
Clinical Pharmacokinetics of Approved RNA Therapeutics


Abstract
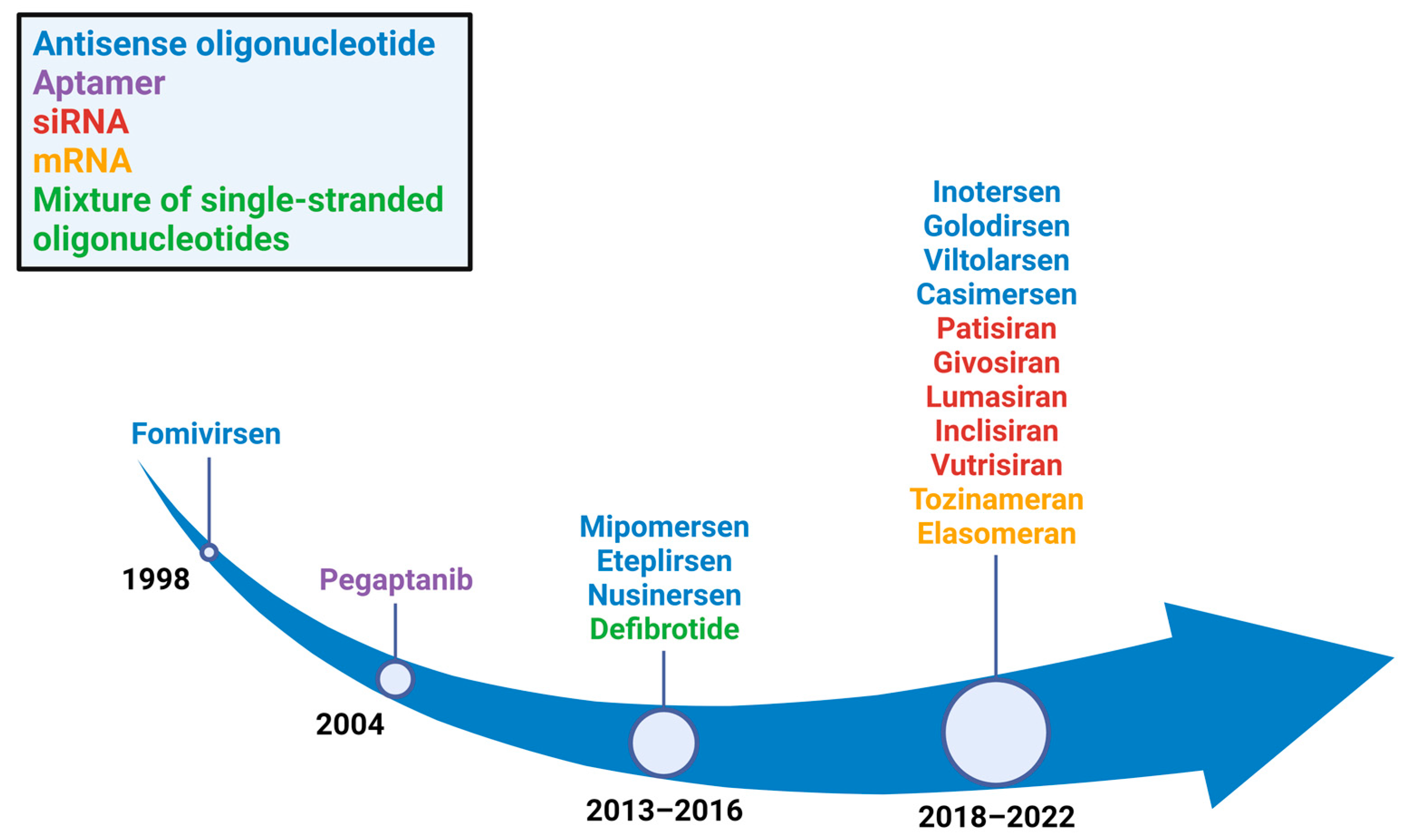
1. Introduction
2. RNA Therapeutics Structure and Base Modification
3. Absorption and Administration Routes
4. Distribution
5. Metabolism
6. Elimination
7. Drug-Drug Interactions
8. Pharmacokinetic-Pharmacodynamic (PK/PD) Relationships
9. Population Pharmacokinetic Analysis
10. The Drawbacks of RNA Therapeutics
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kole, R.; Krainer, A.R.; Altman, S. RNA therapeutics: Beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug Discov. 2012, 11, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Aagaard, L.; Rossi, J.J. RNAi therapeutics: Principles, prospects and challenges. Adv. Drug Deliv. Rev. 2007, 59, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K. RNA therapy: Rich history, various applications and unlimited future prospects. Exp. Mol. Med. 2022, 54, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Park, J.; Pei, Y.; Xu, J.; Yeo, Y. Pharmacokinetics and biodistribution of recently-developed siRNA nanomedicines. Adv. Drug Deliv. Rev. 2016, 104, 93–109. [Google Scholar] [CrossRef]
- Migliorati, J.M.; Liu, S.; Liu, A.; Gogate, A.; Nair, S.; Bahal, R.; Rasmussen, T.P.; Manautou, J.E.; Zhong, X.B. Absorption, distribution, metabolism, and excretion of US Food and Drug Administration-approved antisense oligonucleotide drugs. Drug Metab. Dispos. 2022, 50, 888–897. [Google Scholar] [CrossRef]
- Paunovska, K.; Loughrey, D.; Dahlman, J.E. Drug delivery systems for RNA therapeutics. Nat. Rev. Genet. 2022, 23, 265–280. [Google Scholar] [CrossRef]
- Colquitt, J.L.; Jones, J.; Tan, S.C.; Takeda, A.; Clegg, A.J.; Price, A. Ranibizumab and pegaptanib for the treatment of age-related macular degeneration: A systematic review and economic evaluation. Health Technol. Assess. 2008, 12, 201. [Google Scholar] [CrossRef]
- Espinosa, G.; Berman, H.; Cervera, R. Management of refractory cases of catastrophic antiphospholipid syndrome. Autoimmun. Rev. 2011, 10, 664–668. [Google Scholar] [CrossRef]
- Baker, D.E.; Demaris, K. Defibrotide. Hosp. Pharm. 2016, 51, 847–854. [Google Scholar] [CrossRef]
- Marxreiter, F.; Stemick, J.; Kohl, Z. Huntingtin lowering strategies. Int. J. Mol. Sci. 2020, 21, 2146. [Google Scholar] [CrossRef]
- Damase, T.R.; Sukhovershin, R.; Boada, C.; Taraballi, F.; Pettigrew, R.I.; Cooke, J.P. The limitless future of RNA therapeutics. Front. Bioeng. Biotechnol. 2021, 9, 628137. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, S.C.; Davis, J.A.; Iqbal, S.; Kamel, A.; Kulmatycki, K.; Lao, Y.; Liu, X.; Rodgers, J.; Snoeys, J.; Vigil, A.; et al. Considerations and recommendations for assessment of plasma protein binding and drug-drug interactions for siRNA therapeutics. Nucleic Acids Res. 2022, 50, 6020–6037. [Google Scholar] [CrossRef] [PubMed]
- US FDA. Multi-Discipline Review: Patisiran. 2018. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/210922Orig1s000MultiR.pdf (accessed on 18 December 2022).
- Zhang, X.; Goel, V.; Robbie, G.J. Pharmacokinetics of patisiran, the first approved RNA interference therapy in patients with hereditary transthyretin-mediated amyloidosis. J. Clin. Pharmacol. 2020, 60, 573–585. [Google Scholar] [CrossRef]
- Kilanowska, A.; Studzinska, S. In vivo and in vitro studies of antisense oligonucleotides-a review. RSC Adv. 2020, 10, 34501–34516. [Google Scholar] [CrossRef]
- Dowdy, S.F. Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 2017, 35, 222–229. [Google Scholar] [CrossRef]
- Bost, J.P.; Barriga, H.; Holme, M.N.; Gallud, A.; Maugeri, M.; Gupta, D.; Lehto, T.; Valadi, H.; Esbjorner, E.K.; Stevens, M.M.; et al. Delivery of oligonucleotide therapeutics: Chemical modifications, lipid nanoparticles, and extracellular vesicles. ACS Nano 2021, 15, 13993–14021. [Google Scholar] [CrossRef] [PubMed]
- Terrazas, M.; Kool, E.T. RNA major groove modifications improve siRNA stability and biological activity. Nucleic Acids Res. 2009, 37, 346–353. [Google Scholar] [CrossRef]
- Geary, R.S.; Norris, D.; Yu, R.; Bennett, C.F. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug Deliv. Rev. 2015, 87, 46–51. [Google Scholar] [CrossRef]
- Eckstein, F. Phosphorothioates, essential components of therapeutic oligonucleotides. Nucleic Acid Ther. 2014, 24, 374–387. [Google Scholar] [CrossRef]
- Putney, S.D.; Benkovic, S.J.; Schimmel, P.R. A DNA fragment with an alpha-phosphorothioate nucleotide at one end is asymmetrically blocked from digestion by exonuclease III and can be replicated in vivo. Proc. Natl. Acad. Sci. USA 1981, 78, 7350–7354. [Google Scholar] [CrossRef]
- Nan, Y.; Zhang, Y.J. Antisense Phosphorodiamidate morpholino oligomers as novel antiviral compounds. Front. Microbiol. 2018, 9, 750. [Google Scholar] [CrossRef] [PubMed]
- Ovacik, M.; Lin, K. Tutorial on monoclonal antibody pharmacokinetics and its considerations in early development. Clin. Transl. Sci. 2018, 11, 540–552. [Google Scholar] [CrossRef]
- Keizer, R.J.; Huitema, A.D.; Schellens, J.H.; Beijnen, J.H. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin. Pharmacokinet. 2010, 49, 493–507. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Assessment Report: Leqvio. 2020. Available online: https://www.ema.europa.eu/en/documents/assessment-report/leqvio-epar-public-assessment-report_en.pdf (accessed on 18 December 2022).
- Yu, R.Z.; Grundy, J.S.; Geary, R.S. Clinical pharmacokinetics of second generation antisense oligonucleotides. Expert Opin. Drug Metab. Toxicol. 2013, 9, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, J.; Zhang, X.; Clausen, V.; Tran, C.; Arciprete, M.; Wang, Q.; Rocca, C.; Guan, L.H.; Zhang, G.; et al. Nonclinical pharmacokinetics and absorption, distribution, metabolism, and excretion of givosiran, the first approved N-acetylgalactosamine-conjugated RNA interference therapeutic. Drug Metab. Dispos. 2021, 49, 572–580. [Google Scholar] [CrossRef]
- Leeds, J.M.; Henry, S.P.; Truong, L.; Zutshi, A.; Levin, A.A.; Kornbrust, D. Pharmacokinetics of a potential human cytomegalovirus therapeutic, a phosphorothioate oligonucleotide, after intravitreal injection in the rabbit. Drug Metab. Dispos. 1997, 25, 921–926. [Google Scholar]
- Leeds, J.M.; Henry, S.P.; Bistner, S.; Scherrill, S.; Williams, K.; Levin, A.A. Pharmacokinetics of an antisense oligonucleotide injected intravitreally in monkeys. Drug Metab. Dispos. 1998, 26, 670–675. [Google Scholar]
- Geary, R.S.; Henry, S.P.; Grillone, L.R. Fomivirsen: Clinical pharmacology and potential drug interactions. Clin. Pharmacokinet. 2002, 41, 255–260. [Google Scholar] [CrossRef]
- Nagano, T.; Sakura, S.; Imamachi, N.; Saito, Y. Ultrasound-assisted intrathecal injection of nusinersen in a patient with severe vertebral deformity: A case report. JA Clin. Rep. 2020, 6, 61. [Google Scholar] [CrossRef]
- Khorkova, O.; Wahlestedt, C. Oligonucleotide therapies for disorders of the nervous system. Nat. Biotechnol. 2017, 35, 249–263. [Google Scholar] [CrossRef]
- Chiriboga, C.A.; Swoboda, K.J.; Darras, B.T.; Iannaccone, S.T.; Montes, J.; De Vivo, D.C.; Norris, D.A.; Bennett, C.F.; Bishop, K.M. Results from a phase 1 study of nusinersen (ISIS-SMN(Rx)) in children with spinal muscular atrophy. Neurology 2016, 86, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Rigo, F.; Chun, S.J.; Norris, D.A.; Hung, G.; Lee, S.; Matson, J.; Fey, R.A.; Gaus, H.; Hua, Y.; Grundy, J.S.; et al. Pharmacology of a central nervous system delivered 2’-O-methoxyethyl-modified survival of motor neuron splicing oligonucleotide in mice and nonhuman primates. J. Pharmacol. Exp. Ther. 2014, 350, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, J. Tapping the RNA world for therapeutics. Nat. Struct. Mol. Biol. 2018, 25, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Wanat, K. Biological barriers, and the influence of protein binding on the passage of drugs across them. Mol. Biol. Rep. 2020, 47, 3221–3231. [Google Scholar] [CrossRef] [PubMed]
- McMahon, B.M.; Mays, D.; Lipsky, J.; Stewart, J.A.; Fauq, A.; Richelson, E. Pharmacokinetics and tissue distribution of a peptide nucleic acid after intravenous administration. Antisense Nucleic Acid Drug Dev. 2002, 12, 65–70. [Google Scholar] [CrossRef]
- Amantana, A.; Iversen, P.L. Pharmacokinetics and biodistribution of phosphorodiamidate morpholino antisense oligomers. Curr. Opin. Pharmacol. 2005, 5, 550–555. [Google Scholar] [CrossRef]
- Yu, R.Z.; Kim, T.W.; Hong, A.; Watanabe, T.A.; Gaus, H.J.; Geary, R.S. Cross-species pharmacokinetic comparison from mouse to man of a second-generation antisense oligonucleotide, ISIS 301012, targeting human apolipoprotein B-100. Drug Metab. Dispos. 2007, 35, 460–468. [Google Scholar] [CrossRef]
- Sheng, L.; Rigo, F.; Bennett, C.F.; Krainer, A.R.; Hua, Y. Comparison of the efficacy of MOE and PMO modifications of systemic antisense oligonucleotides in a severe SMA mouse model. Nucleic Acids Res. 2020, 48, 2853–2865. [Google Scholar] [CrossRef]
- US FDA. Vitravene Final Print Label. 2002. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/98/20961_Vitravene_prntlbl.pdf (accessed on 18 December 2022).
- Sands, H.; Gorey-Feret, L.J.; Cocuzza, A.J.; Hobbs, F.W.; Chidester, D.; Trainor, G.L. Biodistribution and metabolism of internally 3H-labeled oligonucleotides. I. Comparison of a phosphodiester and a phosphorothioate. Mol. Pharmacol. 1994, 45, 932–943. [Google Scholar]
- Yu, R.Z.; Geary, R.S.; Monteith, D.K.; Matson, J.; Truong, L.; Fitchett, J.; Levin, A.A. Tissue disposition of 2’-O-(2-methoxy) ethyl modified antisense oligonucleotides in monkeys. J. Pharm. Sci. 2004, 93, 48–59. [Google Scholar] [CrossRef]
- Biessen, E.A.; Vietsch, H.; Rump, E.T.; Fluiter, K.; Kuiper, J.; Bijsterbosch, M.K.; van Berkel, T.J. Targeted delivery of oligodeoxynucleotides to parenchymal liver cells in vivo. Biochem. J. 1999, 340 Pt 3, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.Z.; Graham, M.J.; Post, N.; Riney, S.; Zanardi, T.; Hall, S.; Burkey, J.; Shemesh, C.S.; Prakash, T.P.; Seth, P.P.; et al. Disposition and Pharmacology of a GalNAc3-conjugated ASO targeting human lipoprotein (a) in Mice. Mol. Ther. Nucleic Acids 2016, 5, e317. [Google Scholar] [CrossRef] [PubMed]
- Prakash, T.P.; Graham, M.J.; Yu, J.; Carty, R.; Low, A.; Chappell, A.; Schmidt, K.; Zhao, C.; Aghajan, M.; Murray, H.F.; et al. Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice. Nucleic Acids Res. 2014, 42, 8796–8807. [Google Scholar] [CrossRef]
- Lieberman, A.P.; Yu, Z.; Murray, S.; Peralta, R.; Low, A.; Guo, S.; Yu, X.X.; Cortes, C.J.; Bennett, C.F.; Monia, B.P.; et al. Peripheral androgen receptor gene suppression rescues disease in mouse models of spinal and bulbar muscular atrophy. Cell Rep. 2014, 7, 774–784. [Google Scholar] [CrossRef] [PubMed]
- Cortes, C.J.; Ling, S.C.; Guo, L.T.; Hung, G.; Tsunemi, T.; Ly, L.; Tokunaga, S.; Lopez, E.; Sopher, B.L.; Bennett, C.F.; et al. Muscle expression of mutant androgen receptor accounts for systemic and motor neuron disease phenotypes in spinal and bulbar muscular atrophy. Neuron 2014, 82, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Eder, P.S.; DeVine, R.J.; Dagle, J.M.; Walder, J.A. Substrate specificity and kinetics of degradation of antisense oligonucleotides by a 3’ exonuclease in plasma. Antisense Res. Dev. 1991, 1, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, M.; Cieslak, M.; Stec, W.J.; Goding, J.W.; Koziolkiewicz, M. Nucleotide pyrophosphatase/phosphodiesterase 1 is responsible for degradation of antisense phosphorothioate oligonucleotides. Oligonucleotides 2007, 17, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Dirin, M.; Winkler, J. Influence of diverse chemical modifications on the ADME characteristics and toxicology of antisense oligonucleotides. Expert Opin. Biol. Ther. 2013, 13, 875–888. [Google Scholar] [CrossRef]
- Agarwal, S.; Simon, A.R.; Goel, V.; Habtemariam, B.A.; Clausen, V.A.; Kim, J.B.; Robbie, G.J. Pharmacokinetics and pharmacodynamics of the small interfering ribonucleic acid, givosiran, in patients with acute hepatic porphyria. Clin. Pharmacol. Ther. 2020, 108, 63–72. [Google Scholar] [CrossRef]
- European Medicines Agency. Assessment Report: Oxlumo. 2020. Available online: https://www.ema.europa.eu/en/documents/assessment-report/oxlumo-epar-public-assessment-report_en.pdf (accessed on 18 December 2022).
- Deng, P.; Chen, X.; Zhang, G.; Zhong, D. Bioanalysis of an oligonucleotide and its metabolites by liquid chromatography-tandem mass spectrometry. J. Pharm. Biomed. Anal. 2010, 52, 571–579. [Google Scholar] [CrossRef]
- Gilar, M.; Belenky, A.; Smisek, D.L.; Bourque, A.; Cohen, A.S. Kinetics of phosphorothioate oligonucleotide metabolism in biological fluids. Nucleic Acids Res. 1997, 25, 3615–3620. [Google Scholar] [CrossRef] [PubMed]
- Ramsden, D.; Wu, J.T.; Zerler, B.; Iqbal, S.; Jiang, J.; Clausen, V.; Aluri, K.; Gu, Y.; Dennin, S.; Kim, J.; et al. In vitro drug-drug interaction evaluation of GalNAc conjugated siRNAs against CYP450 enzymes and transporters. Drug Metab. Dispos. 2019, 47, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- US FDA. Clinical Pharmacology Considerations for Human Radiolabeled Mass Balance Studies. 2022. Available online: https://www.fda.gov/media/158178/download (accessed on 18 December 2022).
- Coppola, P.; Andersson, A.; Cole, S. The importance of the human mass balance study in regulatory submissions. CPT Pharmacomet. Syst. Pharm. 2019, 8, 792–804. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. CHMP Assessment Report: Exondys. 2018. Available online: https://www.ema.europa.eu/en/documents/assessment-report/exondys-epar-refusal-public-assessment-report_en.pdf (accessed on 18 December 2022).
- Bell, D.A.; Hooper, A.J.; Burnett, J.R. Mipomersen, an antisense apolipoprotein B synthesis inhibitor. Expert Opin. Investig. Drugs 2011, 20, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T.; Geary, R.S. Clinical pharmacological properties of mipomersen (Kynamro), a second generation antisense inhibitor of apolipoprotein B. Br. J. Clin. Pharmacol. 2013, 76, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.A.; Scheinberg, M.; Waddington-Cruz, M.; Heitner, S.B.; Karam, C.; Drachman, B.; Khella, S.; Whelan, C.; Obici, L. Inotersen for the treatment of adults with polyneuropathy caused by hereditary transthyretin-mediated amyloidosis. Expert Rev. Clin. Pharmacol. 2019, 12, 701–711. [Google Scholar] [CrossRef]
- Neil, E.E.; Bisaccia, E.K. Nusinersen: A novel antisense oligonucleotide for the treatment of spinal muscular atrophy. J. Pediatr. Pharmacol. Ther. 2019, 24, 194–203. [Google Scholar] [CrossRef]
- US FDA. Clinical Pharmacology and Biopharmaceutics Review(s): Casimersen. 2021. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2021/213026Orig1s000ClinPharmR.pdf (accessed on 18 December 2022).
- US FDA. Clinical Pharmacology Review(s): Golodirsen. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/211970Orig1s000ClinPharmR.pdf (accessed on 18 December 2022).
- US FDA. Clinical Pharmacology and Biopharmaceutics Review(s): Viltolarsen. 2020. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/212154Orig1s000ClinPharmR.pdf (accessed on 18 December 2022).
- European Medicines Agency. Assessment Report: Givlaari. 2020. Available online: https://www.ema.europa.eu/en/documents/assessment-report/givlaari-epar-public-assessment-report_en.pdf (accessed on 18 December 2022).
- Scicchitano, P.; Milo, M.; Mallamaci, R.; De Palo, M.; Caldarola, P.; Massari, F.; Gabrielli, D.; Colivicchi, F.; Ciccone, M.M. Inclisiran in lipid management: A Literature overview and future perspectives. Biomed. Pharm. 2021, 143, 112227. [Google Scholar] [CrossRef]
- Frishberg, Y.; Deschenes, G.; Groothoff, J.W.; Hulton, S.A.; Magen, D.; Harambat, J.; Van’t Hoff, W.G.; Lorch, U.; Milliner, D.S.; Lieske, J.C.; et al. Phase 1/2 Study of lumasiran for treatment of primary hyperoxaluria type 1: A placebo-controlled randomized clinical trial. Clin. J. Am. Soc. Nephrol. 2021, 16, 1025–1036. [Google Scholar] [CrossRef]
- Habtemariam, B.A.; Karsten, V.; Attarwala, H.; Goel, V.; Melch, M.; Clausen, V.A.; Garg, P.; Vaishnaw, A.K.; Sweetser, M.T.; Robbie, G.J.; et al. Single-dose pharmacokinetics and pharmacodynamics of transthyretin targeting N-acetylgalactosamine-small interfering ribonucleic acid conjugate, vutrisiran, in healthy subjects. Clin. Pharmacol. Ther. 2021, 109, 372–382. [Google Scholar] [CrossRef]
- Glund, S.; Gan, G.; Moschetti, V.; Reilly, P.; Honickel, M.; Grottke, O.; Van Ryn, J. The Renal elimination pathways of the dabigatran reversal agent idarucizumab and its impact on dabigatran elimination. Clin. Appl. Thromb. Hemost. 2018, 24, 724–733. [Google Scholar] [CrossRef] [PubMed]
- Tapia, E.; Zatarain-Barron, Z.L.; Hernandez-Pando, R.; Zarco-Marquez, G.; Molina-Jijon, E.; Cristobal-Garcia, M.; Santamaria, J.; Pedraza-Chaverri, J. Curcumin reverses glomerular hemodynamic alterations and oxidant stress in 5/6 nephrectomized rats. Phytomedicine 2013, 20, 359–366. [Google Scholar] [CrossRef] [PubMed]
- McDougall, R.; Ramsden, D.; Agarwal, S.; Agarwal, S.; Aluri, K.; Arciprete, M.; Brown, C.; Castellanos-Rizaldos, E.; Charisse, K.; Chong, S.; et al. The nonclinical disposition and pharmacokinetic/pharmacodynamic properties of N-acetylgalactosamine-conjugated small interfering RNA are highly predictable and build confidence in translation to human. Drug Metab. Dispos. 2022, 50, 781–797. [Google Scholar] [CrossRef] [PubMed]
- Wright, R.S.; Collins, M.G.; Stoekenbroek, R.M.; Robson, R.; Wijngaard, P.L.J.; Landmesser, U.; Leiter, L.A.; Kastelein, J.J.P.; Ray, K.K.; Kallend, D. Effects of renal impairment on the pharmacokinetics, efficacy, and safety of inclisiran: An analysis of the ORION-7 and ORION-1 studies. Mayo Clin. Proc. 2020, 95, 77–89. [Google Scholar] [CrossRef]
- Vassiliou, D.; Sardh, E.; Harper, P.; Simon, A.R.; Clausen, V.A.; Najafian, N.; Robbie, G.J.; Agarwal, S. A Drug-drug interaction study evaluating the effect of givosiran, a small interfering ribonucleic acid, on cytochrome P450 activity in the liver. Clin. Pharmacol. Ther. 2021, 110, 1250–1260. [Google Scholar] [CrossRef]
- Shugarts, S.; Benet, L.Z. The role of transporters in the pharmacokinetics of orally administered drugs. Pharm. Res. 2009, 26, 2039–2054. [Google Scholar] [CrossRef]
- Terada, T.; Hira, D. Intestinal and hepatic drug transporters: Pharmacokinetic, pathophysiological, and pharmacogenetic roles. J. Gastroenterol. 2015, 50, 508–519. [Google Scholar] [CrossRef]
- Lee, S.C.; Arya, V.; Yang, X.; Volpe, D.A.; Zhang, L. Evaluation of transporters in drug development: Current status and contemporary issues. Adv. Drug Deliv. Rev. 2017, 116, 100–118. [Google Scholar] [CrossRef]
- Ellens, H.; Deng, S.; Coleman, J.; Bentz, J.; Taub, M.E.; Ragueneau-Majlessi, I.; Chung, S.P.; Heredi-Szabo, K.; Neuhoff, S.; Palm, J.; et al. Application of receiver operating characteristic analysis to refine the prediction of potential digoxin drug interactions. Drug Metab. Dispos. 2013, 41, 1367–1374. [Google Scholar] [CrossRef]
- US FDA. Clinical Pharmacology and Biopharmaceutics Review(s): Inotersen. 2018. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/211172Orig1s000ClinPharmR.pdf (accessed on 18 December 2022).
- European Medicines Agency. Assessment Report: Kynamro. 2011. Available online: https://www.ema.europa.eu/en/documents/assessment-report/kynamro-epar-public-assessment-report_en.pdf (accessed on 18 December 2022).
- US FDA. Clinical Pharmacology and Biopharmaceutics Review(s): Nusinersen. 2016. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/209531Orig1s000ClinPharmR.pdf (accessed on 18 December 2022).
- McElnay, J.C.; D’Arcy, P.F. Protein binding displacement interactions and their clinical importance. Drugs 1983, 25, 495–513. [Google Scholar] [CrossRef]
- Crooke, S.T.; Vickers, T.A.; Liang, X.H. Phosphorothioate modified oligonucleotide-protein interactions. Nucleic Acids Res. 2020, 48, 5235–5253. [Google Scholar] [CrossRef] [PubMed]
- Gaus, H.J.; Gupta, R.; Chappell, A.E.; Ostergaard, M.E.; Swayze, E.E.; Seth, P.P. Characterization of the interactions of chemically-modified therapeutic nucleic acids with plasma proteins using a fluorescence polarization assay. Nucleic Acids Res. 2019, 47, 1110–1122. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, R.Z.; Henry, S.; Geary, R.S. Pharmacokinetics and clinical pharmacology considerations of GalNAc3-conjugated antisense oligonucleotides. Expert Opin. Drug Metab. Toxicol. 2019, 15, 475–485. [Google Scholar] [CrossRef]
- Yu, R.Z.; Gunawan, R.; Post, N.; Zanardi, T.; Hall, S.; Burkey, J.; Kim, T.W.; Graham, M.J.; Prakash, T.P.; Seth, P.P.; et al. Disposition and pharmacokinetics of a GalNAc3-conjugated antisense oligonucleotide targeting human lipoprotein (a) in monkeys. Nucleic Acid Ther. 2016, 26, 372–380. [Google Scholar] [CrossRef]
- Derendorf, H.; Meibohm, B. Modeling of pharmacokinetic/pharmacodynamic (PK/PD) relationships: Concepts and perspectives. Pharm. Res. 1999, 16, 176–185. [Google Scholar] [CrossRef]
- European Medicines Agency. Assessment Report: Tegsedi. 2018. Available online: https://www.ema.europa.eu/en/documents/assessment-report/tegsedi-epar-public-assessment-report_en.pdf (accessed on 18 December 2022).
- An, G. Concept of pharmacologic target-mediated drug disposition in large-molecule and small-molecule compounds. J. Clin. Pharmacol. 2020, 60, 149–163. [Google Scholar] [CrossRef]
- Ayyar, V.S.; Song, D.; Zheng, S.; Carpenter, T.; Heald, D.L. Minimal physiologically based pharmacokinetic-pharmacodynamic (mPBPK-PD) model of N-Acetylgalactosamine-conjugated small interfering RNA disposition and gene silencing in preclinical species and humans. J. Pharmacol. Exp. Ther. 2021, 379, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.R.; Kuntz, N.L.; Koenig, E.; East, L.; Upadhyay, S.; Han, B.; Shieh, P.B. Safety, tolerability, and pharmacokinetics of casimersen in patients with Duchenne muscular dystrophy amenable to exon 45 skipping: A randomized, double-blind, placebo-controlled, dose-titration trial. Muscle Nerve 2021, 64, 285–292. [Google Scholar] [CrossRef]
- Shimizu, R.; Kitade, M.; Kobayashi, T.; Hori, S.; Watanabe, A. Pharmacokinetic-pharmacodynamic modeling for reduction of hepatic apolipoprotein B mRNA and plasma total cholesterol after administration of antisense oligonucleotide in mice. J. Pharmacokinet. Pharmacodyn. 2015, 42, 67–77. [Google Scholar] [CrossRef]
- Mahmood, I.; Duan, J. Population pharmacokinetics with a very small sample size. Drug Metab. Drug Interact. 2009, 24, 259–274. [Google Scholar] [CrossRef]
- Yu, R.Z.; Collins, J.W.; Hall, S.; Ackermann, E.J.; Geary, R.S.; Monia, B.P.; Henry, S.P.; Wang, Y. Population pharmacokinetic-pharmacodynamic modeling of inotersen, an antisense oligonucleotide for treatment of patients with hereditary transthyretin amyloidosis. Nucleic Acid Ther. 2020, 30, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Goel, V.; Gosselin, N.H.; Jomphe, C.; Zhang, X.; Marier, J.F.; Robbie, G.J. Population pharmacokinetic-pharmacodynamic model of serum transthyretin following patisiran administration. Nucleic Acid Ther. 2020, 30, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Roth, C.M. Molecular and cellular barriers limiting the effectiveness of antisense oligonucleotides. Biophys. J. 2005, 89, 2286–2295. [Google Scholar] [CrossRef]
- Kulkarni, J.A.; Darjuan, M.M.; Mercer, J.E.; Chen, S.; van der Meel, R.; Thewalt, J.L.; Tam, Y.Y.C.; Cullis, P.R. On the formation and morphology of lipid nanoparticles containing ionizable cationic lipids and siRNA. ACS Nano 2018, 12, 4787–4795. [Google Scholar] [CrossRef]
- Rai, R.; Alwani, S.; Badea, I. Polymeric nanoparticles in gene therapy: New avenues of design and optimization for delivery applications. Polymers 2019, 11, 745. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.; Abu Lila, A.S.; Shimizu, T.; Alaaeldin, E.; Hussein, A.; Sarhan, H.A.; Szebeni, J.; Ishida, T. PEGylated liposomes: Immunological responses. Sci. Technol. Adv. Mater. 2019, 20, 710–724. [Google Scholar] [CrossRef] [PubMed]
- Kozma, G.T.; Shimizu, T.; Ishida, T.; Szebeni, J. Anti-PEG antibodies: Properties, formation, testing and role in adverse immune reactions to PEGylated nano-biopharmaceuticals. Adv. Drug Deliv. Rev. 2020, 154–155, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.M.; Cheng, T.L.; Roffler, S.R. Polyethylene glycol immunogenicity: Theoretical, clinical, and practical aspects of anti-polyethylene glycol antibodies. ACS Nano 2021, 15, 14022–14048. [Google Scholar] [CrossRef]
- Zhang, X.; Goel, V.; Attarwala, H.; Sweetser, M.T.; Clausen, V.A.; Robbie, G.J. Patisiran pharmacokinetics, pharmacodynamics, and exposure-response analyses in the phase 3 APOLLO trial in patients with gereditary transthyretin-mediated (hATTR) amyloidosis. J. Clin. Pharmacol. 2020, 60, 37–49. [Google Scholar] [CrossRef]
- Kobiyama, K.; Ishii, K.J. Making innate sense of mRNA vaccine adjuvanticity. Nat. Immunol. 2022, 23, 474–476. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Type | Drug | FDA Approval | Indication | Administration Route | Chemistry |
|---|---|---|---|---|---|
| ASO | Fomivirsen | 1998 | Cytomegalovirus retinitis in immunocompromised patients | Intravitreal | Phosphorothioate linkage |
| Mipomersen | 2013 | Homozygous familial hypercholesterolemia | SC | 2′-O-methoxyethylribonucleotides Deoxyribonucleotides Phosphorothioate linkage | |
| Nusinersen | 2016 | Spinal muscular atrophy | Intrathecal | 2′-O-methoxyethylribonucleotides Phosphorothioate linkage | |
| Eteplirsen | 2016 | Duchenne muscular dystrophy | IV | Phosphorodiamidate morpholino oligomer | |
| Defibrotide | 2016 | Veno-occlusive disease | IV | Mixture of single and double stranded oligonucleotides | |
| Inotersen | 2018 | Hereditary transthyretin-mediated amyloidosis | SC | 2′-O-methoxyethylribonucleotides Deoxyribonucleotides Phosphorothioate linkage | |
| Golodirsen | 2019 | Duchenne muscular dystrophy | IV | Phosphorodiamidate morpholino oligomer | |
| Viltolarsen | 2020 | Duchenne muscular dystrophy | IV | Phosphorodiamidate morpholino oligomer | |
| Casimersen | 2021 | Duchenne muscular dystrophy | IV | Phosphorodiamidate morpholino oligomer | |
| siRNA | Patisiran | 2018 | Hereditary transthyretin-mediated amyloidosis | IV | Deoxyribonucleotides 2′-OMe ribonucleotides Lipid nanoparticle |
| Givosiran | 2019 | Acute hepatic porphyria | SC | 2′-F ribonucleotides 2′-OMe ribonucleotides Phosphorothioate linkage GalNAc moiety | |
| Lumasiran | 2020 | Primary hyperoxaluria type 1 | SC | 2′-F ribonucleotides 2′-OMe ribonucleotides Phosphorothioate linkage GalNAc moiety | |
| Inclisiran | 2021 | Primary hypercholesterolemia | SC | 2′-F ribonucleotides 2′-OMe ribonucleotides Phosphorothioate linkage GalNAc moiety | |
| Vutrisiran | 2022 | Hereditary transthyretin-mediated amyloidosis | SC | 2′-F ribonucleotides 2′-OMe ribonucleotides Phosphorothioate linkage GalNAc moiety |
| Type | Drug | Tmax (h) a | Volume of Distribution | Protein Binding (%) | t1/2 |
|---|---|---|---|---|---|
| ASO | Fomivirsen | - | - | 40 (vitreous) | 55 h |
| Mipomersen | - | 48.3 L/kg | >85 | 30 d | |
| Nusinersen | 1–6 | CSF: 0.4 L Plasma: 29 L | CSF: <25 Plasma: >94 | CSF: 133–177 d Plasma: 68–87 d | |
| Eteplirsen | - | 0.601 L/kg | 6.1–16.5 | 1.6–3.6 h | |
| Defibrotide | - | 8.1–9.1 L | 93 | 2 h | |
| Inotersen | 2–4 | 293 L | 94 | 32.3 d | |
| Golodirsen | - | 0.668 L/kg | 33–39 | 3.5 h | |
| Viltolarsen | - | 0.300 L/kg | 39–40 | 2.5 h | |
| Casimersen | - | 0.367 L/kg | 8.4–31.6 | 3.5 h | |
| siRNA | Patisiran | - | 0.26 L/kg | 2.1 | 3.2 d |
| Givosiran | 3 (0.5–8) | 10.4 L | 90 | 6 h | |
| Lumasiran | 4 (0.5–12) | 4.9 L | 85 | 5.2 h | |
| Inclisiran | 4 | 500 L | 87 | 9 h | |
| Vutrisiran | 4 | 10.1 L | 80 | 5.2 h |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jo, S.J.; Chae, S.U.; Lee, C.B.; Bae, S.K. Clinical Pharmacokinetics of Approved RNA Therapeutics. Int. J. Mol. Sci. 2023, 24, 746. https://doi.org/10.3390/ijms24010746
Jo SJ, Chae SU, Lee CB, Bae SK. Clinical Pharmacokinetics of Approved RNA Therapeutics. International Journal of Molecular Sciences. 2023; 24(1):746. https://doi.org/10.3390/ijms24010746
Chicago/Turabian StyleJo, Seong Jun, Soon Uk Chae, Chae Bin Lee, and Soo Kyung Bae. 2023. "Clinical Pharmacokinetics of Approved RNA Therapeutics" International Journal of Molecular Sciences 24, no. 1: 746. https://doi.org/10.3390/ijms24010746
APA StyleJo, S. J., Chae, S. U., Lee, C. B., & Bae, S. K. (2023). Clinical Pharmacokinetics of Approved RNA Therapeutics. International Journal of Molecular Sciences, 24(1), 746. https://doi.org/10.3390/ijms24010746