
Dysregulation of ErbB4 Signaling Pathway in the Dorsal Hippocampus after Neonatal Hypoxia-Ischemia and Late Deficits in PV+ Interneurons, Synaptic Plasticity and Working Memory
 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
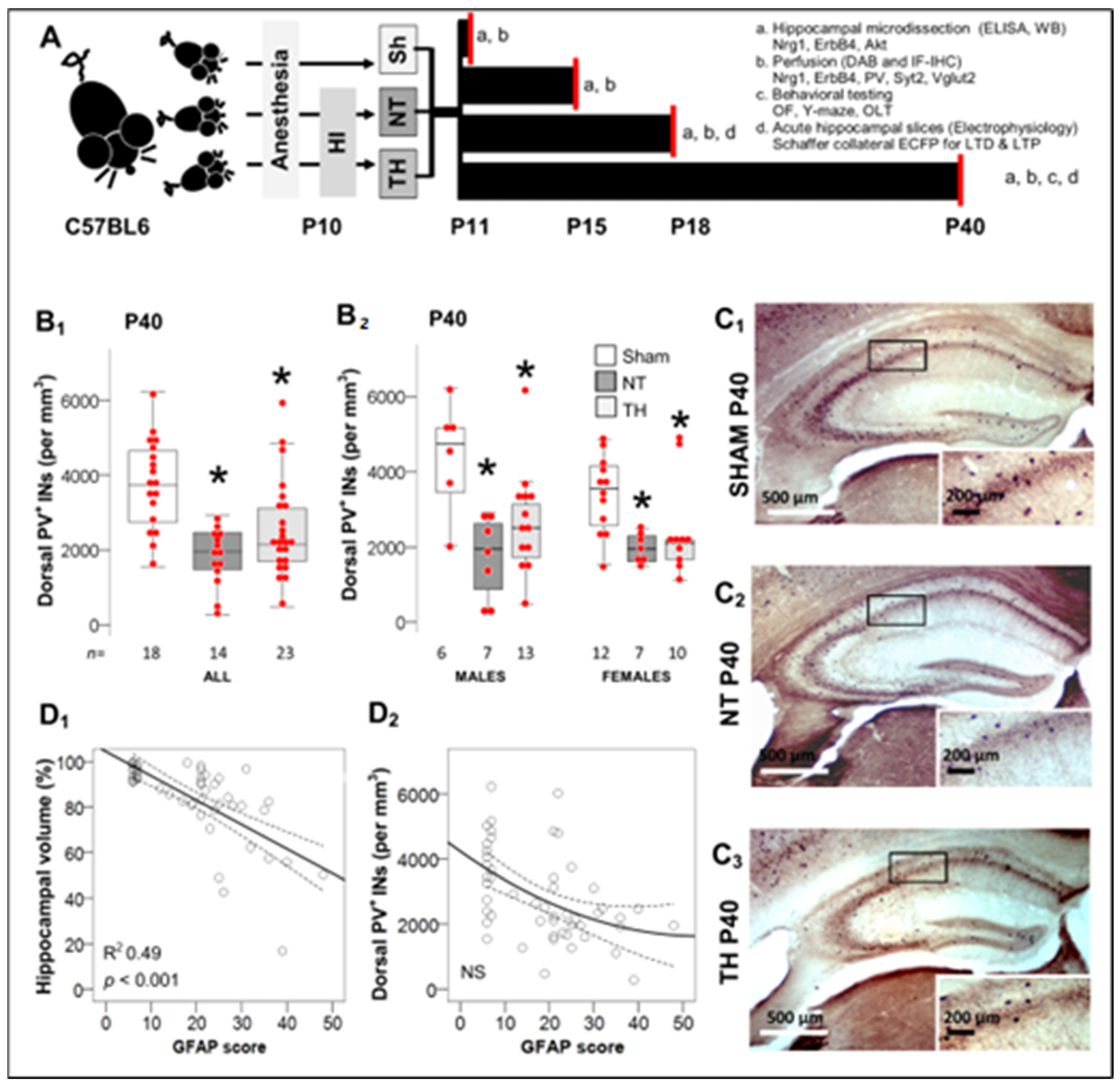
2.1. Deficit in PV+ IN Counts in Dorsal Hippocampus after Moderate-Severe Neonatal HI Persists to Early Adulhood
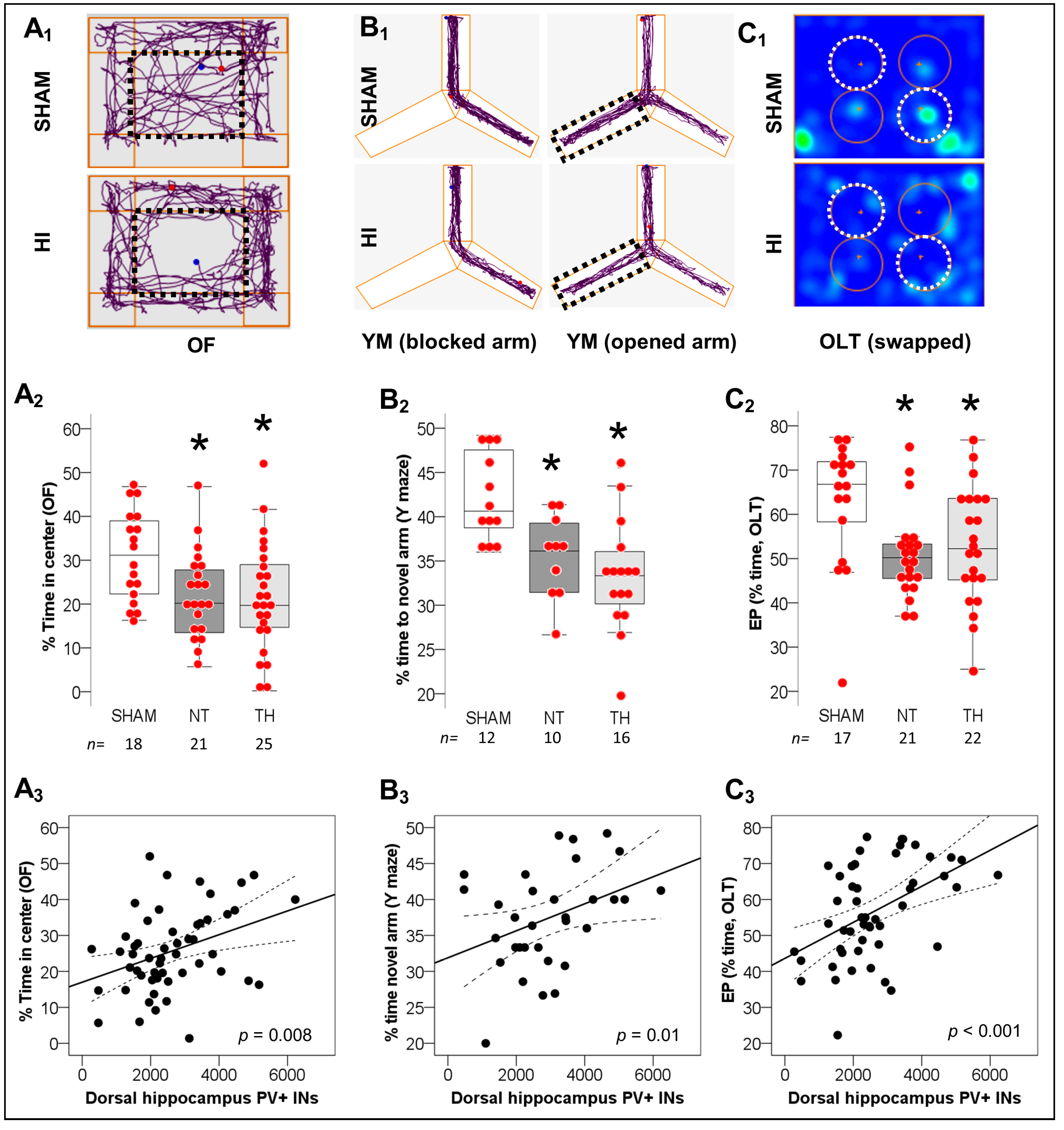
2.2. Spatial Working Memory Impairments after Neonatal HI and Correlation with Deficits in PV+ IN Counts in Dorsal Hippocampus
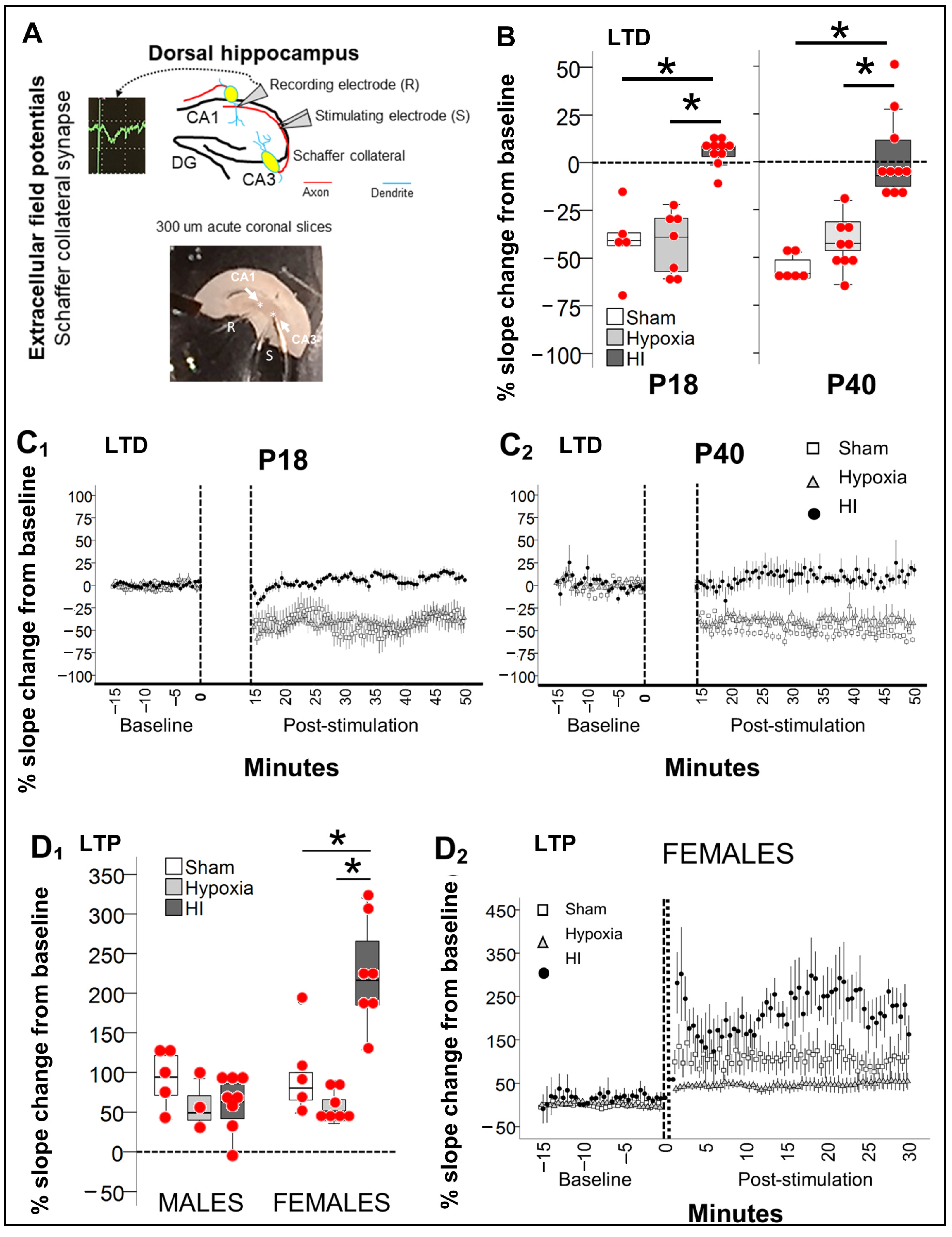
2.3. Disturbed Mechanisms of Synaptic Plasticity 8 and 30 Days after Neonatal HI
2.3.1. Persistently Blunted LTD in Schaffer Collateral Synapse after Neonatal HI
2.3.2. Preserved Schaffer Collateral LTP in Male Hippocampus Contrasts with Heightened LTP in Female Hippocampus 30 Days after HI
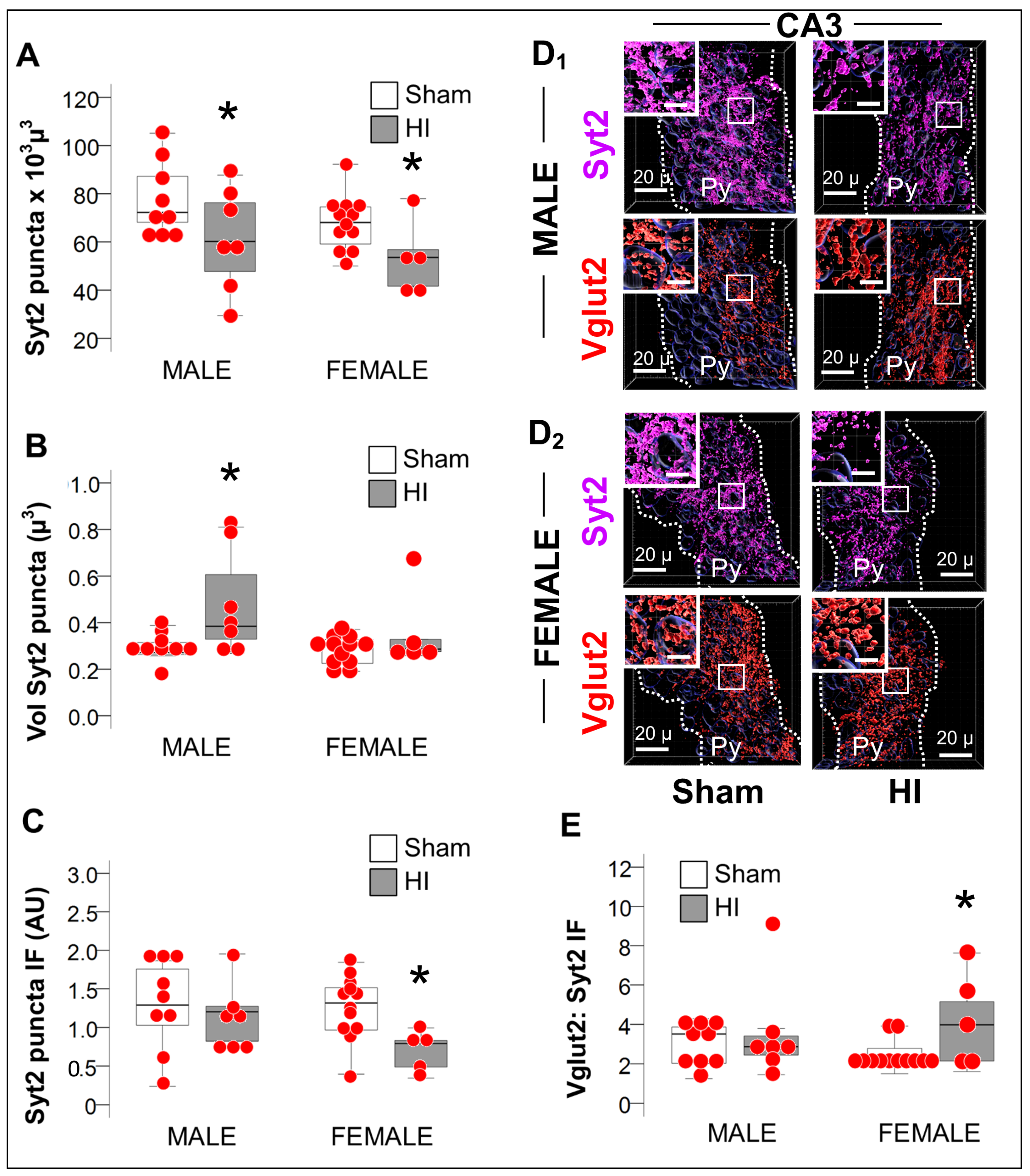
2.3.3. Increase in Vglut2/Syt2 Ratio in HI-Injured Dorsal Hippocampus Supports Relative Deficit in PV Inhibitory Input
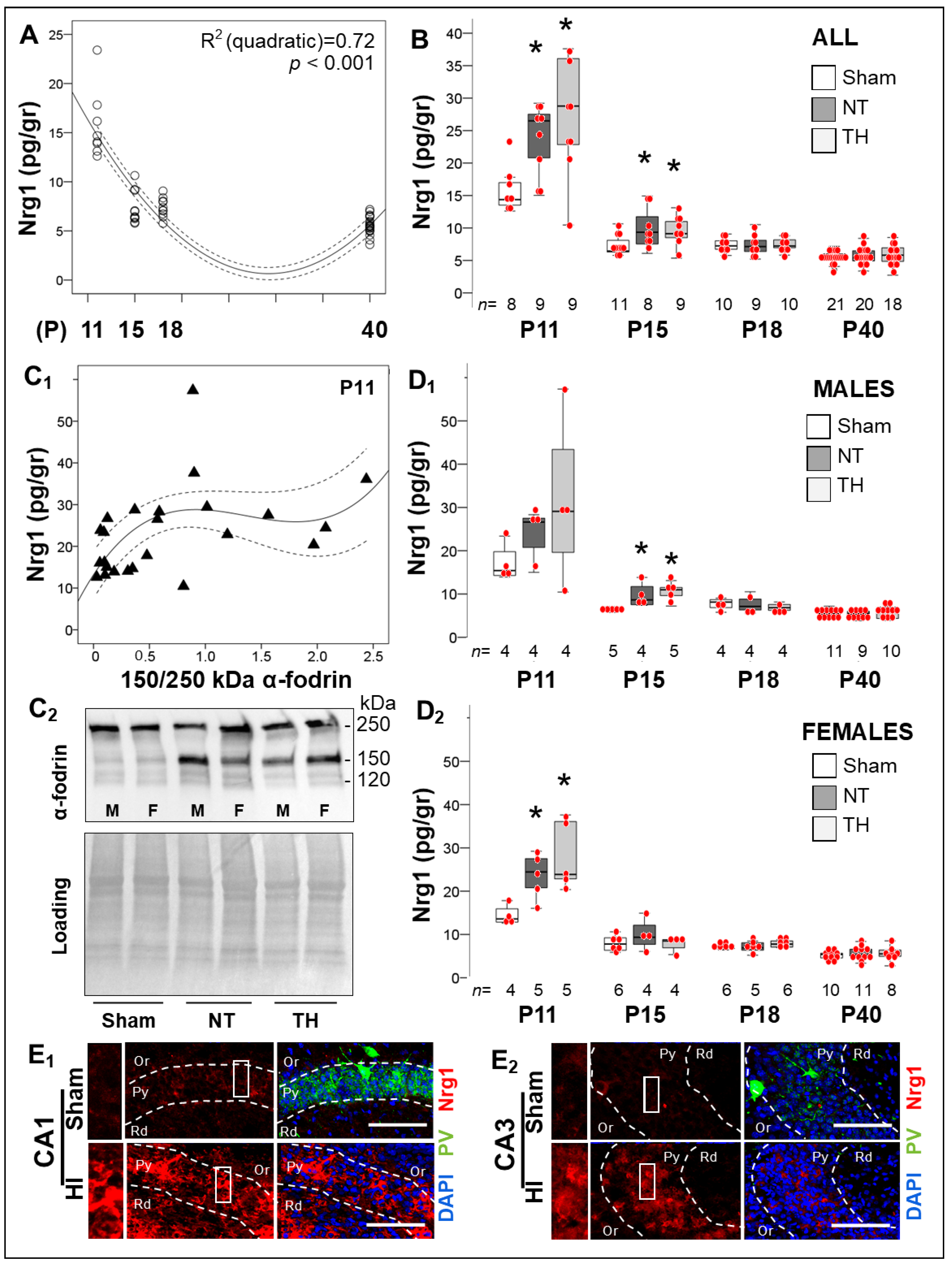
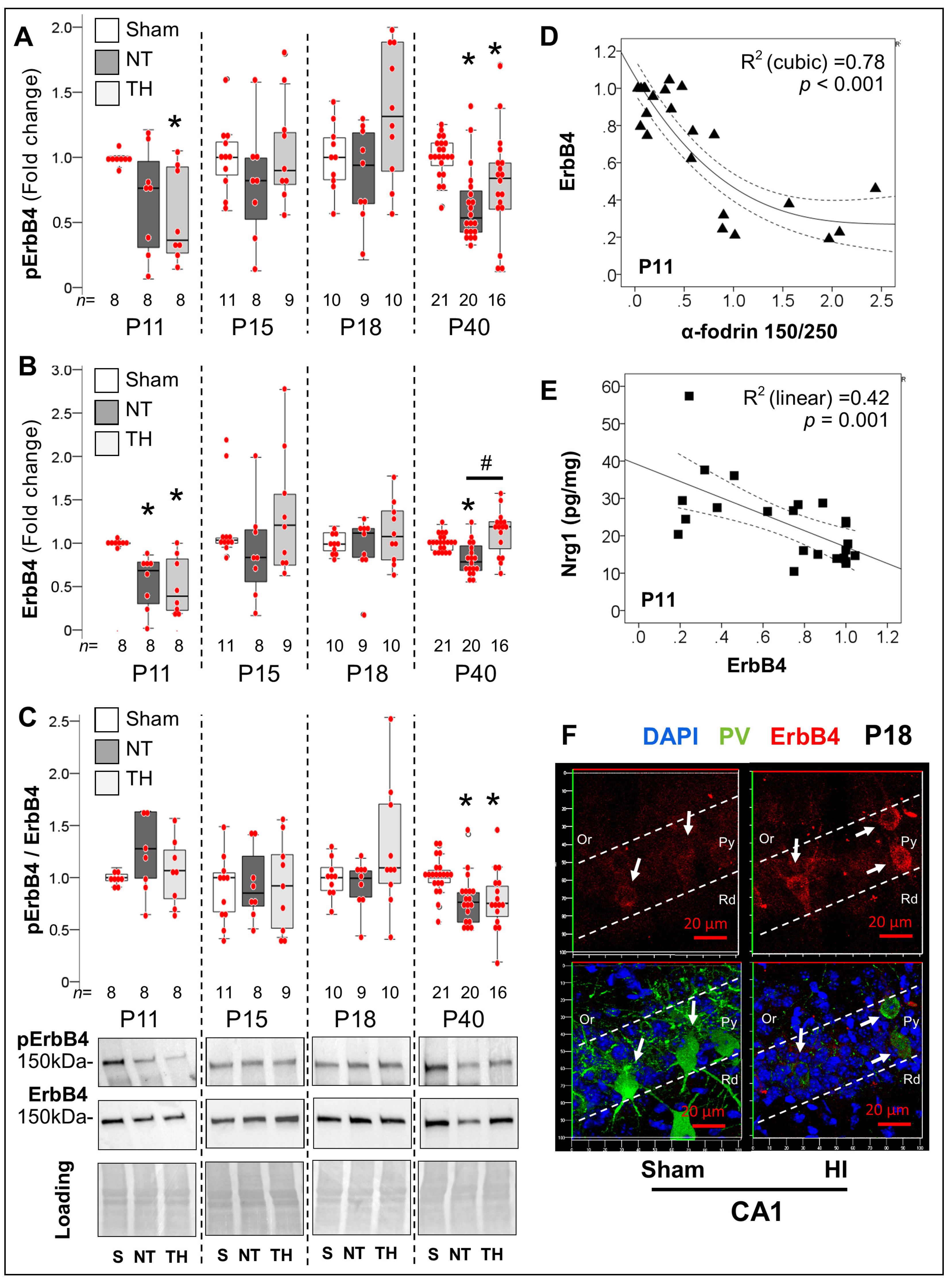
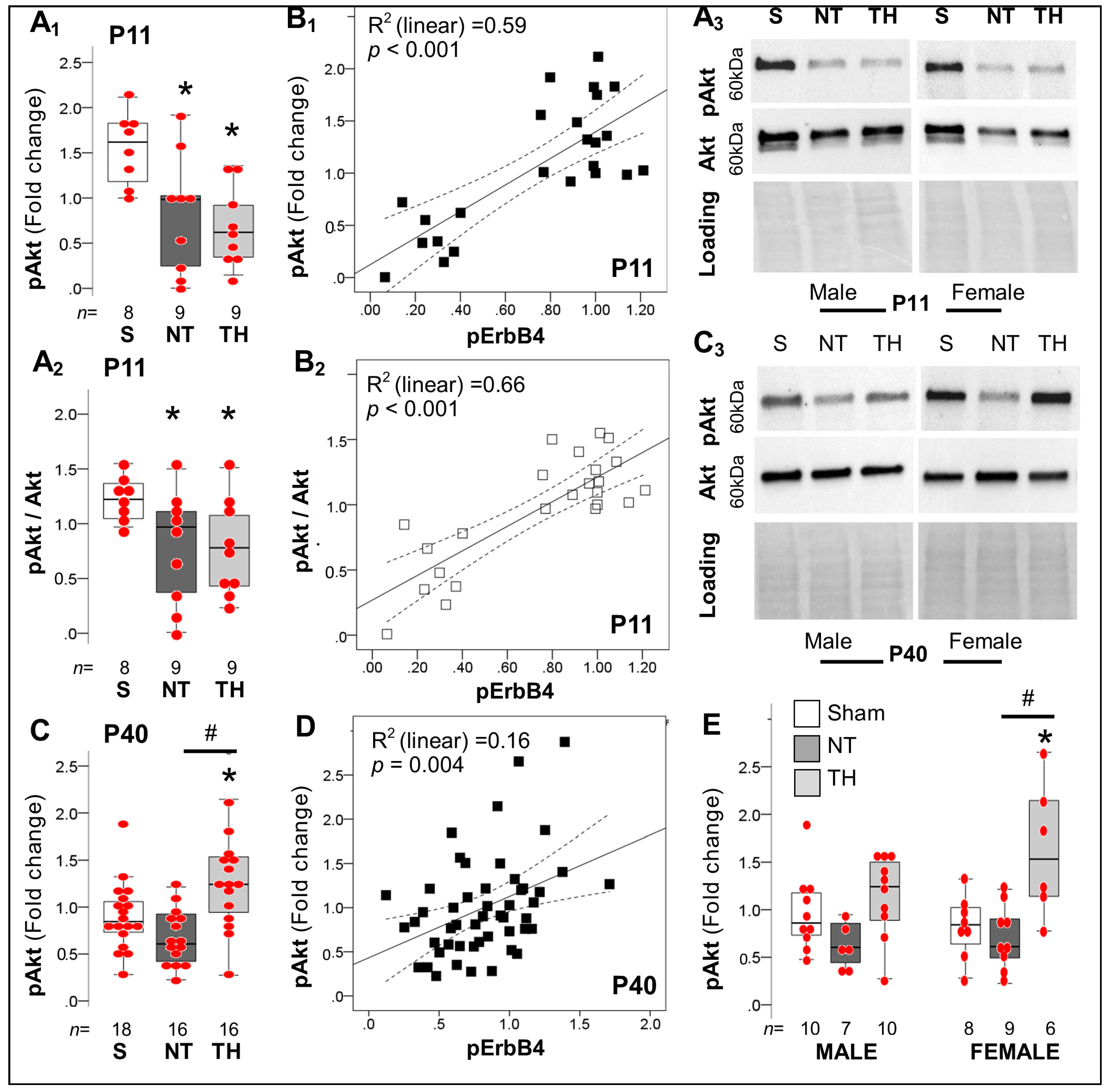
2.4. Persistent Dysregulation of Nrg1/ErbB4 Pathway after Neonatal HI Injury of the Hippocampus
2.4.1. Delayed Developmental Decline in Hippocampal Nrg1 Levels in Response to Neonatal HI
2.4.2. Acute and Late Deficits in ErbB4 Receptor Expression and Activation after Neonatal HI
2.4.3. Akt-Dependent Cascade after Neonatal HI
3. Discussion
Limitations
4. Materials and Methods
4.1. Animals and Experimental Design
4.2. Brain Processing
4.3. Immunohistochemistry (IHC)
4.3.1. GFAP-Derived Injury Scoring System
4.3.2. DAB-Based IHC for PV
4.3.3. Multichannel Immunofluorescent (IF)-IHC
4.3.4. Primary Antibodies for IF-IHC (Supplementary Table S1)
- (i)
- PV (Novus, Littleton, CO, USA, NBP2-50036; RRID: AB_2814697) Chicken polyclonal IgY antibody raised against recombinant full-length human PV (2.5 μg/mL)
- (ii)
- Nrg1 (ProteinTech Group, Rosemont, IL, USA, 66492-1-Ig; RRID: AB_2881857) Mouse monoclonal IgG1 antibody raised against NRG1 Fusion Protein [ProteinTech Group Arg0803] (4 μg/mL)
- (iii)
- ErbB4 (GeneTex, Irvine, CA, USA, GTX80811; RRID: AB_625545) Mouse monoclonal IgG2b antibody raised against an unconjugated synthetic peptide encompassing amino acids 1250-1264 (3 μg/mL)
- (iv)
- Syt2 (Developmental Studies Hydridoma Bank, Iowa City, IA, USA, 808402; RRID: AB_2315626) Mouse monoclonal IgG2a antibody raised against zebrafish synaptotagmin-2 [69]
- (v)
- VGlut2 (Abcam, Cambridge, UK, ab216463; RRID: AB_2893024) Rabbit monoclonal IgG antibody raised against synthetic VGlut2 peptide
4.3.5. Negative Controls
4.4. Confocal Microscopy and Image Processing
4.5. Semi-Quantitative Assessment of Hippocampal Atrophy
- S = hippocampal area (mm2)
- I = section position in anterior-posterior axis
- n = number of sum repeats
4.6. Nrg1 Enzyme-Linked Immunosorbent Assay (ELISA)
4.7. Western Blotting (WB)
Antibodies for WB (Supplementary Table S1)
- (i)
- ErbB4 (Cell Signaling, 4795S; RRID: AB_2099883) Rabbit monoclonal IgG antibody raised against synthetic peptide corresponding to residues near the carboxy-terminus of human ErbB4 (1 μg/mL), detects a single band at ~150 kD
- (ii)
- phosphorylated ErbB4 (pErbB4) (ThermoFischer, PA5-12606; RRID: AB_10990353) Rabbit polyclonal IgG antibody raised against a KLH conjugated synthetic phosphopeptide corresponding to amino acid residues surrounding Tyr1162 of human ErbB4 (2 μg/mL), detecting a single band at ~150 kDa
- (iii)
- Akt (Cell Signaling, 2920S; RRID: AB_1147620) Mouse monoclonal IgG1 antibody raised against synthetic peptide at the carboxy-terminal sequence of human Akt (1 μg/mL), detecting a band at ~ 60kDa
- (iv)
- phosphorylated Akt (pAkt) (Cell Signaling, 9271S; RRID: AB_329825) Rabbit polyclonal IgG antibody raised against a synthetic phosphopeptide corresponding to residues surrounding Ser473 of mouse Akt (1 μg/mL), detecting a band at ~60 kDa
- (v)
- α-fodrin (Enzo Life Sciences, Farmingdale, NY, USA, FG6090; RRID: AB_2050678) Mouse monoclonal IgG1 antibody raised against chicken blood cell membranes following hypotonic lysis and mechanical enucleation (0.25 μg/mL), detecting bands at 250, ~150, and ~120 kDa
- (vi)
- GFAP (ProteinTech, 16825-1-AP; RRID: AB_2109646) Rabbit polyclonal IgG antibody raised against GFAP fusion protein (ProteinTECH, Ag10423) (0.2 μg/mL), detecting a band at ~50 kDa.
4.8. EcFP of Schaffer Collateral Synapse
4.9. Behavioral Testing
4.9.1. Open Field (P36)
4.9.2. Object Location Task (P36–P37)
4.9.3. Y-Maze (Phase 1 P34–35 & Phase 2 P38–P39)
4.10. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Campanac, E.; Gasselin, C.; Baude, A.; Rama, S.; Ankri, N.; Debanne, D. Enhanced intrinsic excitability in basket cells maintains excitatory-inhibitory balance in hippocampal circuits. Neuron 2013, 77, 712–722. [Google Scholar] [CrossRef] [PubMed]
- Khoo, G.H.; Lin, Y.-T.; Tsai, T.-C.; Hsu, K.-S. Perineuronal Nets Restrict the Induction of Long-Term Depression in the Mouse Hippocampal CA1 Region. Mol. Neurobiol. 2019, 56, 6436–6450. [Google Scholar] [CrossRef] [PubMed]
- Udakis, M.; Pedrosa, V.; Chamberlain, S.E.L.; Clopath, C.; Mellor, J.R. Interneuron-specific plasticity at parvalbumin and somatostatin inhibitory synapses onto CA1 pyramidal neurons shapes hippocampal output. Nat. Commun. 2020, 11, 4395. [Google Scholar] [CrossRef] [PubMed]
- Buzsáki, G. Hippocampal GABAergic interneurons: A physiological perspective. Neurochem. Res. 2001, 26, 899–905. [Google Scholar] [CrossRef]
- Xu, M.-Y.; Wong, A.H.C. GABAergic inhibitory neurons as therapeutic targets for cognitive impairment in schizophrenia. Acta Pharmacol. Sin. 2018, 39, 733–753. [Google Scholar] [CrossRef]
- Chavez-Valdez, R.; Emerson, P.; Goffigan-Holmes, J.; Kirkwood, A.; Martin, L.J.; Northington, F.J. Delayed injury of hippocampal interneurons after neonatal hypoxia-ischemia and therapeutic hypothermia in a murine model. Hippocampus 2018, 28, 617–630. [Google Scholar] [CrossRef]
- Chavez-Valdez, R.; Lechner, C.; Emerson, P.; Northington, F.J.; Martin, L.J. Accumulation of PSA-NCAM marks nascent neurodegeneration in the dorsal hippocampus after neonatal hypoxic-ischemic brain injury in mice. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2021, 41, 1039–1057. [Google Scholar] [CrossRef]
- Lechner, C.R.; McNally, M.A.; St Pierre, M.; Felling, R.J.; Northington, F.J.; Stafstrom, C.E.; Chavez-Valdez, R. Sex specific correlation between GABAergic disruption in the dorsal hippocampus and flurothyl seizure susceptibility after neonatal hypoxic-ischemic brain injury. Neurobiol. Dis. 2021, 148, 105222. [Google Scholar] [CrossRef]
- Burnsed, J.C.; Chavez-Valdez, R.; Hossain, M.S.; Kesavan, K.; Martin, L.J.; Zhang, J.; Northington, F.J. Hypoxia-ischemia and therapeutic hypothermia in the neonatal mouse brain—A longitudinal study. PLoS ONE 2015, 10, e0118889. [Google Scholar] [CrossRef]
- Diaz, J.; Abiola, S.; Kim, N.; Avaritt, O.; Flock, D.; Yu, J.; Northington, F.J.; Chavez-Valdez, R. Therapeutic Hypothermia Provides Variable Protection against Behavioral Deficits after Neonatal Hypoxia-Ischemia: A Potential Role for Brain-Derived Neurotrophic Factor. Dev. Neurosci. 2017, 39, 257–272. [Google Scholar] [CrossRef]
- Goffigan-Holmes, J.; Sanabria, D.; Diaz, J.; Flock, D.; Chavez-Valdez, R. Calbindin-1 Expression in the Hippocampus following Neonatal Hypoxia-Ischemia and Therapeutic Hypothermia and Deficits in Spatial Memory. Dev. Neurosci. 2018, 40, 508–522. [Google Scholar] [CrossRef] [PubMed]
- Azzopardi, D.; Strohm, B.; Marlow, N.; Brocklehurst, P.; Deierl, A.; Eddama, O.; Goodwin, J.; Halliday, H.L.; Juszczak, E.; Kapellou, O.; et al. Effects of Hypothermia for Perinatal Asphyxia on Childhood Outcomes. N. Engl. J. Med. 2014, 371, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Pelkey, K.A.; Chittajallu, R.; Craig, M.T.; Tricoire, L.; Wester, J.C.; McBain, C.J. Hippocampal GABAergic Inhibitory Interneurons. Physiol. Rev. 2017, 97, 1619–1747. [Google Scholar] [CrossRef]
- Jin, I.; Hawkins, R.D. Presynaptic and postsynaptic mechanisms of a novel form of homosynaptic potentiation at aplysia sensory-motor neuron synapses. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 7288–7297. [Google Scholar] [CrossRef] [PubMed]
- Pozas, E.; Ibáñez, C.F. GDNF and GFRalpha1 promote differentiation and tangential migration of cortical GABAergic neurons. Neuron 2005, 45, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Fazzari, P.; Paternain, A.V.; Valiente, M.; Pla, R.; Luján, R.; Lloyd, K.; Lerma, J.; Marín, O.; Rico, B. Control of cortical GABA circuitry development by Nrg1 and ErbB4 signalling. Nature 2010, 464, 1376–1380. [Google Scholar] [CrossRef]
- Yau, H.-J.; Wang, H.-F.; Lai, C.; Liu, F.-C. Neural development of the neuregulin receptor ErbB4 in the cerebral cortex and the hippocampus: Preferential expression by interneurons tangentially migrating from the ganglionic eminences. Cereb. Cortex 2003, 13, 252–264. [Google Scholar] [CrossRef]
- Li, H.; Chou, S.-J.; Hamasaki, T.; Perez-Garcia, C.G.; O’Leary, D.D.M. Neuregulin repellent signaling via ErbB4 restricts GABAergic interneurons to migratory paths from ganglionic eminence to cortical destinations. Neural Dev. 2012, 7, 10. [Google Scholar] [CrossRef]
- Flames, N.; Long, J.E.; Garratt, A.N.; Fischer, T.M.; Gassmann, M.; Birchmeier, C.; Lai, C.; Rubenstein, J.L.R.; Marín, O. Short- and long-range attraction of cortical GABAergic interneurons by neuregulin-1. Neuron 2004, 44, 251–261. [Google Scholar] [CrossRef]
- Mei, L.; Xiong, W.-C. Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat. Rev. Neurosci. 2008, 9, 437–452. [Google Scholar] [CrossRef]
- Guan, Y.-F.; Wu, C.-Y.; Fang, Y.-Y.; Zeng, Y.-N.; Luo, Z.-Y.; Li, S.-J.; Li, X.-W.; Zhu, X.-H.; Mei, L.; Gao, T.-M. Neuregulin 1 protects against ischemic brain injury via ErbB4 receptors by increasing GABAergic transmission. Neuroscience 2015, 307, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Zhang, M.; Yin, D.-M.; Wen, L.; Ting, A.; Wang, P.; Lu, Y.-S.; Zhu, X.-H.; Li, S.-J.; Wu, C.-Y.; et al. ErbB4 in parvalbumin-positive interneurons is critical for neuregulin 1 regulation of long-term potentiation. Proc. Natl. Acad. Sci. USA 2010, 107, 21818–21823. [Google Scholar] [CrossRef] [PubMed]
- Rong, L.; Ding, K.; Zhang, M.; Guo, Y. Neuregulin1β improves cognitive dysfunction and up-regulates expression of p-ERK1/2 in rats with chronic omethoate poisoning. Behav. Brain Funct. BBF 2015, 11, 5. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vullhorst, D.; Neddens, J.; Karavanova, I.; Tricoire, L.; Petralia, R.S.; McBain, C.J.; Buonanno, A. Selective expression of ErbB4 in interneurons, but not pyramidal cells, of the rodent hippocampus. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 12255–12264. [Google Scholar] [CrossRef] [PubMed]
- López-Bendito, G.; Cautinat, A.; Sánchez, J.A.; Bielle, F.; Flames, N.; Garratt, A.N.; Talmage, D.A.; Role, L.W.; Charnay, P.; Marín, O.; et al. Tangential neuronal migration controls axon guidance: A role for neuregulin-1 in thalamocortical axon navigation. Cell 2006, 125, 127–142. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Woo, R.-S.; Mei, L.; Malinow, R. The neuregulin-1 receptor erbB4 controls glutamatergic synapse maturation and plasticity. Neuron 2007, 54, 583–597. [Google Scholar] [CrossRef]
- Woo, R.-S.; Li, X.-M.; Tao, Y.; Carpenter-Hyland, E.; Huang, Y.Z.; Weber, J.; Neiswender, H.; Dong, X.-P.; Wu, J.; Gassmann, M.; et al. Neuregulin-1 enhances depolarization-induced GABA release. Neuron 2007, 54, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Krivosheya, D.; Tapia, L.; Levinson, J.N.; Huang, K.; Kang, Y.; Hines, R.; Ting, A.K.; Craig, A.M.; Mei, L.; Bamji, S.X.; et al. ErbB4-neuregulin signaling modulates synapse development and dendritic arborization through distinct mechanisms. J. Biol. Chem. 2008, 283, 32944–32956. [Google Scholar] [CrossRef]
- Cahill, M.E.; Jones, K.A.; Rafalovich, I.; Xie, Z.; Barros, C.S.; Müller, U.; Penzes, P. Control of interneuron dendritic growth through NRG1/erbB4-mediated kalirin-7 disinhibition. Mol. Psychiatry 2012, 17, 99–107. [Google Scholar] [CrossRef]
- Zhang, H.; He, X.; Mei, Y.; Ling, Q. Ablation of ErbB4 in parvalbumin-positive interneurons inhibits adult hippocampal neurogenesis through down-regulating BDNF/TrkB expression. J. Comp. Neurol. 2018, 526, 2482–2492. [Google Scholar] [CrossRef]
- Neddens, J.; Buonanno, A. Selective populations of hippocampal interneurons express ErbB4 and their number and distribution is altered in ErbB4 knockout mice. Hippocampus 2010, 20, 724–744. [Google Scholar] [CrossRef] [PubMed]
- Horn, M.E.; Nicoll, R.A. Somatostatin and parvalbumin inhibitory synapses onto hippocampal pyramidal neurons are regulated by distinct mechanisms. Proc. Natl. Acad. Sci. USA 2018, 115, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Luo, F.; Li, B.-M.; Mei, L. NRG1-ErbB4 signaling promotes functional recovery in a murine model of traumatic brain injury via regulation of GABA release. Exp. Brain Res. 2019, 237, 3351–3362. [Google Scholar] [CrossRef] [PubMed]
- Tucker, L.B.; Winston, B.S.; Liu, J.; Velosky, A.G.; Fu, A.H.; Grillakis, A.A.; McCabe, J.T. Sex differences in cued fear responses and parvalbumin cell density in the hippocampus following repetitive concussive brain injuries in C57BL/6J mice. PLoS ONE 2019, 14, e0222153. [Google Scholar] [CrossRef]
- McNally, M.A.; Chavez-Valdez, R.; Felling, R.J.; Flock, D.L.; Northington, F.J.; Stafstrom, C.E. Seizure Susceptibility Correlates with Brain Injury in Male Mice Treated with Hypothermia after Neonatal Hypoxia-Ischemia. Dev. Neurosci. 2018, 40, 576–585. [Google Scholar] [CrossRef]
- de Lecea, L.; del Río, J.A.; Soriano, E. Developmental expression of parvalbumin mRNA in the cerebral cortex and hippocampus of the rat. Brain Res. Mol. Brain Res. 1995, 32, 1–13. [Google Scholar] [CrossRef]
- Wen, L.; Lu, Y.-S.; Zhu, X.-H.; Li, X.-M.; Woo, R.-S.; Chen, Y.-J.; Yin, D.-M.; Lai, C.; Terry, A.V.; Vazdarjanova, A.; et al. Neuregulin 1 regulates pyramidal neuron activity via ErbB4 in parvalbumin-positive interneurons. Proc. Natl. Acad. Sci. USA 2010, 107, 1211–1216. [Google Scholar] [CrossRef]
- Ting, A.K.; Chen, Y.; Wen, L.; Yin, D.-M.; Shen, C.; Tao, Y.; Liu, X.; Xiong, W.-C.; Mei, L. Neuregulin 1 Promotes Excitatory Synapse Development and Function in GABAergic Interneurons. J. Neurosci. 2011, 31, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.Z.; Won, S.; Ali, D.W.; Wang, Q.; Tanowitz, M.; Du, Q.S.; Pelkey, K.A.; Yang, D.J.; Xiong, W.C.; Salter, M.W.; et al. Regulation of neuregulin signaling by PSD-95 interacting with ErbB4 at CNS synapses. Neuron 2000, 26, 443–455. [Google Scholar] [CrossRef]
- Ferguson, B.R.; Gao, W.-J. PV Interneurons: Critical Regulators of E/I Balance for Prefrontal Cortex-Dependent Behavior and Psychiatric Disorders. Front. Neural Circuits 2018, 12, 37. [Google Scholar] [CrossRef]
- Que, L.; Lukacsovich, D.; Luo, W.; Földy, C. Transcriptional and morphological profiling of parvalbumin interneuron subpopulations in the mouse hippocampus. Nat. Commun. 2021, 12, 108. [Google Scholar] [CrossRef] [PubMed]
- Iwakura, Y.; Nawa, H. ErbB1–4-dependent EGF/neuregulin signals and their cross talk in the central nervous system: Pathological implications in schizophrenia and Parkinson’s disease. Front. Cell. Neurosci. 2013, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Mei, L.; Nave, K.-A. Neuregulin-ERBB signaling in the nervous system and neuropsychiatric diseases. Neuron 2014, 83, 27–49. [Google Scholar] [CrossRef] [PubMed]
- Moster, D.; Lie, R.T.; Markestad, T. Joint association of Apgar scores and early neonatal symptoms with minor disabilities at school age. Arch. Dis. Child. Fetal Neonatal Ed. 2002, 86, F16–F21. [Google Scholar] [CrossRef] [PubMed]
- Guillet, R.; Edwards, A.D.; Thoresen, M.; Ferriero, D.M.; Gluckman, P.D.; Whitelaw, A.; Gunn, A.J.; CoolCap Trial Group. Seven- to eight-year follow-up of the CoolCap trial of head cooling for neonatal encephalopathy. Pediatr. Res. 2012, 71, 205–209. [Google Scholar] [CrossRef]
- Pappas, A.; Shankaran, S.; McDonald, S.A.; Vohr, B.R.; Hintz, S.R.; Ehrenkranz, R.A.; Tyson, J.E.; Yolton, K.; Das, A.; Bara, R.; et al. Cognitive outcomes after neonatal encephalopathy. Pediatrics 2015, 135, e624–e634. [Google Scholar] [CrossRef]
- Shankaran, S.; Pappas, A.; McDonald, S.A.; Vohr, B.R.; Hintz, S.R.; Yolton, K.; Gustafson, K.E.; Leach, T.M.; Green, C.; Bara, R.; et al. Childhood Outcomes after Hypothermia for Neonatal Encephalopathy. N. Engl. J. Med. 2012, 366, 2085–2092. [Google Scholar] [CrossRef]
- Ognjanovski, N.; Schaeffer, S.; Wu, J.; Mofakham, S.; Maruyama, D.; Zochowski, M.; Aton, S.J. Parvalbumin-expressing interneurons coordinate hippocampal network dynamics required for memory consolidation. Nat. Commun. 2017, 8, 15039. [Google Scholar] [CrossRef]
- Xia, F.; Richards, B.A.; Tran, M.M.; Josselyn, S.A.; Takehara-Nishiuchi, K.; Frankland, P.W. Parvalbumin-positive interneurons mediate neocortical-hippocampal interactions that are necessary for memory consolidation. eLife 2017, 6, e27868. [Google Scholar] [CrossRef]
- Yang, X.; Yao, C.; Tian, T.; Li, X.; Yan, H.; Wu, J.; Li, H.; Pei, L.; Liu, D.; Tian, Q.; et al. A novel mechanism of memory loss in Alzheimer’s disease mice via the degeneration of entorhinal-CA1 synapses. Mol. Psychiatry 2018, 23, 199–210. [Google Scholar] [CrossRef]
- Abdul-Monim, Z.; Neill, J.C.; Reynolds, G.P. Sub-chronic psychotomimetic phencyclidine induces deficits in reversal learning and alterations in parvalbumin-immunoreactive expression in the rat. J. Psychopharmacol. 2007, 21, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Hayashida, M.; Miyaoka, T.; Tsuchie, K.; Araki, T.; Izuhara, M.; Miura, S.; Kanayama, M.; Ohtsuki, K.; Nagahama, M.; Azis, I.A.; et al. Parvalbumin-positive GABAergic interneurons deficit in the hippocampus in Gunn rats: A possible hyperbilirubinemia-induced animal model of schizophrenia. Heliyon 2019, 5, e02037. [Google Scholar] [CrossRef] [PubMed]
- Lodge, D.J.; Behrens, M.M.; Grace, A.A. A loss of parvalbumin-containing interneurons is associated with diminished oscillatory activity in an animal model of schizophrenia. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 2344–2354. [Google Scholar] [CrossRef] [PubMed]
- Caccavano, A.; Bozzelli, P.L.; Forcelli, P.A.; Pak, D.T.S.; Wu, J.-Y.; Conant, K.; Vicini, S. Inhibitory Parvalbumin Basket Cell Activity is Selectively Reduced during Hippocampal Sharp Wave Ripples in a Mouse Model of Familial Alzheimer’s Disease. J. Neurosci. 2020, 40, 5116–5136. [Google Scholar] [CrossRef]
- Giesers, N.K.; Wirths, O. Loss of Hippocampal Calretinin and Parvalbumin Interneurons in the 5XFAD Mouse Model of Alzheimer’s Disease. ASN Neuro 2020, 12, 175909142092535. [Google Scholar] [CrossRef]
- Mahar, I.; Albuquerque, M.S.; Mondragon-Rodriguez, S.; Cavanagh, C.; Davoli, M.A.; Chabot, J.-G.; Williams, S.; Mechawar, N.; Quirion, R.; Krantic, S. Phenotypic Alterations in Hippocampal NPY- and PV-Expressing Interneurons in a Presymptomatic Transgenic Mouse Model of Alzheimer’s Disease. Front. Aging Neurosci. 2017, 8, 327. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Konradi, C.; Yang, C.K.; Zimmerman, E.I.; Lohmann, K.M.; Gresch, P.; Pantazopoulos, H.; Berretta, S.; Heckers, S. Hippocampal interneurons are abnormal in schizophrenia. Schizophr. Res. 2011, 131, 165–173. [Google Scholar] [CrossRef]
- Lewis, D.A.; Hashimoto, T.; Volk, D.W. Cortical inhibitory neurons and schizophrenia. Nat. Rev. Neurosci. 2005, 6, 312–324. [Google Scholar] [CrossRef]
- Li, Y.; Sun, H.; Chen, Z.; Xu, H.; Bu, G.; Zheng, H. Implications of GABAergic Neurotransmission in Alzheimer’s Disease. Front. Aging Neurosci. 2016, 8, 31. [Google Scholar] [CrossRef]
- Palop, J.J.; Mucke, L. Amyloid-β–induced neuronal dysfunction in Alzheimer’s disease: From synapses toward neural networks. Nat. Neurosci. 2010, 13, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Ramani, M.; Miller, K.; Ambalavanan, N.; McMahon, L.L. Increased Excitability and Heightened Magnitude of Long-Term Potentiation at Hippocampal CA3–CA1 Synapses in a Mouse Model of Neonatal Hyperoxia Exposure. Front. Synaptic Neurosci. 2021, 12, 609903. [Google Scholar] [CrossRef] [PubMed]
- Goussakov, I.; Synowiec, S.; Yarnykh, V.; Drobyshevsky, A. Immediate and delayed decrease of long term potentiation and memory deficits after neonatal intermittent hypoxia. Int. J. Dev. Neurosci. Off. J. Int. Soc. Dev. Neurosci. 2019, 74, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ikrar, T.; Davis, M.F.; Gong, N.; Zheng, X.; Luo, Z.D.; Lai, C.; Mei, L.; Holmes, T.C.; Gandhi, S.P.; et al. Neuregulin-1/ErbB4 Signaling Regulates Visual Cortical Plasticity. Neuron 2016, 92, 160–173. [Google Scholar] [CrossRef]
- Wang, H.; Liu, F.; Chen, W.; Sun, X.; Cui, W.; Dong, Z.; Zhao, K.; Zhang, H.; Li, H.; Xing, G.; et al. Genetic recovery of ErbB4 in adulthood partially restores brain functions in null mice. Proc. Natl. Acad. Sci. USA 2018, 115, 13105–13110. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-B.; Yoo, J.-Y.; Yoo, S.-Y.; Lee, J.-H.; Chang, W.; Kim, H.-S.; Baik, T.-K.; Woo, R.-S. Neuregulin-1 inhibits CoCl2-induced upregulation of excitatory amino acid carrier 1 expression and oxidative stress in SH-SY5Y cells and the hippocampus of mice. Mol. Brain 2020, 13, 153. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.-Y.; Kim, H.-B.; Yoo, S.-Y.; Yoo, H.-I.; Song, D.-Y.; Baik, T.-K.; Lee, J.-H.; Woo, R.-S. Neuregulin 1/ErbB4 signaling attenuates neuronal cell damage under oxygen-glucose deprivation in primary hippocampal neurons. Anat. Cell Biol. 2019, 52, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Fabres, R.B.; Montes, N.L.; Camboim, Y.d.M.; de Souza, S.K.; Nicola, F.; Tassinari, I.D.; Ribeiro, M.F.M.; Netto, C.A.; de Fraga, L.S. Long-Lasting Actions of Progesterone Protect the Neonatal Brain Following Hypoxia-Ischemia. Cell. Mol. Neurobiol. 2020, 40, 1417–1428. [Google Scholar] [CrossRef]
- Trevarrow, B.; Marks, D.L.; Kimmel, C.B. Organization of hindbrain segments in the zebrafish embryo. Neuron 1990, 4, 669–679. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Wang, H.; Ardiles, A.O.; Yang, S.; Tran, T.; Posada-Duque, R.; Valdivia, G.; Baek, M.; Chuang, Y.-A.; Palacios, A.G.; Gallagher, M.; et al. Metabotropic Glutamate Receptors Induce a Form of LTP Controlled by Translation and Arc Signaling in the Hippocampus. J. Neurosci. 2016, 36, 1723–1729. [Google Scholar] [CrossRef] [PubMed]
- Bailey, K.R.; Crawley, J.N. Anxiety-Related Behaviors in Mice. In Methods of Behavior Analysis in Neuroscience; Buccafusco, J.J., Ed.; Frontiers in Neuroscience; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2009; ISBN 978-1-4200-5234-3. [Google Scholar]
- Prut, L.; Belzung, C. The open field as a paradigm to measure the effects of drugs on anxiety-like behaviors: A review. Eur. J. Pharmacol. 2003, 463, 3–33. [Google Scholar] [CrossRef] [PubMed]
- Carlini, V.P.; Ghersi, M.; Gabach, L.; Schiöth, H.B.; Pérez, M.F.; Ramirez, O.A.; Fiol de Cuneo, M.; de Barioglio, S.R. Hippocampal effects of neuronostatin on memory, anxiety-like behavior and food intake in rats. Neuroscience 2011, 197, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Antunes, M.; Biala, G. The novel object recognition memory: Neurobiology, test procedure, and its modifications. Cogn. Process. 2012, 13, 93–110. [Google Scholar] [CrossRef] [PubMed]
- Jablonski, S.A.; Schreiber, W.B.; Westbrook, S.R.; Brennan, L.E.; Stanton, M.E. Determinants of novel object and location recognition during development. Behav. Brain Res. 2013, 256, 140–150. [Google Scholar] [CrossRef]
- Vogel-Ciernia, A.; Wood, M.A. Examining object location and object recognition memory in mice. Curr. Protoc. Neurosci. 2014, 69, 8.31.1–8.31.17. [Google Scholar] [CrossRef]
- Kraeuter, A.-K.; Guest, P.C.; Sarnyai, Z. The Y-Maze for Assessment of Spatial Working and Reference Memory in Mice. Methods Mol. Biol. 2019, 1916, 105–111. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spahic, H.; Parmar, P.; Miller, S.; Emerson, P.C.; Lechner, C.; St. Pierre, M.; Rastogi, N.; Nugent, M.; Duck, S.A.; Kirkwood, A.; et al. Dysregulation of ErbB4 Signaling Pathway in the Dorsal Hippocampus after Neonatal Hypoxia-Ischemia and Late Deficits in PV+ Interneurons, Synaptic Plasticity and Working Memory. Int. J. Mol. Sci. 2023, 24, 508. https://doi.org/10.3390/ijms24010508
Spahic H, Parmar P, Miller S, Emerson PC, Lechner C, St. Pierre M, Rastogi N, Nugent M, Duck SA, Kirkwood A, et al. Dysregulation of ErbB4 Signaling Pathway in the Dorsal Hippocampus after Neonatal Hypoxia-Ischemia and Late Deficits in PV+ Interneurons, Synaptic Plasticity and Working Memory. International Journal of Molecular Sciences. 2023; 24(1):508. https://doi.org/10.3390/ijms24010508
Chicago/Turabian StyleSpahic, Harisa, Pritika Parmar, Sarah Miller, Paul Casey Emerson, Charles Lechner, Mark St. Pierre, Neetika Rastogi, Michael Nugent, Sarah Ann Duck, Alfredo Kirkwood, and et al. 2023. "Dysregulation of ErbB4 Signaling Pathway in the Dorsal Hippocampus after Neonatal Hypoxia-Ischemia and Late Deficits in PV+ Interneurons, Synaptic Plasticity and Working Memory" International Journal of Molecular Sciences 24, no. 1: 508. https://doi.org/10.3390/ijms24010508
APA StyleSpahic, H., Parmar, P., Miller, S., Emerson, P. C., Lechner, C., St. Pierre, M., Rastogi, N., Nugent, M., Duck, S. A., Kirkwood, A., & Chavez-Valdez, R. (2023). Dysregulation of ErbB4 Signaling Pathway in the Dorsal Hippocampus after Neonatal Hypoxia-Ischemia and Late Deficits in PV+ Interneurons, Synaptic Plasticity and Working Memory. International Journal of Molecular Sciences, 24(1), 508. https://doi.org/10.3390/ijms24010508