T.S.; Duerksen-Hughes, P.J. The Human Papillomavirus 16 E6 Protein Binds to Tumor Necrosis Factor (TNF) R1 and Protects Cells from TNF-induced Apoptosis. J. Biol. Chem. 2002, 277, 21730–21739. [Google Scholar] [CrossRef]
- Funk, J.O.; Waga, S.; Harry, J.B.; Espling, E.; Stillman, B.; Galloway, D.A. Inhibition of CDK activity and PCNA-dependent DNA replication by p21 is blocked by interaction with the HPV-16 E7 oncoprotein. Genes Dev. 1997, 11, 2090–2100. [Google Scholar] [CrossRef]
- Jones, D.L.; Alani, R.M.; Münger, K. The human papillomavirus E7 oncoprotein can uncouple cellular differentiation and proliferation in human keratinocytes by abrogating p21<sup>Cip1</sup>-mediated inhibition of cdk2. Genes Dev. 1997, 11, 2101–2111. [Google Scholar]
- Zerfass-Thome, K.; Zwerschke, W.; Mannhardt, B.; Tindle, R.; Botz, J.W.; Jansen-Dürr, P. Inactivation of the cdk inhibitor p27KIP1 by the human papillomavirus type 16 E7 oncoprotein. Oncogene 1996, 13, 2323–2330. [Google Scholar]
- DiMaio, D.; Petti, L.M. The E5 proteins. Virology 2013, 445, 99–114. [Google Scholar] [CrossRef]
- Pim, D.; Collins, M.; Banks, L. Human papillomavirus type 16 E5 gene stimulates the transforming activity of the epidermal growth factor receptor. Oncogene 1992, 7, 27–32. [Google Scholar]
- Straight, S.W.; Hinkle, P.M.; Jewers, R.J.; McCance, D.J. The E5 oncoprotein of human papillomavirus type 16 transforms fibroblasts and effects the downregulation of the epidermal growth factor receptor in keratinocytes. J. Virol. 1993, 67, 4521–4532. [Google Scholar] [CrossRef]
- Barbaresi, S.; Cortese, M.S.; Quinn, J.; Ashrafi, G.H.; Graham, S.V.; Campo, M.S. Effects of human papillomavirus type 16 E5 deletion mutants on epithelial morphology: Functional characterization of each transmembrane domain. J. Gen. Virol. 2010, 91, 521–530. [Google Scholar] [CrossRef]
- Belleudi, F.; Leone, L.; Purpura, V.; Cannella, F.; Scrofani, C.; Torrisi, M.R. HPV16 E5 affects the KGFR/FGFR2b-mediated epithelial growth through alteration of the receptor expression, signaling and endocytic traffic. Oncogene 2011, 30, 4963–4976. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HPV Type | Binding Site (BS) | E2 Protein | Binding Site Sequence | Kd (nM) |
|---|---|---|---|---|
| A. E2 Binding Affinities for HPV11 and HPV16 Binding Sites | ||||
| HPV11 | BS1 | HPV11 E2 | ACCG AAAA CGGT | 5.8 ± 0.7 |
| HPV11 | BS2 | HPV11 E2 | ACCG AAAA CGGT | 5.3 ± 0.4 |
| HPV11 | BS3 | HPV11 E2 | ACCG GTTT CGGT | 8.9 ± 0.9 |
| HPV11 | BS4 | HPV11 E2 | ACCG TTTT CGGT | 3.9 ± 0.5 |
| HPV16 | BS1 | HPV16 E2 | ACCG AAAC CGGT | 7.7 ± 0.7 |
| HPV16 | BS2 | HPV16 E2 | ACCG AAAT CGGT | 4.2 ± 0.4 |
| HPV16, 66 | BS3 | HPV16 E2 | ACCG TTTT gGGT | ≥18 |
| HPV16 | BS4 | HPV16 E2 | ACCG AATT CGGT | 3.8 ± 0.4 |
| HPV16 | BS3 (reversed) | HPV16 E2 | ACCG TTTT CGGT | 7.0 ± 0.3 |
| HPV16 | BS3 | HPV11 E2 | ACCG TTTT gGGT | ≥21 |
| B. E2 Binding Affinities for Variant Binding Sites | ||||
| † HPV18, 30, 33, 39, 45, 51, 53, 59, 68, 70, 97 | Various | HPV16 E2 | ACCG TTTT aGGT | 27 ± 2 |
| HPV31 | BS2 | HPV16 E2 | ACCG TTTT aGGT | 30 ± 3 |
| † HPV34, 35, 56, 58, 73 | Various | HPV16 E2 | ACCG TTTT gGaT | 67 ± 5 |
| † HPV26, 69, 82 | BS3 | HPV16 E2 | ACCG TTTT gtGT | No binding |
| † HPV16, 66, 67, 82 (BS2) | Various | HPV16 E2 | ACCG TTTT gGGT | ≥18 |
| HPV69 (BS2) | BS2 | HPV16 E2 | ACCG AAG CGGT | No binding |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evande, R.; Rana, A.; Biswas-Fiss, E.E.; Biswas, S.B. Protein–DNA Interactions Regulate Human Papillomavirus DNA Replication, Transcription, and Oncogenesis. Int. J. Mol. Sci. 2023, 24, 8493. https://doi.org/10.3390/ijms24108493
Evande R, Rana A, Biswas-Fiss EE, Biswas SB. Protein–DNA Interactions Regulate Human Papillomavirus DNA Replication, Transcription, and Oncogenesis. International Journal of Molecular Sciences. 2023; 24(10):8493. https://doi.org/10.3390/ijms24108493
Chicago/Turabian StyleEvande, Roxanne, Anshul Rana, Esther E. Biswas-Fiss, and Subhasis B. Biswas. 2023. "Protein–DNA Interactions Regulate Human Papillomavirus DNA Replication, Transcription, and Oncogenesis" International Journal of Molecular Sciences 24, no. 10: 8493. https://doi.org/10.3390/ijms24108493
APA StyleEvande, R., Rana, A., Biswas-Fiss, E. E., & Biswas, S. B. (2023). Protein–DNA Interactions Regulate Human Papillomavirus DNA Replication, Transcription, and Oncogenesis. International Journal of Molecular Sciences, 24(10), 8493. https://doi.org/10.3390/ijms24108493