
Current Understanding on the Role of Lipids in Macrophages and Associated Diseases

Abstract

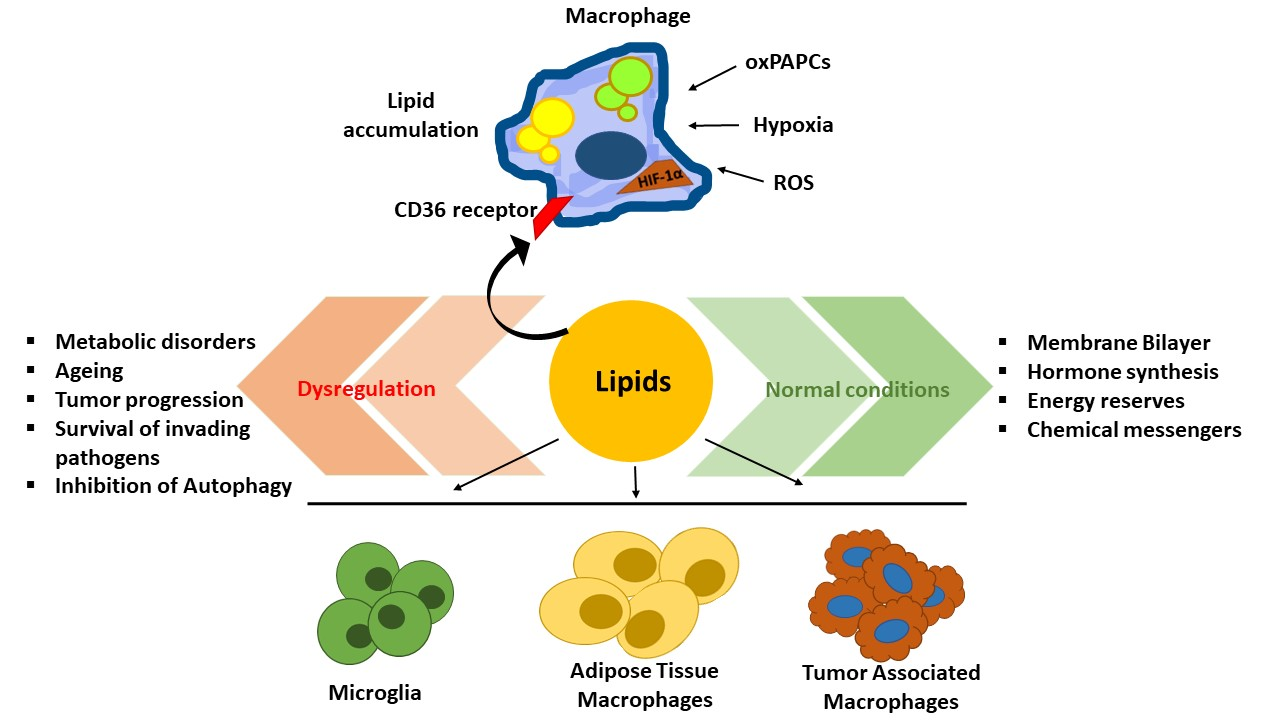
1. Introduction
2. Functions of Lipids in Macrophages of Different Tissue Location
2.1. Lipids and Microglia
2.2. Lipids in Adipose Tissue Macrophages (ATMs)
2.3. Lipids in Tumor Associated Macrophages (TAMs)
2.4. Lipids in Phagocytic Function of Macrophages
2.5. Oxidized Phosphocholines in the Immune Function of Macrophages
3. Modulators of Lipid Metabolism at a Glance
4. The Network of Lipids and ROS in Macrophages
5. HIF-1α, a Master Regulator of Lipid Metabolism
6. Integration of Autophagy in Lipid Metabolism
6.1. PPARα
6.2. JNK
6.3. AMPK
6.4. TFEB
6.5. TAK1
6.6. NF-κB
7. The Lysosomal Lipid Handling in Macrophages
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Xu, J.; Taubert, S. Beyond Proteostasis: Lipid Metabolism as a New Player in Er Homeostasis. Metabolites 2021, 11, 52. [Google Scholar] [CrossRef] [PubMed]
- Jacquemyn, J.; Cascalho, A.; Goodchild, R.E. The Ins and Outs of Endoplasmic Reticulum-controlled Lipid Biosynthesis. EMBO Rep. 2017, 18, 1905–1921. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Watkins, S.M.; Hotamisligil, G.S. The Role of Endoplasmic Reticulum in Hepatic Lipid Homeostasis and Stress Signaling. Cell Metab. 2012, 15, 623–634. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. Cholesterol Feedback: From Schoenheimer’s Bottle to Scap’s MELADL. J. Lipid Res. 2009, 50, S15–S27. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.W. The Enzymes, Regulation, and Genetics of Bile Acid Synthesis. Annu. Rev. Biochem. 2003, 72, 137–174. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Parton, R.G. Lipid Droplets: A Unified View of a Dynamic Organelle. Nat. Rev. Mol. Cell Biol. 2006, 7, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, M.; Ntambi, J.M. Fatty Acid Desaturation and Chain Elongation in Mammals. In Biochemistry of Lipids, Lipoproteins and Membranes; Elsevier: Amsterdam, The Netherlands, 2008; pp. 191–211. ISBN 9780444532190. [Google Scholar]
- Dorotea, D.; Koya, D.; Ha, H. Recent Insights Into SREBP as a Direct Mediator of Kidney Fibrosis via Lipid-Independent Pathways. Front. Pharmacol. 2020, 11, 265. [Google Scholar] [CrossRef]
- Shimano, H.; Horton, J.D.; Shimomura, I.; Hammer, R.E.; Brown, M.S.; Goldstein, J.L. Isoform 1c of Sterol Regulatory Element Binding Protein Is Less Active than Isoform 1a in Livers of Transgenic Mice and in Cultured Cells. J. Clin. Investig. 1997, 99, 846–854. [Google Scholar] [CrossRef]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the Complete Program of Cholesterol and Fatty Acid Synthesis in the Liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Horton, J.D. Sterol Regulatory Element-Binding Proteins: Transcriptional Activators of Lipid Synthesis. Biochem. Soc. Trans 2002, 30, 1091–1095. [Google Scholar] [CrossRef]
- Wilfling, F.; Wang, H.; Haas, J.T.; Krahmer, N.; Gould, T.J.; Uchida, A.; Cheng, J.X.; Graham, M.; Christiano, R.; Fröhlich, F.; et al. Triacylglycerol Synthesis Enzymes Mediate Lipid Droplet Growth by Relocalizing from the ER to Lipid Droplets. Dev. Cell 2013, 24, 384–399. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Reue, K. Lipin Proteins and Glycerolipid Metabolism: Roles at the ER Membrane and beyond. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1583–1595. [Google Scholar] [CrossRef] [PubMed]
- Fagone, P.; Jackowski, S. Membrane Phospholipid Synthesis and Endoplasmic Reticulum Function. J. Lipid Res. 2009, 50. [Google Scholar] [CrossRef] [PubMed]
- Appenzeller-Herzog, C.; Hauri, H.P. The ER-Golgi Intermediate Compartment (ERGIC): In Search of Its Identity and Function. J. Cell Sci. 2006, 119, 2173–2183. [Google Scholar] [CrossRef]
- Farese, R.V.; Walther, T.C. Lipid Droplets Finally Get a Little R-E-S-P-E-C-T. Cell 2009, 139, 855–860. [Google Scholar] [CrossRef]
- McFie, P.J.; Banman, S.L.; Kary, S.; Stone, S.J. Murine Diacylglycerol Acyltransferase-2 (DGAT2) Can Catalyze Triacylglycerol Synthesis and Promote Lipid Droplet Formation Independent of Its Localization to the Endoplasmic Reticulum. J. Biol. Chem. 2011, 286, 28235–28246. [Google Scholar] [CrossRef]
- Salo, V.T.; Li, S.; Vihinen, H.; Hölttä-Vuori, M.; Szkalisity, A.; Horvath, P.; Belevich, I.; Peränen, J.; Thiele, C.; Somerharju, P.; et al. Seipin Facilitates Triglyceride Flow to Lipid Droplet and Counteracts Droplet Ripening via Endoplasmic Reticulum Contact. Dev. Cell 2019, 50, 478–493.e9. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, D.; Liu, X.; Li, X.; Liu, F.; Yu, Y.; Jia, S.; Zhou, Y.; Zhao, Y. Endoplasmic Reticulum Stress Affects Lipid Metabolism in Atherosclerosis Via CHOP Activation and Over-Expression of miR-33. Cell. Physiol. Biochem. 2018, 48, 1995–2010. [Google Scholar] [CrossRef]
- Fu, S.; Yang, L.; Li, P.; Hofmann, O.; Dicker, L.; Hide, W.; Lin, X.; Watkins, S.M.; Ivanov, A.R.; Hotamisligil, G.S. Aberrant Lipid Metabolism Disrupts Calcium Homeostasis Causing Liver Endoplasmic Reticulum Stress in Obesity. Nature 2011, 473, 528–531. [Google Scholar] [CrossRef]
- Röhrl, C.; Eigner, K.; Winter, K.; Korbelius, M.; Obrowsky, S.; Kratky, D.; Kovacs, W.J.; Stangl, H. Endoplasmic Reticulum Stress Impairs Cholesterol Efflux and Synthesis in Hepatic Cells. J. Lipid Res. 2014, 55, 94–103. [Google Scholar] [CrossRef]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian Transcription Factor ATF6 Is Synthesized as a Transmembrane Protein and Activated by Proteolysis in Response to Endoplasmic Reticulum Stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. A Proteolytic Pathway That Controls the Cholesterol Content of Membranes, Cells, and Blood. Proc. Natl. Acad. Sci. USA 1999, 96, 11041–11048. [Google Scholar] [CrossRef]
- Colgan, S.M.; Al-Hashimi, A.A.; Austin, R.C. Endoplasmic Reticulum Stress and Lipid Dysregulation. Expert Rev. Mol. Med. 2011, 13. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Liu, R. ER Stress and Hepatic Lipid Metabolism. Front. Genet. 2014, 5. [Google Scholar] [CrossRef]
- Lee, J.N.; Ye, J. Proteolytic Activation of Sterol Regulatory Element-Binding Protein Induced by Cellular Stress through Depletion of Insig-1. J. Biol. Chem. 2004, 279, 45257–45265. [Google Scholar] [CrossRef]
- Han, J.; Kaufman, R.J. The Role of ER Stress in Lipid Metabolism and Lipotoxicity. J. Lipid Res. 2016, 57, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Oyadomari, S.; Harding, H.P.; Zhang, Y.; Oyadomari, M.; Ron, D. Dephosphorylation of Translation Initiation Factor 2α Enhances Glucose Tolerance and Attenuates Hepatosteatosis in Mice. Cell Metab. 2008, 7, 520–532. [Google Scholar] [CrossRef]
- Rutkowski, D.T.; Wu, J.; Back, S.H.; Callaghan, M.U.; Ferris, S.P.; Iqbal, J.; Clark, R.; Miao, H.; Hassler, J.R.; Fornek, J.; et al. UPR Pathways Combine to Prevent Hepatic Steatosis Caused by ER Stress-Mediated Suppression of Transcriptional Master Regulators. Dev. Cell 2008, 15, 829–840. [Google Scholar] [CrossRef]
- Fei, W.; Wang, H.; Bielby, C.; Yang, H. Conditions of Endoplasmic Reticulum Stress Stimulate Lipid Droplet Formation in Saccharomyces Cerevisiae. Biochem. J. 2009, 424, 61–67. [Google Scholar] [CrossRef]
- Li, H.; Meng, Q.; Xiao, F.; Chen, S.; Du, Y.; Yu, J.; Wang, C.; Guo, F. ATF4 Deficiency Protects Mice from High-Carbohydrate-Diet-Induced Liver Steatosis. Biochem. J. 2011, 438, 283–289. [Google Scholar] [CrossRef]
- Xiao, G.; Zhang, T.; Yu, S.; Lee, S.; Calabuig-Navarro, V.; Yamauchi, J.; Ringquist, S.; Dong, H.H. ATF4 Protein Deficiency Protects against High Fructose-Induced Hypertriglyceridemia in Mice. J. Biol. Chem. 2013, 288, 25350–25361. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Huang, Z.; Du, Y.; Cheng, Y.; Chen, S.; Guo, F. ATF4 Regulates Lipid Metabolism and Thermogenesis. Cell Res. 2010, 20, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Yeh, K.Y.; Lai, C.Y.; Lin, C.Y.; Hsu, C.C.; Lo, C.P.; Her, G.M. ATF4 Overexpression Induces Early Onset of Hyperlipidaemia and Hepatic Steatosis and Enhances Adipogenesis in Zebrafish. Sci. Rep. 2017, 7, 16362. [Google Scholar] [CrossRef]
- Chikka, M.R.; McCabe, D.D.; Tyra, H.M.; Rutkowski, D.T. C/EBP Homologous Protein (CHOP) Contributes to Suppression of Metabolic Genes during Endoplasmic Reticulum Stress in the Liver. J. Biol. Chem. 2013, 288, 4405–4415. [Google Scholar] [CrossRef]
- Bobrovnikova-Marjon, E.; Hatzivassiliou, G.; Grigoriadou, C.; Romero, M.; Cavener, D.R.; Thompson, C.B.; Diehl, J.A. PERK-Dependent Regulation of Lipogenesis during Mouse Mammary Gland Development and Adipocyte Differentiation. Proc. Natl. Acad. Sci. USA 2008, 105, 16314–16319. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Zhao, P.; Huang, P.; Xu, T.; Xiang, X.; Sun, Y.; Liu, J.; Yan, C.; Wang, L.; Gao, J.; Cui, S.; et al. Fat Body Ire1 Regulates Lipid Homeostasis through the Xbp1s-FoxO Axis in Drosophila. iScience 2021, 24, 102819. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA Is Induced by ATF6 and Spliced by IRE1 in Response to ER Stress to Produce a Highly Active Transcription Factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Huang, S.; Xing, Y.; Liu, Y. Emerging Roles for the ER Stress Sensor IRE1 in Metabolic Regulation and Disease. J. Biol. Chem. 2019, 294, 18726–18741. [Google Scholar] [CrossRef]
- Lee, A.H.; Scapa, E.F.; Cohen, D.E.; Glimcher, L.H. Regulation of Hepatic Lipogenesis by the Transcription Factor XBP1. Science 2008, 320, 1492–1496. [Google Scholar] [CrossRef]
- Zhang, K.; Wang, S.; Malhotra, J.; Hassler, J.R.; Back, S.H.; Wang, G.; Chang, L.; Xu, W.; Miao, H.; Leonardi, R.; et al. The Unfolded Protein Response Transducer IRE1Î ± Prevents ER Stress-Induced Hepatic Steatosis. EMBO J. 2011, 30, 1357–1375. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Tang, C.H.A.; Song, J.H.; Mancuso, A.; Del Valle, J.R.; Cao, J.; Xiang, Y.; Dang, C.V.; Lan, R.; Sanchez, D.J.; et al. IRE1α RNase-Dependent Lipid Homeostasis Promotes Survival in Myc-Transformed Cancers. J. Clin. Investig. 2018, 128, 1300–1316. [Google Scholar] [CrossRef] [PubMed]
- So, J.S.; Hur, K.Y.; Tarrio, M.; Ruda, V.; Frank-Kamenetsky, M.; Fitzgerald, K.; Koteliansky, V.; Lichtman, A.H.; Iwawaki, T.; Glimcher, L.H.; et al. Silencing of Lipid Metabolism Genes through ire1α-Mediated Mrna Decay Lowers Plasma Lipids in Mice. Cell Metab. 2012, 16, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Basseri, S.; Austin, R.C. Endoplasmic Reticulum Stress and Lipid Metabolism: Mechanisms and Therapeutic Potential. Biochem. Res. Int. 2012, 2012, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Schiattarella, G.G.; Altamirano, F.; Kim, S.Y.; Tong, D.; Ferdous, A.; Piristine, H.; Dasgupta, S.; Wang, X.; French, K.M.; Villalobos, E.; et al. Xbp1s-FoxO1 Axis Governs Lipid Accumulation and Contractile Performance in Heart Failure with Preserved Ejection Fraction. Nat. Commun. 2021, 12, 1684. [Google Scholar] [CrossRef]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER Stress Regulation of ATF6 Localization by Dissociation of BiP/GRP78 Binding and Unmasking of Golgi Localization Signals. Dev. Cell 2002, 3, 99–111. [Google Scholar] [CrossRef]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Davé, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER Stress Induces Cleavage of Membrane-Bound ATF6 by the Same Proteases That Process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Wu, J.; Rutkowski, D.T.; Dubois, M.; Swathirajan, J.; Saunders, T.; Wang, J.; Song, B.; Yau, G.D.Y.; Kaufman, R.J. ATF6α Optimizes Long-Term Endoplasmic Reticulum Function to Protect Cells from Chronic Stress. Dev. Cell 2007, 13, 351–364. [Google Scholar] [CrossRef]
- Shoulders, M.D.; Ryno, L.M.; Genereux, J.C.; Moresco, J.J.; Tu, P.G.; Wu, C.; Yates, J.R.; Su, A.I.; Kelly, J.W.; Wiseman, R.L. Stress-Independent Activation of XBP1s And/or ATF6 Reveals Three Functionally Diverse ER Proteostasis Environments. Cell Rep. 2013, 3, 1279–1292. [Google Scholar] [CrossRef]
- Radanović, T.; Ernst, R. The Unfolded Protein Response as a Guardian of the Secretory Pathway. Cells 2021, 10, 2965. [Google Scholar] [CrossRef]
- Bommiasamy, H.; Back, S.H.; Fagone, P.; Lee, K.; Meshinchi, S.; Vink, E.; Sriburi, R.; Frank, M.; Jackowski, S.; Kaufman, R.J.; et al. ATF6α Induces XBP1-Independent Expansion of the Endoplasmic Reticulum. J. Cell Sci. 2009, 122, 1626–1636. [Google Scholar] [CrossRef] [PubMed]
- Maiuolo, J.; Bulotta, S.; Verderio, C.; Benfante, R.; Borgese, N. Selective Activation of the Transcription Factor ATF6 Mediates Endoplasmic Reticulum Proliferation Triggered by a Membrane Protein. Proc. Natl. Acad. Sci. USA 2011, 108, 7832–7837. [Google Scholar] [CrossRef]
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional Induction of Mammalian ER Quality Control Proteins Is Mediated by Single or Combined Action of ATF6α and XBP1. Dev. Cell 2007, 13, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Moncan, M.; Mnich, K.; Blomme, A.; Almanza, A.; Samali, A.; Gorman, A.M. Regulation of Lipid Metabolism by the Unfolded Protein Response. J. Cell. Mol. Med. 2021, 25, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Lu, M.; Mori, K.; Luo, S.; Lee, A.S.; Zhu, Y.; Shyy, J.Y.J. ATF6 Modulates SREBP2-Mediated Lipogenesis. EMBO J. 2004, 23, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, F.; Gong, Q.; Cui, A.; Zhuo, S.; Hu, Z.; Han, Y.; Gao, J.; Sun, Y.; Liu, Z.; et al. Hepatic ATF6 Increases Fatty Acid Oxidation to Attenuate Hepatic Steatosis in Mice through Peroxisome Proliferator-Activated Receptor α. Diabetes 2016, 65, 1904–1915. [Google Scholar] [CrossRef]
- DeZwaan-McCabe, D.; Sheldon, R.D.; Gorecki, M.C.; Guo, D.F.; Gansemer, E.R.; Kaufman, R.J.; Rahmouni, K.; Gillum, M.P.; Taylor, E.B.; Teesch, L.M.; et al. ER Stress Inhibits Liver Fatty Acid Oxidation While Unmitigated Stress Leads to Anorexia-Induced Lipolysis and Both Liver and Kidney Steatosis. Cell Rep. 2017, 19, 1794–1806. [Google Scholar] [CrossRef]
- Marschallinger, J.; Iram, T.; Zardeneta, M.; Lee, S.E.; Lehallier, B.; Haney, M.S.; Pluvinage, J.V.; Mathur, V.; Hahn, O.; Morgens, D.W.; et al. Lipid-Droplet-Accumulating Microglia Represent a Dysfunctional and Proinflammatory State in the Aging Brain. Nat. Neurosci. 2020, 23, 194–208. [Google Scholar] [CrossRef]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP Mediates Rapid Microglial Response to Local Brain Injury in Vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Neuroscience: Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma in Vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate Mapping Analysis Reveals That Adult Microglia Derive from Primitive Macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Bachiller, S.; Jiménez-Ferrer, I.; Paulus, A.; Yang, Y.; Swanberg, M.; Deierborg, T.; Boza-Serrano, A. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front. Cell. Neurosci. 2018, 12, 488. [Google Scholar] [CrossRef] [PubMed]
- Wake, H.; Moorhouse, A.J.; Nabekura, J. Functions of Microglia in the Central Nervous System-beyond the Immune Response. Neuron Glia Biol. 2012, 7, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.E.; Kingery, N.D.; Ohsumi, T.K.; Borowsky, M.L.; Wang, L.C.; Means, T.K.; El Khoury, J. The Microglial Sensome Revealed by Direct RNA Sequencing. Nat. Neurosci. 2013, 16, 1896–1905. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef]
- Stephan, A.H.; Barres, B.A.; Stevens, B. The Complement System: An Unexpected Role in Synaptic Pruning during Development and Disease. Annu. Rev. Neurosci. 2012, 35, 369–389. [Google Scholar] [CrossRef]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R.; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.B. Microglia Promote Learning-Dependent Synapse Formation through Brain-Derived Neurotrophic Factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef]
- Ueno, M.; Fujita, Y.; Tanaka, T.; Nakamura, Y.; Kikuta, J.; Ishii, M.; Yamashita, T. Layer v Cortical Neurons Require Microglial Support for Survival during Postnatal Development. Nat. Neurosci. 2013, 16, 543–551. [Google Scholar] [CrossRef]
- Hagemeyer, N.; Hanft, K.M.; Akriditou, M.A.; Unger, N.; Park, E.S.; Stanley, E.R.; Staszewski, O.; Dimou, L.; Prinz, M. Microglia Contribute to Normal Myelinogenesis and to Oligodendrocyte Progenitor Maintenance during Adulthood. Acta Neuropathol. 2017, 134, 441–458. [Google Scholar] [CrossRef]
- Wlodarczyk, A.; Holtman, I.R.; Krueger, M.; Yogev, N.; Bruttger, J.; Khorooshi, R.; Benmamar-Badel, A.; Boer-Bergsma, J.J.; Martin, N.A.; Karram, K.; et al. A Novel Microglial Subset Plays a Key Role in Myelinogenesis in Developing Brain. EMBO J. 2017, 36, 3292–3308. [Google Scholar] [CrossRef]
- Loving, B.A.; Tang, M.; Neal, M.C.; Gorkhali, S.; Murphy, R.; Eckel, R.H.; Bruce, K.D. Lipoprotein Lipase Regulates Microglial Lipid Droplet Accumulation. Cells 2021, 10, 198. [Google Scholar] [CrossRef] [PubMed]
- Bruce, K.D.; Gorkhali, S.; Given, K.; Coates, A.M.; Boyle, K.E.; Macklin, W.B.; Eckel, R.H. Lipoprotein Lipase Is a Feature of Alternatively-Activated Microglia and May Facilitate Lipid Uptake in the CNS during Demyelination. Front. Mol. Neurosci. 2018, 11, 57. [Google Scholar] [CrossRef]
- Lloyd, A.F.; Miron, V.E. The pro-Remyelination Properties of Microglia in the Central Nervous System. Nat. Rev. Neurol. 2019, 15, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Cantuti-Castelvetri, L.; Fitzner, D.; Bosch-Queralt, M.; Weil, M.T.; Su, M.; Sen, P.; Ruhwedel, T.; Mitkovski, M.; Trendelenburg, G.; Lütjohann, D.; et al. Defective Cholesterol Clearance Limits Remyelination in the Aged Central Nervous System. Science 2018, 359, 684–688. [Google Scholar] [CrossRef]
- Nadjar, A. Role of Metabolic Programming in the Modulation of Microglia Phagocytosis by Lipids. Prostaglandins Leukot. Essent. Fat. Acids 2018, 135, 63–73. [Google Scholar] [CrossRef]
- Khatchadourian, A.; Bourque, S.D.; Richard, V.R.; Titorenko, V.I.; Maysinger, D. Dynamics and Regulation of Lipid Droplet Formation in Lipopolysaccharide (LPS)-Stimulated Microglia. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2012, 1821, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Chandak, P.G.; Radović, B.; Aflaki, E.; Kolb, D.; Buchebner, M.; Fröhlich, E.; Magnes, C.; Sinner, F.; Haemmerle, G.; Zechner, R.; et al. Efficient Phagocytosis Requires Triacylglycerol Hydrolysis by Adipose Triglyceride Lipase. J. Biol. Chem. 2010, 285, 20192–20201. [Google Scholar] [CrossRef]
- Patel, T.; Carnwath, T.P.; Wang, X.; Allen, M.; Lincoln, S.J.; Lewis-Tuffin, L.J.; Quicksall, Z.S.; Lin, S.; Tutor-New, F.Q.; Ho, C.C.G.; et al. Transcriptional Landscape of Human Microglia Implicates Age, Sex, and APOE-Related Immunometabolic Pathway Perturbations. Aging Cell 2022, 21, e13606. [Google Scholar] [CrossRef]
- Atagi, Y.; Liu, C.C.; Painter, M.M.; Chen, X.F.; Verbeeck, C.; Zheng, H.; Li, X.; Rademakers, R.; Kang, S.S.; Xu, H.; et al. Apolipoprotein E Is a Ligand for Triggering Receptor Expressed on Myeloid Cells 2 (TREM2). J. Biol. Chem. 2015, 290, 26043–26050. [Google Scholar] [CrossRef]
- Nugent, A.A.; Lin, K.; van Lengerich, B.; Lianoglou, S.; Przybyla, L.; Davis, S.S.; Llapashtica, C.; Wang, J.; Kim, D.J.; Xia, D.; et al. TREM2 Regulates Microglial Cholesterol Metabolism upon Chronic Phagocytic Challenge. Neuron 2020, 105, 837–854.e9. [Google Scholar] [CrossRef]
- Damisah, E.C.; Rai, A.; Grutzendler, J. TREM2: Modulator of Lipid Metabolism in Microglia. Neuron 2020, 105, 759–761. [Google Scholar] [CrossRef]
- Colombo, A.; Dinkel, L.; Müller, S.A.; Sebastian Monasor, L.; Schifferer, M.; Cantuti-Castelvetri, L.; König, J.; Vidatic, L.; Bremova-Ertl, T.; Lieberman, A.P.; et al. Loss of NPC1 Enhances Phagocytic Uptake and Impairs Lipid Trafficking in Microglia. Nat. Commun. 2021, 12, 1158. [Google Scholar] [CrossRef] [PubMed]
- Hassnain Waqas, S.F.; Noble, A.; Hoang, A.C.; Ampem, G.; Popp, M.; Strauß, S.; Guille, M.; Röszer, T. Adipose Tissue Macrophages Develop from Bone Marrow–independent Progenitors in Xenopus Laevis and Mouse. J. Leukoc. Biol. 2017, 102, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Prieur, X.; Mok, C.Y.L.; Velagapudi, V.R.; Núñez, V.; Fuentes, L.; Montaner, D.; Ishikawa, K.; Camacho, A.; Barbarroja, N.; O’Rahilly, S.; et al. Differential Lipid Partitioning between Adipocytes and Tissue Macrophages Modulates Macrophage Lipotoxicity and M2/M1 Polarization in Obese Mice. Diabetes 2011, 60, 797–809. [Google Scholar] [CrossRef] [PubMed]
- Morgan, P.K.; Huynh, K.; Pernes, G.; Miotto, P.M.; Mellett, N.A.; Giles, C.; Meikle, P.J.; Murphy, A.J.; Lancaster, G.I. Macrophage Polarization State Affects Lipid Composition and the Channeling of Exogenous Fatty Acids into Endogenous Lipid Pools. J. Biol. Chem. 2021, 297, 101341. [Google Scholar] [CrossRef]
- Aouadi, M.; Vangala, P.; Yawe, J.C.; Tencerova, M.; Nicoloro, S.M.; Cohen, J.L.; Shen, Y.; Czech, M.P. Lipid Storage by Adipose Tissue Macrophages Regulates Systemic Glucose Tolerance. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E374–E383. [Google Scholar] [CrossRef]
- Jaitin, D.A.; Adlung, L.; Thaiss, C.A.; Weiner, A.; Li, B.; Descamps, H.; Lundgren, P.; Bleriot, C.; Liu, Z.; Deczkowska, A.; et al. Lipid-Associated Macrophages Control Metabolic Homeostasis in a Trem2-Dependent Manner. Cell 2019, 178, 686–698.e14. [Google Scholar] [CrossRef]
- Timperi, E.; Gueguen, P.; Molgora, M.; Magagna, I.; Kieffer, Y.; Lopez-Lastra, S.; Sirven, P.; Baudrin, L.G.; Baulande, S.; Nicolas, A.; et al. Lipid-Associated Macrophages Are Induced by Cancer-Associated Fibroblasts and Mediate Immune Suppression in Breast Cancer. Cancer Res. 2022, 82, 3291–3306. [Google Scholar] [CrossRef]
- Kim, M.J.; Wainwright, H.C.; Locketz, M.; Bekker, L.G.; Walther, G.B.; Dittrich, C.; Visser, A.; Wang, W.; Hsu, F.F.; Wiehart, U.; et al. Caseation of Human Tuberculosis Granulomas Correlates with Elevated Host Lipid Metabolism. EMBO Mol. Med. 2010, 2, 258–274. [Google Scholar] [CrossRef]
- Zheng, X.; Mansouri, S.; Krager, A.; Grimminger, F.; Seeger, W.; Pullamsetti, S.S.; Wheelock, C.E.; Savai, R. Metabolism in Tumour-Associated Macrophages: A Quid pro Quo with the Tumour Microenvironment. Eur. Respir. Rev. 2020, 29, 200134. [Google Scholar] [CrossRef]
- Su, P.; Wang, Q.; Bi, E.; Ma, X.; Liu, L.; Yang, M.; Qian, J.; Yi, Q. Enhanced Lipid Accumulation and Metabolism Are Required for the Differentiation and Activation of Tumor-Associated Macrophages. Cancer Res. 2020, 80, 1438–1450. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Lei, Q.; Yang, L.; Qin, G.; Liu, S.; Wang, D.; Ping, Y.; Zhang, Y. Contradictory Roles of Lipid Metabolism in Immune Response within the Tumor Microenvironment. J. Hematol. Oncol. 2021, 14. [Google Scholar] [CrossRef] [PubMed]
- Masetti, M.; Carriero, R.; Portale, F.; Marelli, G.; Morina, N.; Pandini, M.; Iovino, M.; Partini, B.; Erreni, M.; Ponzetta, A.; et al. Lipid-Loaded Tumor-Associated Macrophages Sustain Tumor Growth and Invasiveness in Prostate Cancer. J. Exp. Med. 2021, 219, e20210564. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Han, Y.; Rodriguez Sillke, Y.; Deng, H.; Siddiqui, S.; Treese, C.; Schmidt, F.; Friedrich, M.; Keye, J.; Wan, J.; et al. Lipid Droplet-dependent Fatty Acid Metabolism Controls the Immune Suppressive Phenotype of Tumor-associated Macrophages. EMBO Mol. Med. 2019, 11, e10698. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Zheng, N.; Jiang, L.; Wang, T.; Zhang, P.; Liu, Y.; Zheng, P.; Wang, W.; Xie, G.; Chen, L.; et al. Lipid Accumulation in Macrophages Confers Protumorigenic Polarization and Immunity in Gastric Cancer. Cancer Sci. 2020, 111, 4000–4011. [Google Scholar] [CrossRef] [PubMed]
- Di Conza, G.; Tsai, C.H.; Gallart-Ayala, H.; Yu, Y.R.; Franco, F.; Zaffalon, L.; Xie, X.; Li, X.; Xiao, Z.; Raines, L.N.; et al. Tumor-Induced Reshuffling of Lipid Composition on the Endoplasmic Reticulum Membrane Sustains Macrophage Survival and pro-Tumorigenic Activity. Nat. Immunol. 2021, 22, 1403–1415. [Google Scholar] [CrossRef]
- D’Avila, H.; Freire-de-Lima, C.G.; Roque, N.R.; Teixeira, L.; Barja-Fidalgo, C.; Silva, A.R.; Melo, R.C.N.; DosReis, G.A.; Castro-Faria-Neto, H.C.; Bozza, P.T. Host Cell Lipid Bodies Triggered by Trypanosoma Cruzi Infection and Enhanced by the Uptake of Apoptotic Cells Are Associated with Prostaglandin E2 Generation and Increased Parasite Growth. J. Infect. Dis. 2011, 204, 951–961. [Google Scholar] [CrossRef]
- Melo, R.C.N.; Fabrino, D.L.; Dias, F.F.; Parreira, G.G. Lipid Bodies: Structural Markers of Inflammatory Macrophages in Innate Immunity. Inflamm. Res. 2006, 55, 342–348. [Google Scholar] [CrossRef]
- D’Avila, H.; Melo, R.C.N.; Parreira, G.G.; Werneck-Barroso, E.; Castro-Faria-Neto, H.C.; Bozza, P.T. Mycobacterium Bovis Bacillus Calmette-Guérin Induces TLR2-Mediated Formation of Lipid Bodies: Intracellular Domains for Eicosanoid Synthesis In Vivo. J. Immunol. 2006, 176, 3087–3097. [Google Scholar] [CrossRef]
- Peyron, P.; Vaubourgeix, J.; Poquet, Y.; Levillain, F.; Botanch, C.; Bardou, F.; Daffé, M.; Emile, J.F.; Marchou, B.; Cardona, P.J.; et al. Foamy Macrophages from Tuberculous Patients’ Granulomas Constitute a Nutrient-Rich Reservoir for M. Tuberculosis Persistence. PLoS Pathog. 2008, 4, e1000204. [Google Scholar] [CrossRef]
- Cocchiaro, J.L.; Kumar, Y.; Fischer, E.R.; Hackstadt, T.; Valdivia, R.H. Cytoplasmic Lipid Droplets Are Translocated into the Lumen of the Chlamydia Trachomatis Parasitophorous Vacuole. Proc. Natl. Acad. Sci. USA 2008, 105, 9379–9384. [Google Scholar] [CrossRef] [PubMed]
- Febbraio, M.; Hajjar, D.P.; Silverstein, R.L. CD36: A Class B Scavenger Receptor Involved in Angiogenesis, Atherosclerosis, Inflammation, and Lipid Metabolism. J. Clin. Investig. 2001, 108, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Zanoni, I.; Tan, Y.; Di Gioia, M.; Springstead, J.R.; Kagan, J.C. By Capturing Inflammatory Lipids Released from Dying Cells, the Receptor CD14 Induces Inflammasome-Dependent Phagocyte Hyperactivation. Immunity 2017, 47, 697–709.e3. [Google Scholar] [CrossRef] [PubMed]
- Bochkov, V.; Gesslbauer, B.; Mauerhofer, C.; Philippova, M.; Erne, P.; Oskolkova, O.V. Pleiotropic Effects of Oxidized Phospholipids. Free Radic. Biol. Med. 2017, 111, 6–24. [Google Scholar] [CrossRef]
- Knapp, S.; Matt, U.; Leitinger, N.; van der Poll, T. Oxidized Phospholipids Inhibit Phagocytosis and Impair Outcome in Gram-Negative Sepsis In Vivo. J. Immunol. 2007, 178, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, M.E.; Sun, M.; Zhang, R.; Febbraio, M.; Silverstein, R.; Hazen, S.L. Oxidized phosphatidylserine–CD36 Interactions Play an Essential Role in Macrophage-Dependent Phagocytosis of Apoptotic Cells. J. Cell Biol. 2006, 203, 2613–2625. [Google Scholar] [CrossRef]
- Zhivaki, D.; Kagan, J.C. Innate Immune Detection of Lipid Oxidation as a Threat Assessment Strategy. Nat. Rev. Immunol. 2022, 22, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Di Gioia, M.; Zanoni, I. Dooming Phagocyte Responses: Inflammatory Effects of Endogenous Oxidized Phospholipids. Front. Endocrinol. (Lausanne) 2021, 12, 626842. [Google Scholar] [CrossRef] [PubMed]
- Erridge, C.; Kennedy, S.; Spickett, C.M.; Webb, D.J. Oxidized Phospholipid Inhibition of Toll-Like Receptor (TLR) Signaling Is Restricted to TLR2 and TLR4: Roles for CD14, LPS-Binding Protein, and MD2 as Targets for Specificity of Inhibition. J. Biol. Chem. 2008, 283, 24748–24759. [Google Scholar] [CrossRef]
- Yang, J.B.; Duan, Z.J.; Yao, W.; Lee, O.; Yang, L.; Yang, X.Y.; Sun, X.; Chang, C.C.Y.; Chang, T.Y.; Li, B.L. Synergistic Transcriptional Activation of Human Acyl-Coenzyme A: Cholesterol Acyltransterase-1 Gene by Interferon-γ and All-Trans-Retinoic Acid THP-1 Cells. J. Biol. Chem. 2001, 276, 20989–20998. [Google Scholar] [CrossRef] [PubMed]
- Larigauderie, G.; Furman, C.; Jaye, M.; Lasselin, C.; Copin, C.; Fruchart, J.C.; Castro, G.; Rouis, M. Adipophilin Enhances Lipid Accumulation and Prevents Lipid Efflux from THP-1 Macrophages: Potential Role in Atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Tsui, L.; Chang, S.F.; Huang, H.P.; Fong, T.H.; Wang, I.J. YC-1 Induces Lipid Droplet Formation in RAW 264.7 Macrophages. J. Biomed. Sci. 2016, 23, 2. [Google Scholar] [CrossRef]
- Joy, T.R.; Hegele, R.A. The Failure of Torcetrapib: What Have We Learned? Br. J. Pharmacol. 2008, 154, 1379–1381. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. Keeping the Home Fires Burning †: AMP-Activated Protein Kinase. J. R. Soc. Interface 2018, 15. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Ballina, M.; Guan, X.L.; Schmidt, A.; Bumann, D. Classical Activation of Macrophages Leads to Lipid Droplet Formation without de Novo Fatty Acid Synthesis. Front. Immunol. 2020, 11, 131. [Google Scholar] [CrossRef] [PubMed]
- Alonso, R.; Cuevas, A.; Mata, P. Lomitapide: A Review of Its Clinical Use, Efficacy, and Tolerability. Core Evid. 2019, 14, 19–30. [Google Scholar] [CrossRef]
- Chambergo-Michilot, D.; Alur, A.; Kulkarni, S.; Agarwala, A. Mipomersen in Familial Hypercholesterolemia: An Update on Health-Related Quality of Life and Patient-Reported Outcomes. Vasc. Health Risk Manag. 2022, 18, 73–80. [Google Scholar] [CrossRef]
- Muthuramu, I.; Amin, R.; Postnov, A.; Mishra, M.; Aboumsallem, J.P.; Dresselaers, T.; Himmelreich, U.; Van Veldhoven, P.P.; Gheysens, O.; Jacobs, F.; et al. Cholesterol-Lowering Gene Therapy Counteracts the Development of Non-Ischemic Cardiomyopathy in Mice. Mol. Ther. 2017, 25, 2513–2525. [Google Scholar] [CrossRef]
- Merćep, I.; Friščić, N.; Strikić, D.; Reiner, Ž. Advantages and Disadvantages of Inclisiran: A Small Interfering Ribonucleic Acid Molecule Targeting PCSK9-A Narrative Review. Cardiovasc. Ther. 2022, 2022. [Google Scholar] [CrossRef]
- Agarwala, A.; Goldberg, A.C. Bempedoic Acid: A Promising Novel Agent for LDL-C Lowering. Future Cardiol. 2020, 16, 361–371. [Google Scholar] [CrossRef]
- Gaudet, D.; Durst, R.; Lepor, N.; Bakker-Arkema, R.; Bisgaier, C.; Masson, L.; Golden, L.; Kastelein, J.J.; Hegele, R.A.; Stein, E. Usefulness of Gemcabene in Homozygous Familial Hypercholesterolemia (from COBALT-1). Am. J. Cardiol. 2019, 124, 1876–1880. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.A.K.; Cornicelli, J.A.; Markham, B.; Bisgaier, C.L. Gemcabene, a First-in-Class Lipid-Lowering Agent in Late-Stage Development, down-Regulates Acute-Phase C-Reactive Protein via C/EBP-δ-Mediated Transcriptional Mechanism. Mol. Cell. Biochem. 2018, 449, 167–183. [Google Scholar] [CrossRef] [PubMed]
- Westphal, S.P. First Gene Therapy Approved. New Sci. 2003, 180, 13. [Google Scholar]
- Meyers, C.D.; Tremblay, K.; Amer, A.; Chen, J.; Jiang, L.; Gaudet, D. Effect of the DGAT1 Inhibitor Pradigastat on Triglyceride and apoB48 Levels in Patients with Familial Chylomicronemia Syndrome. Lipids Health Dis. 2015, 14, 8. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, D.; Digenio, A.; Alexander, V.J.; Arca, M.; Jones, A.F.; Stroes, E.; Bergeron, J.; Civeira, F.; Hemphill, L.; Blom, D.J.; et al. The APPROACH Study: A Randomized, Double-Blind, Placebo-Controlled, Phase 3 Study of Volanesorsen Administered Subcutaneously to Patients with Familial Chylomicronemia Syndrome (FCS). J. Clin. Lipidol. 2017, 11, 814–815. [Google Scholar] [CrossRef]
- Bays, H.; Jones, P.H. Colesevelam Hydrochloride: Reducing Atherosclerotic Coronary Heart Disease Risk Factors. Vasc. Health Risk Manag. 2007, 3, 733–742. [Google Scholar]
- Llaverías, G.; Laguna, J.C.; Alegret, M. Pharmacology of the ACAT Inhibitor Avasimibe (CI-1011). Cardiovasc. Drug Rev. 2003, 21, 33–50. [Google Scholar] [CrossRef]
- Ueshima, K.; Akihisa-Umeno, H.; Nagayoshi, A.; Takakura, S.; Matsuo, M.; Mutoh, S. Implitapide, a Microsomal Triglyceride Transfer Protein Inhibitor, Reduces Progression of Atherosclerosis in Apolipoprotein E Knockout Mice Fed a Western-Type Diet: Involvement of the Inhibition of Postprandial Triglyceride Elevation. Biol. Pharm. Bull. 2005, 28, 247–252. [Google Scholar] [CrossRef]
- Capuzzi, D.M.; Morgan, J.M.; Brusco, O.A.; Intenzo, C.M. Niacin Dosing: Relationship to Benefits and Adverse Effects. Curr. Atheroscler. Rep. 2000, 2, 64–71. [Google Scholar] [CrossRef]
- Patel, J.; Sheehan, V.; Gurk-Turner, C. Ezetimibe (Zetia): A New Type of Lipid-Lowering Agent. Baylor Univ. Med. Cent. Proc. 2003, 16, 354–358. [Google Scholar] [CrossRef]
- Pintó, X.; Zúñiga, M. Treatment of Hypercholesterolemia and Prevention of Cardiovascular Diseases through Inhibition of Bile Acid Reabsorption with Resin Cholestyramine. Clin. Investig. Arterioscler. 2011, 23, 9–16. [Google Scholar] [CrossRef]
- Gaw, A.; Packard, C.J.; Lindsay, G.M.; Murray, E.F.; Griffin, B.A.; Caslake, M.J.; Colquhoun, I.; Wheatley, D.J.; Ross Lorimer, A.; Shepherd, J. Effects of Colestipol Alone and in Combination with Simvastatin on Apolipoprotein B Metabolism. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 236–249. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.P.; Tsang, M.; Wright, J.M. Atorvastatin for Lowering Lipids. Cochrane Database Syst. Rev. 2015, 2015, CD008226. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.P.; Sekhon, S.S.; Tsang, M.; Wright, J.M. Fluvastatin for Lowering Lipids. Cochrane Database Syst. Rev. 2018, 2018, CD012282. [Google Scholar] [CrossRef] [PubMed]
- McKenney, J.M. Lovastatin: A New Cholesterol-Lowering Agent. Clin. Pharm. 1988, 7, 21–36. [Google Scholar] [CrossRef]
- Adams, S.P.; Alaeiilkhchi, N.; Wright, J.M. Pitavastatin for Lowering Lipids. Cochrane Database Syst. Rev. 2020, 2020, CD012735. [Google Scholar] [CrossRef]
- Adams, S.P.; Sekhon, S.S.; Wright, J.M. Lipid-Lowering Efficacy of Rosuvastatin. Cochrane Database Syst. Rev. 2014, 2014, CD010254. [Google Scholar]
- Baigent, C.; Landray, M.J.; Reith, C.; Emberson, J.; Wheeler, D.C.; Tomson, C.; Wanner, C.; Krane, V.; Cass, A.; Craig, J.; et al. The Effects of Lowering LDL Cholesterol with Simvastatin plus Ezetimibe in Patients with Chronic Kidney Disease (Study of Heart and Renal Protection): A Randomised Placebo-Controlled Trial. Lancet 2011, 377, 2181–2192. [Google Scholar] [CrossRef]
- Kaesemeyer, W.H.; Caldwell, R.B.; Huang, J.; William Caldwell, R. Pravastatin Sodium Activates Endothelial Nitric Oxide Synthase Independent of Its Cholesterol-Lowering Actions. J. Am. Coll. Cardiol. 1999, 33, 234–241. [Google Scholar] [CrossRef]
- Roy, A.; Pahan, K. Gemfibrozil, Stretching Arms beyond Lipid Lowering. Immunopharmacol. Immunotoxicol. 2009, 31, 339–351. [Google Scholar] [CrossRef]
- Nishi, K.; Oda, T.; Takabuchi, S.; Oda, S.; Fukuda, K.; Adachi, T.; Semenza, G.L.; Shingu, K.; Hirota, K. LPS Induces Hypoxia-Inducible Factor 1 Activation in Macrophage- Differentiated Cells in a Reactive Oxygen Species-Dependent Manner. Antioxidants Redox Signal. 2008, 10, 983–995. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Choksi, S.; Chen, K.; Pobezinskaya, Y.; Linnoila, I.; Liu, Z.G. ROS Play a Critical Role in the Differentiation of Alternatively Activated Macrophages and the Occurrence of Tumor-Associated Macrophages. Cell Res. 2013, 23, 898–914. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.Y.; Wang, N.; Li, S.; Hong, M.; Wang, X.; Feng, Y. The Reactive Oxygen Species in Macrophage Polarization: Reflecting Its Dual Role in Progression and Treatment of Human Diseases. Oxid. Med. Cell. Longev. 2016, 2016, 2795090. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.I.K.; Bergmark, C.; Laurila, A.; Hörkkö, S.; Han, K.H.; Friedman, P.; Dennis, E.A.; Witztum, J.L. Monoclonal Antibodies against Oxidized Low-Density Lipoprotein Bind to Apoptotic Cells and Inhibit Their Phagocytosis by Elicited Macrophages: Evidence That Oxidation-Specific Epitopes Mediate Macrophage Recognition. Proc. Natl. Acad. Sci. USA 1999, 96, 6353–6358. [Google Scholar] [CrossRef]
- Lee, H.N.; Surh, Y.J. Resolvin D1-Mediated NOX2 Inactivation Rescues Macrophages Undertaking Efferocytosis from Oxidative Stress-Induced Apoptosis. Biochem. Pharmacol. 2013, 86, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Liu, H.; Lian, G.; Zhang, S.Y.; Wang, X.; Jiang, C. HIF1 α -Induced Glycolysis Metabolism Is Essential to the Activation of Inflammatory Macrophages. Mediators Inflamm. 2017, 2017, 9029327. [Google Scholar] [CrossRef]
- Hirai, K.; Furusho, H.; Hirota, K.; Sasaki, H. Activation of Hypoxia-Inducible Factor 1 Attenuates Periapical Inflammation and Bone Loss. Int. J. Oral Sci. 2018, 10, 12. [Google Scholar] [CrossRef]
- Li, C.; Wang, Y.; Li, Y.; Yu, Q.; Jin, X.; Wang, X.; Jia, A.; Hu, Y.; Han, L.; Wang, J.; et al. HIF1α-Dependent Glycolysis Promotes Macrophage Functional Activities in Protecting against Bacterial and Fungal Infection. Sci. Rep. 2018, 8, 3603. [Google Scholar] [CrossRef]
- Woods, P.S.; Kimmig, L.M.; Sun, K.A.; Meliton, A.Y.; Shamaa, O.R.; Tian, Y.; Cetin-Atalay, R.; Sharp, W.W.; Hamanaka, R.B.; Mutlu, G.M. HIF-1α Induces Glycolytic Reprograming in Tissue-Resident Alveolar Macrophages to Promote Cell Survival during Acute Lung Injury. Elife 2022, 11, e77457. [Google Scholar] [CrossRef]
- Watts, E.R.; Walmsley, S.R. Inflammation and Hypoxia: HIF and PHD Isoform Selectivity. Trends Mol. Med. 2019, 25, 33–46. [Google Scholar] [CrossRef]
- Aarup, A.; Pedersen, T.X.; Junker, N.; Christoffersen, C.; Bartels, E.D.; Madsen, M.; Nielsen, C.H.; Nielsen, L.B. Hypoxia-Inducible Factor-1α Expression in Macrophages Promotes Development of Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1782–1790. [Google Scholar] [CrossRef] [PubMed]
- Parathath, S.; Mick, S.L.; Feig, J.E.; Joaquin, V.; Grauer, L.; Habiel, D.M.; Gassmann, M.; Gardner, L.B.; Fisher, E.A. Hypoxia Is Present in Murine Atherosclerotic Plaques and Has Multiple Adverse Effects on Macrophage Lipid Metabolism. Circ. Res. 2011, 109, 1141–1152. [Google Scholar] [CrossRef] [PubMed]
- Vink, A.; Schoneveld, A.H.; Lamers, D.; Houben, A.J.S.; van der Groep, P.; van Diest, P.J.; Pasterkamp, G. HIF-1alpha Expression Is Associated with an Atheromatous Inflammatory Plaque Phenotype and Upregulated in Activated Macrophages. Atherosclerosis 2007, 195, e69–e75. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.L.; et al. Fatty Acid Uptake and Lipid Storage Induced by HIF-1α Contribute to Cell Growth and Survival after Hypoxia-Reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [CrossRef] [PubMed]
- Maier, A.; Wu, H.; Cordasic, N.; Oefner, P.; DIetel, B.; Thiele, C.; Weidemann, A.; Eckardt, K.U.; Warnecke, C. Hypoxia-Inducible Protein 2 Hig2/Hilpda Mediates Neutral Lipid Accumulation in Macrophages and Contributes to Atherosclerosis in Apolipoprotein E-Deficient Mice. FASEB J. 2017, 31, 4971–4984. [Google Scholar] [CrossRef]
- Boström, P.; Magnusson, B.; Svensson, P.A.; Wiklund, O.; Borén, J.; Carlsson, L.M.S.; Ståhlman, M.; Olofsson, S.O.; Hultén, L.M. Hypoxia Converts Human Macrophages into Triglyceride-Loaded Foam Cells. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1871–1876. [Google Scholar] [CrossRef]
- Singh, R.; Cuervo, A.M. Lipophagy: Connecting Autophagy and Lipid Metabolism. Int. J. Cell Biol. 2012. [CrossRef]
- Berger, J.; Moller, D.E. The Mechanisms of Action of PPARs. Annu. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef]
- Chan, S.M.H.; Sun, R.Q.; Zeng, X.Y.; Choong, Z.H.; Wang, H.; Watt, M.J.; Ye, J.M. Activation of PPARα Ameliorates Hepatic Insulin Resistance and Steatosis in High Fructose-Fed Mice despite Increased Endoplasmic Reticulum Stress. Diabetes 2013, 62, 2095–2105. [Google Scholar] [CrossRef]
- Saito, T.; Kuma, A.; Sugiura, Y.; Ichimura, Y.; Obata, M.; Kitamura, H.; Okuda, S.; Lee, H.C.; Ikeda, K.; Kanegae, Y.; et al. Autophagy Regulates Lipid Metabolism through Selective Turnover of NCoR1. Nat. Commun. 2019, 10, 1567. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, X.; Tian, Y.; Fan, Z.; Li, W.; Liu, M.; Hu, J.; Duan, Z.; Jin, R.; Ren, F. The Critical Role of PPARα in the Binary Switch between Life and Death Induced by Endoplasmic Reticulum Stress. Cell Death Dis. 2020, 11, 691. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Han, S.L.; Lu, D.L.; Li, L.Y.; Limbu, S.M.; Li, D.L.; Zhang, M.L.; Du, Z.Y. Inhibited Lipophagy Suppresses Lipid Metabolism in Zebrafish Liver Cells. Front. Physiol. 2019, 10, 1077. [Google Scholar] [CrossRef] [PubMed]
- Grabacka, M.; Reiss, K. Anticancer Properties of PPARα-Effects on Cellular Metabolism and Inflammation. PPAR Res. 2008. [CrossRef]
- Vernia, S.; Cavanagh-Kyros, J.; Garcia-Haro, L.; Sabio, G.; Barrett, T.; Jung, D.Y.; Kim, J.K.; Xu, J.; Shulha, H.P.; Garber, M.; et al. The PPARα-FGF21 Hormone Axis Contributes to Metabolic Regulation by the Hepatic JNK Signaling Pathway. Cell Metab. 2014, 20, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.T.A.; Satoh, H.; Favelyukis, S.; Babendure, J.L.; Imamura, T.; Sbodio, J.I.; Zalevsky, J.; Dahiyat, B.I.; Chi, N.W.; Olefsky, J.M. JNK and Tumor Necrosis Factor-α Mediate Free Fatty Acid-Induced Insulin Resistance in 3T3-L1 Adipocytes. J. Biol. Chem. 2005, 280, 35361–35371. [Google Scholar] [CrossRef]
- Malhi, H.; Bronk, S.F.; Werneburg, N.W.; Gores, G.J. Free Fatty Acids Induce JNK-Dependent Hepatocyte Lipoapoptosis. J. Biol. Chem. 2006, 281, 12093–12101. [Google Scholar] [CrossRef]
- Gathercole, L.L.; Morgan, S.A.; Tomlinson, J.W. Hormonal Regulation of Lipogenesis. Vitam. Horm. 2013, 91, 1–27. [Google Scholar]
- Kim, S.-J.; Tang, T.; Abbott, M.; Viscarra, J.A.; Wang, Y.; Sul, H.S. AMPK Phosphorylates Desnutrin/ATGL and Hormone-Sensitive Lipase To Regulate Lipolysis and Fatty Acid Oxidation within Adipose Tissue. Mol. Cell. Biol. 2016, 36, 1961–1976. [Google Scholar] [CrossRef]
- Zhang, X.; Song, Y.; Feng, M.; Zhou, X.; Lu, Y.; Gao, L.; Yu, C.; Jiang, X.; Zhao, J. Thyroid-Stimulating Hormone Decreases HMG-CoA Reductase Phosphorylation via AMP-Activated Protein Kinase in the Liver. J. Lipid Res. 2015, 56, 963–971. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, S.; Zhai, A.; Zhang, B.; Tian, G. AMPK-Mediated Regulation of Lipid Metabolism by Phosphorylation. Biol. Pharm. Bull. 2018, 41, 985–993. [Google Scholar] [CrossRef]
- Foretz, M.; Even, P.C.; Viollet, B. AMPK Activation Reduces Hepatic Lipid Content by Increasing Fat Oxidation in Vivo. Int. J. Mol. Sci. 2018, 19, 2826. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Min, Q.; Ouyang, C.; Lee, J.; He, C.; Zou, M.H.; Xie, Z. AMPK Activation Prevents Excess Nutrient-Induced Hepatic Lipid Accumulation by Inhibiting mTORC1 Signaling and Endoplasmic Reticulum Stress Response. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1844–1854. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; De Cegli, R.; Mansueto, G.; Saha, P.K.; Vetrini, F.; Visvikis, O.; Huynh, T.; Carissimo, A.; Palmer, D.; Jürgen Klisch, T.; et al. TFEB Controls Cellular Lipid Metabolism through a Starvation-Induced Autoregulatory Loop. Nat. Cell Biol. 2013, 15, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Arencibia, M.G.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB Links Autophagy to Lysosomal Biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef]
- Li, M.; Wang, Z.; Wang, P.; Li, H.; Yang, L. TFEB: A Emerging Regulator in Lipid Homeostasis for Atherosclerosis. Front. Physiol. 2021, 12. [Google Scholar] [CrossRef]
- Shin, J.H.; Min, S.H.; Kim, S.J.; Kim, Y.I.; Park, J.; Lee, H.K.; Yoo, O.J. TAK1 Regulates Autophagic Cell Death by Suppressing the Phosphorylation of p70 S6 Kinase 1. Sci. Rep. 2013, 3, 1561. [Google Scholar] [CrossRef]
- Inokuchi-Shimizu, S.; Park, E.J.; Roh, Y.S.; Yang, L.; Zhang, B.; Song, J.; Liang, S.; Pimienta, M.; Taniguchi, K.; Wu, X.; et al. TAK1-Mediated Autophagy and Fatty Acid Oxidation Prevent Hepatosteatosis and Tumorigenesis. J. Clin. Investig. 2014, 124, 3566–3578. [Google Scholar] [CrossRef]
- Gallot, Y.S.; McMillan, J.D.; Xiong, G.; Bohnert, K.R.; Straughn, A.R.; Hill, B.G.; Kumar, A. Distinct Roles of TRAF6 and TAK1 in the Regulation of Adipocyte Survival, Thermogenesis Program, and High-Fat Diet-Induced Obesity. Oncotarget 2017, 8, 112565–112583. [Google Scholar] [CrossRef]
- Molaei, M.; Vandehoef, C.; Karpac, J. NF-κB Shapes Metabolic Adaptation by Attenuating Foxo-Mediated Lipolysis in Drosophila. Dev. Cell 2019, 49, 802–810.e6. [Google Scholar] [CrossRef]
- Yu, X.H.; Zheng, X.L.; Tang, C.K. Nuclear Factor-κB Activation as a Pathological Mechanism of Lipid Metabolism and Atherosclerosis, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2015; Volume 70. [Google Scholar]
- Planavila, A.; Laguna, J.C.; Vázquez-Carrera, M. Nuclear Factor-κB Activation Leads to down-Regulation of Fatty Acid Oxidation during Cardiac Hypertrophy. J. Biol. Chem. 2005, 280, 17464–17471. [Google Scholar] [CrossRef]
- Zeng, T.; Zhou, J.; He, L.; Zheng, J.; Chen, L.; Wu, C.; Xia, W. Blocking Nuclear Factor-Kappa B Protects against Diet-Induced Hepatic Steatosis and Insulin Resistance in Mice. PLoS ONE 2016, 11, e0149677. [Google Scholar] [CrossRef] [PubMed]
- Bonam, S.R.; Wang, F.; Muller, S. Lysosomes as a Therapeutic Target. Nat. Rev. Drug Discov. 2019, 18, 923–948. [Google Scholar] [CrossRef] [PubMed]
- Marques, A.R.A.; Ramos, C.; Machado-Oliveira, G.; Vieira, O.V. Lysosome (Dys)function in Atherosclerosis—A Big Weight on the Shoulders of a Small Organelle. Front. Cell Dev. Biol. 2021, 9, 658995. [Google Scholar] [CrossRef] [PubMed]
- Thelen, A.M.; Zoncu, R. Emerging Roles for the Lysosome in Lipid Metabolism. Trends Cell Biol. 2017, 27, 833–850. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Brown, M.S. A Century of Cholesterol and Coronaries: From Plaques to Genes to Statins. Cell 2015, 161, 161–172. [Google Scholar] [CrossRef]
- Li, J.; Pfeffer, S.R. Lysosomal Membrane Glycoproteins Bind Cholesterol and Contribute to Lysosomal Cholesterol Export. Elife 2016, 5, 21635. [Google Scholar] [CrossRef]
- Dubland, J.A.; Francis, G.A. Lysosomal Acid Lipase: At the Crossroads of Normal and Atherogenic Cholesterol Metabolism. Front. Cell Dev. Biol. 2015, 3, 3. [Google Scholar] [CrossRef]
- Cox, B.E.; Griffin, E.E.; Ullery, J.C.; Jerome, W.G. Effects of Cellular Cholesterol Loading on Macrophage Foam Cell Lysosome Acidification. J. Lipid Res. 2007, 48, 1012–1021. [Google Scholar] [CrossRef]
- Dubland, J.A.; Allahverdian, S.; Besler, K.J.; Ortega, C.; Wang, Y.; Pryma, C.S.; Boukais, K.; Chan, T.; Seidman, M.A.; Francis, G.A. Low LAL (Lysosomal Acid Lipase) Expression by Smooth Muscle Cells Relative to Macrophages as a Mechanism for Arterial Foam Cell Formation. Arterioscler. Thromb. Vasc. Biol. 2021, 41, E354–E368. [Google Scholar] [CrossRef]
- Yancey, P.G.; Gray Jerome, W. Lysosomal Cholesterol Derived from Mildly Oxidized Low Density Lipoprotein Is Resistant to Efflux. J. Lipid Res. 2001, 42, 317–327. [Google Scholar] [CrossRef]
- Xu, X.; Grijalva, A.; Skowronski, A.; Van Eijk, M.; Serlie, M.J.; Ferrante, A.W. Obesity Activates a Program of Lysosomal-Dependent Lipid Metabolism in Adipose Tissue Macrophages Independently of Classic Activation. Cell Metab. 2013, 18, 816–830. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.C.C.; Everts, B.; Ivanova, Y.; O’Sullivan, D.; Nascimento, M.; Smith, A.M.; Beatty, W.; Love-Gregory, L.; Lam, W.Y.; O’Neill, C.M.; et al. Cell-Intrinsic Lysosomal Lipolysis Is Essential for Alternative Activation of Macrophages. Nat. Immunol. 2014, 15, 846–855. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SNo | Modulator | Mechanism | Effect |
|---|---|---|---|
| 1 | IFNγ [116] | ACAT1 mRNA expression | Lipid accumulation |
| 2 | Adipophilin [112] | Promotes TG and cholesterol storage | Increased cholesterol accumulation and reduced efflux |
| 3 | 3-(5′-Hydroxymethyl-2′-furyl)-1-benzylindazole (YC-1) [113] | sGC/cGMP/PKG signaling pathway | Induces lipid accumulation |
| 4 | Lomitapide [117] | Inhibition of microsomal triglyceride transfer protein (MTP) | Lowers LDL cholesterol |
| 5 | Mipomersen [118] | Selective degradation of the apoB-100 mRNA-antisense oligonucleotide | Reduction in LDL-C and other lipoprotein levels |
| 6 | AAV8 (Adeno-associated viral serotype 8) TBG.hLDLR [119] | (AAV8)-low-density lipoprotein receptor (AAV8-LDLr) gene therapy | Reduction of plasma cholesterol levels |
| 7 | Inclisiran [120] | PCSK9 targeting siRNA | Decreased LDL-c levels |
| 8 | Bempedoic acid [121] | Inhibits ATP-citrate lyase | Lowers LDL-c in hypercholesterolaemia and established atherosclerosis |
| 9 | Gemcabene [122,123] | Down regulation of CRP (C-reactive protein) | Lipid lowering activity |
| 10 | Alipogene tiparvovec [124] | Adeno-associated viruses (AAVs) targeting lipoprotein lipase (LPL)- gene therapy | Reduce circulating plasma triglyceride levels |
| 11 | Pradigastat [125] | Specific inhibition of diacylglycerol acyltransferase 1 (DGAT1) blocking chylomicron triglyceride (TG) synthesis | Reduced TG levels in FCS (familial chylomicronemia syndrome) |
| 12 | Volanesorsen [126] | Antisense oligonucleotides targeting ApoC3 mRNA | Reducing triglyceride levels in patients with hypertriglyceridemia or familial chylomicronemia syndrome |
| 13 | Colesevelam HCl [127] | Accelerates cholesterol 7-α-hydroxylase mediated conversion of bile acids | Reduces total and low-density lipoprotein (LDL) cholesterol levels |
| 14 | Torcetrapib [114] | Inhibits cholesteryl ester-transfer protein (CETP) | Increased HDL-cholesterol and apolipoprotein A-I levels and decreased apolipoprotein B levels |
| 15 | Avasimibe [128] | Enhances cholesterol efflux and reduces LDL uptake | Reduces foam cell formation |
| 16 | Implitapide [129] | Inhibition of microsomal triglyceride transfer protein (MTP) | Reduction in TG levels and total cholesterol in plasma |
| 17 | Niacin [130] | Inhibits synthesis of apolipoprotein B-100 required for synthesis of VLDL | Increases HDL cholesterol levels and lowers LDL, VLDL and lipoprotein |
| 18 | Ezetimibe [131] | Reduces intestinal absorption of cholesterol | Reduces LDL cholesterol levels in patients with primary hypercholesterolemia |
| 19 | Cholestyramine [132] | Limits the reabsorption of bile acids in GI tract by forming a resin | Reduction in LDL level in primary hypercholesterolemia |
| 20 | Cholestipol [133] | Formation of complex with bile acids | Elimination of apoB-containing lipoproteins and LDL |
| 21 | Atorvastatin [134] | HMG-CoA reductase inhibition | Lowers blood total cholesterol and LDL |
| 22 | Fluvastatin [135] | Competitive inhibition of hydroxymethylglutaryl-coenzyme A (HMG-CoA) reductase | Decreases total cholesterol, LDL and triglyceride |
| 23 | Lovastatin [136] | HMG-CoA reductase inhibtion | Decreases triglyceride, VLDL and increases HDL |
| 24 | Pitavastatin sodium [137] | HMG-CoA reductase inhibtion | Decreases triglyceride, VLDL and increases HDL |
| 25 | Rosuvastatin [138] | Inhibition of HMG-CoA reductase | Lowers LDL-cholesterol, non-HDL cholesterol and total cholesterol |
| 26 | Simvastatin [139] | Competitive and reversible inhibition of HMG-CoA reductase enzyme | Reduced plasma LDL cholesterol levels |
| 27 | Pravastatin sodium [140] | HMG-CoA reductase inhibition | Reduces plasma LDL |
| 28 | Gernfibrosil [141] | Reduces TG production in liver | Reduction in plasma TG levels |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Florance, I.; Ramasubbu, S. Current Understanding on the Role of Lipids in Macrophages and Associated Diseases. Int. J. Mol. Sci. 2023, 24, 589. https://doi.org/10.3390/ijms24010589
Florance I, Ramasubbu S. Current Understanding on the Role of Lipids in Macrophages and Associated Diseases. International Journal of Molecular Sciences. 2023; 24(1):589. https://doi.org/10.3390/ijms24010589
Chicago/Turabian StyleFlorance, Ida, and Seenivasan Ramasubbu. 2023. "Current Understanding on the Role of Lipids in Macrophages and Associated Diseases" International Journal of Molecular Sciences 24, no. 1: 589. https://doi.org/10.3390/ijms24010589
APA StyleFlorance, I., & Ramasubbu, S. (2023). Current Understanding on the Role of Lipids in Macrophages and Associated Diseases. International Journal of Molecular Sciences, 24(1), 589. https://doi.org/10.3390/ijms24010589