
Targeting MYC Regulation with Polypurine Reverse Hoogsteen Oligonucleotides
 , , ,
, , , 
Abstract
1. Introduction
2. Results
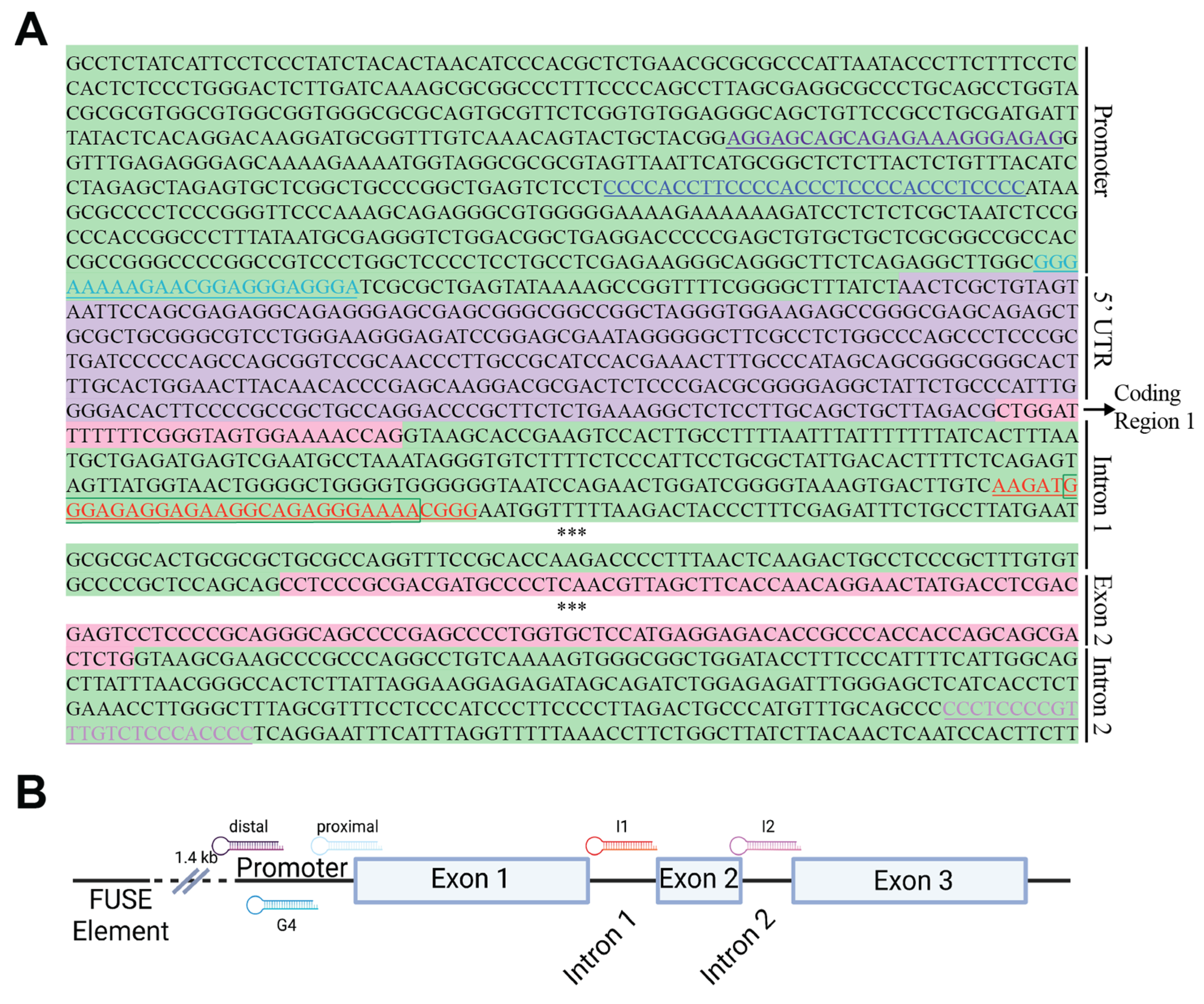
2.1. PPRH Target Selection and Sequence Design
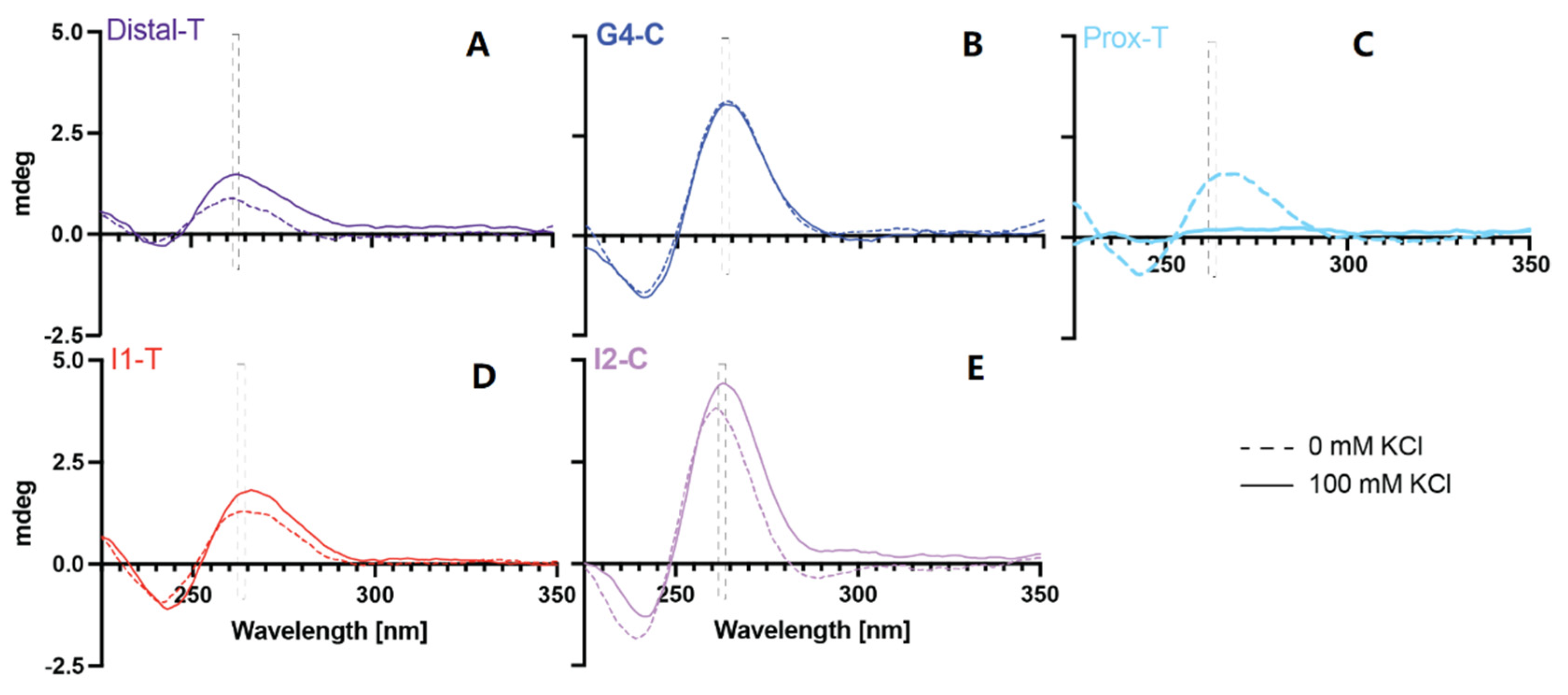
2.2. Examination of G4 Formation within the Target Sequences
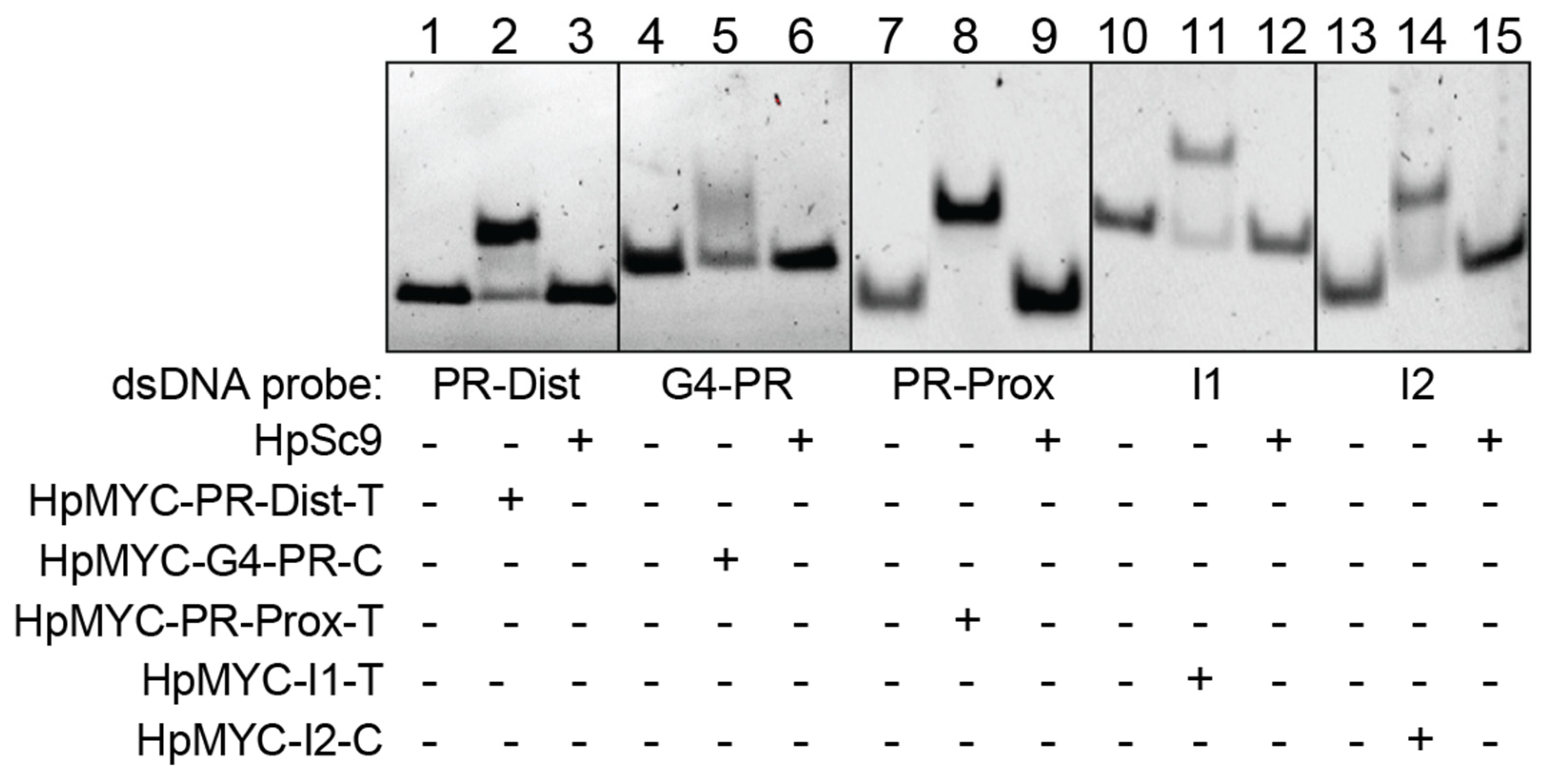
2.3. PPRHs Binding to MYC Target Sequences
2.4. Polypurine Strand Displacement
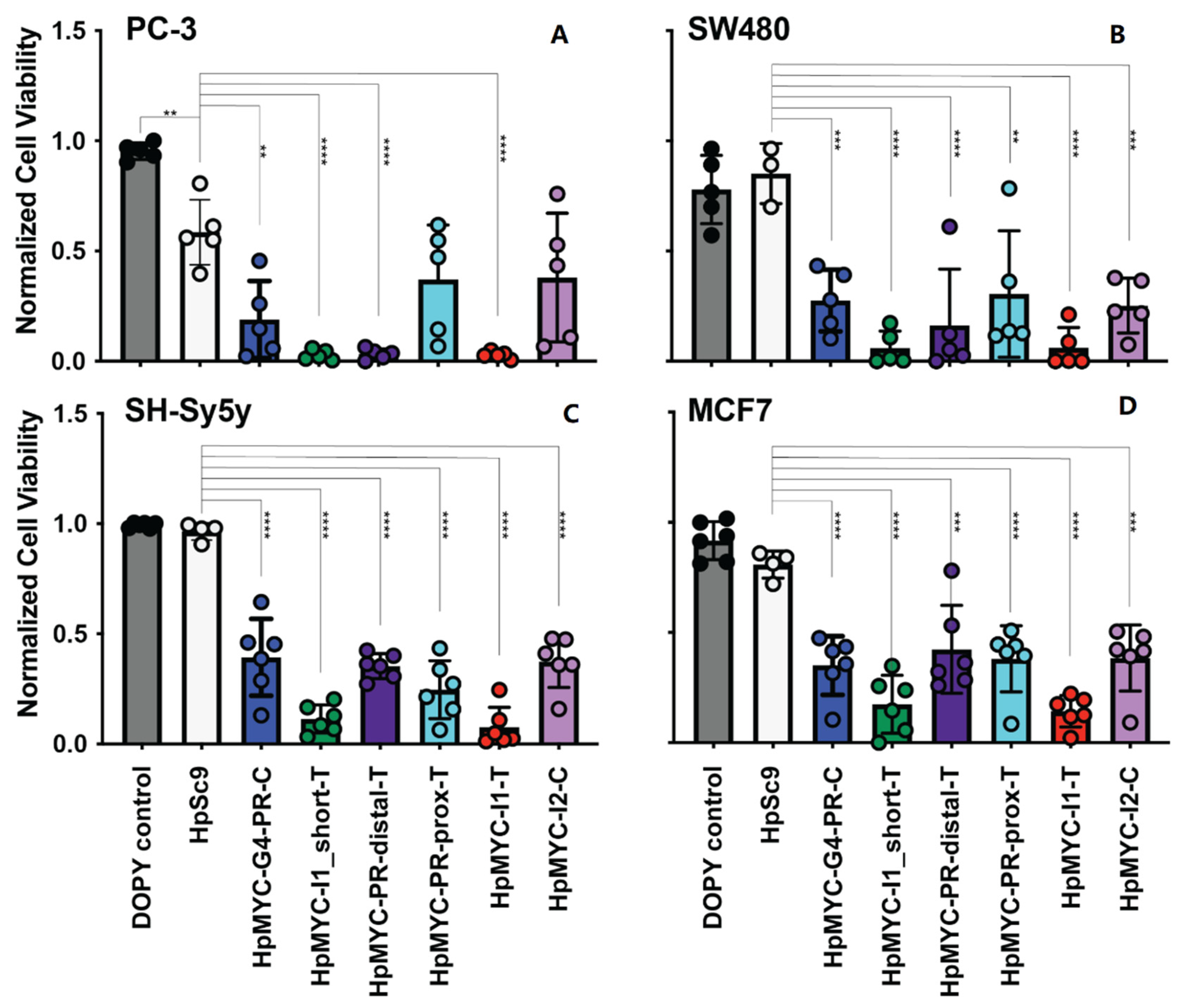
2.5. Effect of MYC-Targeting PPRHs on Promoter Activity, Cell Growth and Viability, and Correlating Changes in Transcription and Translation
3. Discussion
4. Materials and Methods
4.1. Design of Polypurine Reverse Hoogsteen Hairpins
4.2. Electronic Circular Dichroism (ECD)
4.3. Melting Temperature Assay
4.4. Electrophoretic Mobility Shift Assay (EMSA)
4.5. Strand Displacement Assay upon PPRH Incubation
4.6. Cell Cultures
4.7. Cellular Viability and Cell Growth Studies
4.8. Luciferase Assays
4.9. RT-qPCR
4.10. Western Blot Analyses
4.11. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shen, L.; O’Shea, J.M.; Kaadige, M.R.; Cunha, S.; Wilde, B.R.; Cohen, A.L.; Welm, A.L.; Ayer, D.E. Metabolic reprogramming in triple-negative breast cancer through Myc suppression of TXNIP. Proc. Natl. Acad. Sci. USA 2015, 112, 5425–5430. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; O’Donnell, K.A.; Zeller, K.I.; Nguyen, T.; Osthus, R.C.; Li, F. The c-Myc target gene network. Semin. Cancer Biol. 2006, 16, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.K.; Burley, S.K. X-ray Structures of Myc-Max and Mad-Max Recognizing DNA: Molecular Bases of Regulation by Proto-Oncogenic Transcription Factors. Cell 2003, 112, 193–205. [Google Scholar] [CrossRef]
- Nanbru, C.; Lafon, I.; Audigier, S.; Gensac, M.-C.; Vagner, S.; Huez, G.; Prats, A.-C. Alternative Translation of the Proto-oncogene c-mycby an Internal Ribosome Entry Site. J. Biol. Chem. 1997, 272, 32061–32066. [Google Scholar] [CrossRef]
- Ro-Choi, T.S.; Choi, Y.C. The Analysis of Structure of c-Myc Gene Transcript. J. Mol. Genet. 2019, 2, 1–7. [Google Scholar]
- Levens, D. How the c-myc Promoter Works and Why It Sometimes Does Not. J. Natl. Cancer Inst. Monogr. 2008, 2008, 41–43. [Google Scholar] [CrossRef] [PubMed]
- Herrick, D.J.; Ross, J. The half-life of c-myc mRNA in growing and serum-stimulated cells: Influence of the coding and 3’ untranslated regions and role of ribosome translocation. Mol. Cell. Biol. 1994, 14, 2119–2128. [Google Scholar] [CrossRef] [PubMed]
- Schleger, C.; Verbeke, C.; Hildenbrand, R.; Zentgraf, H.; Bleyl, U. c-MYC Activation in Primary and Metastatic Ductal Adenocarcinoma of the Pancreas: Incidence, Mechanisms, and Clinical Significance. Mod. Pathol. 2002, 15, 462–469. [Google Scholar] [CrossRef]
- Madden, S.K.; de Araujo, A.D.; Gerhardt, M.; Fairlie, D.P.; Mason, J.M. Taking the Myc out of cancer: Toward therapeutic strategies to directly inhibit c-Myc. Mol. Cancer 2021, 20, 1–18. [Google Scholar] [CrossRef]
- Han, G.; Wang, Y.; Bi, W. c-Myc Overexpression Promotes Osteosarcoma Cell Invasion Via Activation of MEK-ERK Pathway. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2012, 20, 149–156. [Google Scholar] [CrossRef]
- Ala, M. Target c-Myc to treat pancreatic cancer. Cancer Biol. Ther. 2022, 23, 34–50. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Boufaied, N.; Hallal, T.; Feit, A.; de Polo, A.; Luoma, A.M.; Alahmadi, W.; Larocque, J.; Zadra, G.; Xie, Y.; et al. MYC drives aggressive prostate cancer by disrupting transcriptional pause release at androgen receptor targets. Nat. Commun. 2022, 13, 1–17. [Google Scholar] [CrossRef]
- Mossafa, H.; Damotte, D.; Jenabian, A.; Delarue, R.; Vincenneau, A.; Amouroux, I.; Jeandel, R.; Khoury, E.; Martelli, J.M.; Samson, T.; et al. Non-Hodgkin’s lymphomas with Burkitt-like cells are associated with c-Myc amplification and poor prognosis. Leuk. Lymphoma 2006, 47, 1885–1893. [Google Scholar] [CrossRef] [PubMed]
- Vita, M.; Henriksson, M. The Myc oncoprotein as a therapeutic target for human cancer. Semin. Cancer Biol. 2006, 16, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Bochman, M.L.; Paeschke, K.; Zakian, V.A. DNA secondary structures: Stability and function of G-quadruplex structures. Nat. Rev. Genet. 2012, 13, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Cahoon, L.A.; Seifert, H.S. An Alternative DNA Structure Is Necessary for Pilin Antigenic Variation in Neisseria gonorrhoeae. Science 2009, 325, 764–767. [Google Scholar] [CrossRef] [PubMed]
- Paeschke, K.; Juranek, S.; Simonsson, T.; Hempel, A.; Rhodes, D.; Lipps, H.J. Telomerase recruitment by the telomere end binding protein-β facilitates G-quadruplex DNA unfolding in ciliates. Nat. Struct. Mol. Biol. 2008, 15, 598–604. [Google Scholar] [CrossRef]
- Postberg, J.; Tsytlonok, M.; Sparvoli, D.; Rhodes, D.; Lipps, H.J. A telomerase-associated RecQ protein-like helicase resolves telomeric G-quadruplex structures during replication. Gene 2012, 497, 147–154. [Google Scholar] [CrossRef]
- Beaudoin, J.-D.; Perreault, J.-P. Exploring mRNA 3′-UTR G-quadruplexes: Evidence of roles in both alternative polyadenylation and mRNA shortening. Nucleic Acids Res. 2013, 41, 5898–5911. [Google Scholar] [CrossRef]
- Huppert, J.L. Four-stranded nucleic acids: Structure, function and targeting of G-quadruplexes. Chem. Soc. Rev. 2008, 37, 1375–1384. [Google Scholar] [CrossRef]
- Balasubramanian, S.; Hurley, L.H.; Neidle, S. Targeting G-quadruplexes in gene promoters: A novel anticancer strategy? Nat. Rev. Drug Discov. 2011, 10, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Simonsson, T.; Pribylovab, M.; Vorlickova, M. A Nuclease Hypersensitive Element in the Human c-myc Promoter Adopts Several Distinct i-Tetraplex Structures. Biochem. Biophys. Res. Commun. 2000, 278, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Brooks, T.A.; Hurley, L.H. Targeting MYC Expression through G-Quadruplexes. Genes Cancer 2010, 1, 641–649. [Google Scholar] [CrossRef]
- Simonsson, T.; Kubista, M.; Pečinka, P. DNA tetraplex formation in the control region of c-myc. Nucleic Acids Res. 1998, 26, 1167–1172. [Google Scholar] [CrossRef]
- Zyner, K.G.; Mulhearn, D.S.; Adhikari, S.; Cuesta, S.M.; Di Antonio, M.; Erard, N.; Hannon, G.J.; Tannahill, D.; Balasubramanian, S. Genetic interactions of G-quadruplexes in humans. eLife 2019, 8, e46793. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.K.L.; Liao, J.Z.; Verheyen, E.M. A positive feedback loop between Myc and aerobic glycolysis sustains tumor growth in a Drosophila tumor model. eLife 2019, 8, e46315. [Google Scholar] [CrossRef] [PubMed]
- Aubets, E.; Chillon, M.; Ciudad, C.J.; Noé, V. PolyPurine Reverse Hoogsteen Hairpins Work as RNA Species for Gene Silencing. Int. J. Mol. Sci. 2021, 22, 10025. [Google Scholar] [CrossRef]
- Aviñó, A.; Cuestas-Ayllón, C.; Gutiérrez-Capitán, M.; Vilaplana, L.; Grazu, V.; Noé, V.; Balada, E.; Baldi, A.; Félix, A.J.; Aubets, E.; et al. Detection of SARS-CoV-2 Virus by Triplex Enhanced Nucleic Acid Detection Assay (TENADA). Int. J. Mol. Sci. 2022, 23, 15258. [Google Scholar] [CrossRef]
- Coma, S.; Noe, V.; Eritja, R.; Ciudad, C. Strand Displacement of Double-Stranded DNA by Triplex-Forming Antiparallel Purine-Hairpins. Oligonucleotides 2005, 15, 269–283. [Google Scholar] [CrossRef]
- de Almagro, M.C.; Coma, S.; Noé, V.; Ciudad, C.J. Polypurine Hairpins Directed against the Template Strand of DNA Knock Down the Expression of Mammalian Genes. J. Biol. Chem. 2009, 284, 11579–11589. [Google Scholar] [CrossRef]
- de Almagro, M.C.; Mencia, N.; Noé, V.; Ciudad, C.J. Coding Polypurine Hairpins Cause Target-Induced Cell Death in Breast Cancer Cells. Hum. Gene Ther. 2011, 22, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Bener, G.; Félix, A.J.; Sánchez de Diego, C.; Fabregat, I.P.; Ciudad, C.J.; Noé, V. Silencing of CD47 and SIRPα by Polypurine reverse Hoogsteen hairpins to promote MCF-7 breast cancer cells death by PMA-differentiated THP-1 cells. BMC Immunol. 2016, 17, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ciudad, C.J.; Enriquez, M.M.M.; Félix, A.J.; Bener, G.; Noé, V. Silencing PD-1 and PD-L1: The potential of PolyPurine Reverse Hoogsteen hairpins for the elimination of tumor cells. Immunotherapy 2019, 11, 369–372. [Google Scholar] [CrossRef] [PubMed]
- Huertas, C.S.; Aviñó, A.; Kurachi, C.; Piqué, A.; Sandoval, J.; Eritja, R.; Esteller, M.; Lechuga, L.M. Label-free DNA-methylation detection by direct ds-DNA fragment screening using poly-purine hairpins. Biosens. Bioelectron. 2018, 120, 47–54. [Google Scholar] [CrossRef]
- Noé, V.; Aubets, E.; Félix, A.J.; Ciudad, C.J. Nucleic acids therapeutics using PolyPurine Reverse Hoogsteen hairpins. Biochem. Pharmacol. 2020, 189, 114371. [Google Scholar] [CrossRef]
- Solé, A.; Ciudad, C.J.; Chasin, L.A.; Noé, V. Correction of point mutations at the endogenous locus of the dihydrofolate reductase gene using repair-PolyPurine Reverse Hoogsteen hairpins in mammalian cells. Biochem. Pharmacol. 2016, 110-111, 16–24. [Google Scholar] [CrossRef]
- Félix, A.J.; Ciudad, C.J.; Noé, V. Correction of the aprt Gene Using Repair-Polypurine Reverse Hoogsteen Hairpins in Mammalian Cells. Mol. Ther. -Nucleic Acids 2019, 19, 683–695. [Google Scholar] [CrossRef]
- Aubets, E.; Noé, V.; Ciudad, C.J. Targeting replication stress response using polypurine reverse hoogsteen hairpins directed against WEE1 and CHK1 genes in human cancer cells. Biochem. Pharmacol. 2020, 175, 113911. [Google Scholar] [CrossRef]
- Villalobos, X.; Rodríguez, L.; Solé, A.; Lliberós, C.; Mencia, N.; Ciudad, C.; Noe, V. Effect of Polypurine Reverse Hoogsteen Hairpins on Relevant Cancer Target Genes in Different Human Cell Lines. Nucleic Acid Ther. 2015, 25, 198–208. [Google Scholar] [CrossRef]
- Aubets, E.; Félix, A.J.; Garavís, M.; Reyes, L.; Aviñó, A.; Eritja, R.; Ciudad, C.J.; Noé, V. Detection of a G-Quadruplex as a Regulatory Element in Thymidylate Synthase for Gene Silencing Using Polypurine Reverse Hoogsteen Hairpins. Int. J. Mol. Sci. 2020, 21, 5028. [Google Scholar] [CrossRef]
- Psaras, A.M.; Valiuska, S.; Noé, V.; Ciudad, C.J.; Brooks, T.A. Targeting KRAS Regulation with PolyPurine Reverse Hoogsteen Oligonucleotides. Int. J. Mol. Sci. 2022, 23, 2097. [Google Scholar] [CrossRef] [PubMed]
- Brooks, T.A.; Hurley, L.H. The role of supercoiling in transcriptional control of MYC and its importance in molecular therapeutics. Nat. Rev. Cancer 2009, 9, 849–861. [Google Scholar] [CrossRef] [PubMed]
- Aubets, E.; Griera, R.; Felix, A.J.; Rigol, G.; Sikorski, C.; Limón, D.; Mastrorosa, C.; Busquets, M.A.; Pérez-García, L.; Noé, V.; et al. Synthesis and validation of DOPY: A new gemini dioleylbispyridinium based amphiphile for nucleic acid transfection. Eur. J. Pharm. Biopharm. 2021, 165, 279–292. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, J.; Yin, J.; Gan, Y.; Xu, S.; Gu, Y.; Huang, W. Alternative approaches to target Myc for cancer treatment. Signal Transduct. Target. Ther. 2021, 6, 1–14. [Google Scholar] [CrossRef]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct. Target. Ther. 2018, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Villalobos, X.; Rodríguez, L.; Prévot, J.; Oleaga, C.; Ciudad, C.; Noé, V. Stability and Immunogenicity Properties of the Gene-Silencing Polypurine Reverse Hoogsteen Hairpins. Mol. Pharm. 2013, 11, 254–264. [Google Scholar] [CrossRef]
- Dumas, L.; Herviou, P.; Dassi, E.; Cammas, A.; Millevoi, S. G-Quadruplexes in RNA Biology: Recent Advances and Future Directions. Trends Biochem. Sci. 2020, 46, 270–283. [Google Scholar] [CrossRef]
- He, T.-C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of c- MYC as a Target of the APC Pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef]
- Brown, R.V.; Danford, F.L.; Gokhale, V.; Hurley, L.H.; Brooks, T.A. Demonstration that Drug-targeted Down-regulation of MYC in Non-Hodgkins Lymphoma Is Directly Mediated through the Promoter G-quadruplex. J. Biol. Chem. 2011, 286, 41018–41027. [Google Scholar] [CrossRef]
- Morales-Martinez, M.; Valencia, A.; Vega, G.; Neri, N.; Nambo, M.J.; Alvarado, I.; Cuadra, I.; Duran-Padilla, M.A.; Martinez-Maza, O.; Huerta-Yepez, S.; et al. Regulation of Krüppel-Like Factor 4 (KLF4) expression through the transcription factor Yin-Yang 1 (YY1) in non-Hodgkin B-cell lymphoma. Oncotarget 2019, 10, 2173–2188. [Google Scholar] [CrossRef]
- Guo, P.; Dong, X.-Y.; Zhao, K.; Sun, X.; Li, Q.; Dong, J.-T. Opposing Effects of KLF5 on the Transcription of MYC in Epithelial Proliferation in the Context of Transforming Growth Factor β. J. Biol. Chem. 2009, 284, 28243–28252. [Google Scholar] [CrossRef] [PubMed]
- Tsai, L.-H.; Wu, J.-Y.; Cheng, Y.-W.; Chen, C.-Y.; Sheu, G.-T.; Wu, T.-C.; Lee, H. The MZF1/c-MYC axis mediates lung adenocarcinoma progression caused by wild-type lkb1 loss. Oncogene 2015, 34, 1641–1649. [Google Scholar] [CrossRef] [PubMed]
- Castro-Mondragon, J.A.; Riudavets-Puig, R.; Rauluseviciute, I.; Lemma, R.B.; Turchi, L.; Blanc-Mathieu, R.; Lucas, J.; Boddie, P.; Khan, A.; Pérez, N.M.; et al. JASPAR 2022: The 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2021, 50, D165–D173. [Google Scholar] [CrossRef] [PubMed]
- Aviñó, A.; Eritja, R.; Ciudad, C.J.; Noé, V. Parallel Clamps and Polypurine Hairpins (PPRH) for Gene Silencing and Triplex-Affinity Capture: Design, Synthesis, and Use. Curr. Protoc. Nucleic Acid Chem. 2019, 77, e78. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MYC Region | Length | G-rich PPRH Target Sequence (5′-3′) | G-Score |
|---|---|---|---|
| G4-Pr-C | 46 [31] | GCGCTTAT[GGGGAGGGTGGGGAGGGTGGGGAAGGTGGGG]AGGAGAC | 42 |
| I1_T | 26 [21] | GAT[GGGAGAGGAGAAGGCAGAGGG]AA | 21 |
| PR-distal-T | 24 | TCCTCGTCGTCTCTTTCCCTCTC | 0 |
| MYC-PR-prox-T | 27 [22] | GC[GGGAAAAAGAACGGAGGGAGGG]ATC | 14 |
| MYC-I2-C | 30 [27] | TGA[GGGGTGGGAGACAAACGGGGAGGGGGG] | 36 |
| MYC Region | TM (°C) | ΔTM (°C) | Complex (PPY+) |
|---|---|---|---|
| PR-Prox | 73.34 | 44.88 | HpMYC-PR-Prox |
| 28.46 | HpSc9 | ||
| G4-PR | 88.90 | 60.92 | HpMYC-G4-PR |
| 27.98 | HpSc9 | ||
| PR-Distal | 71.89 | 41.96 | HpMYC-PR-distal |
| 29.93 | HpSc9 | ||
| I1 | 78.36 | 50.00 | HpMYC-I1-T |
| 28.36 | HpSc9 | ||
| I1_short | 73.80 | 46.47 | HpMYC-I1_short |
| 27.33 | HpSc9 | ||
| I2 | 80.53 | 52.63 | HpMYC-I2 |
| 27.90 | HpSc9 |
 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valiuska, S.; Psaras, A.M.; Noé, V.; Brooks, T.A.; Ciudad, C.J. Targeting MYC Regulation with Polypurine Reverse Hoogsteen Oligonucleotides. Int. J. Mol. Sci. 2023, 24, 378. https://doi.org/10.3390/ijms24010378
Valiuska S, Psaras AM, Noé V, Brooks TA, Ciudad CJ. Targeting MYC Regulation with Polypurine Reverse Hoogsteen Oligonucleotides. International Journal of Molecular Sciences. 2023; 24(1):378. https://doi.org/10.3390/ijms24010378
Chicago/Turabian StyleValiuska, Simonas, Alexandra Maria Psaras, Véronique Noé, Tracy A. Brooks, and Carlos J. Ciudad. 2023. "Targeting MYC Regulation with Polypurine Reverse Hoogsteen Oligonucleotides" International Journal of Molecular Sciences 24, no. 1: 378. https://doi.org/10.3390/ijms24010378
APA StyleValiuska, S., Psaras, A. M., Noé, V., Brooks, T. A., & Ciudad, C. J. (2023). Targeting MYC Regulation with Polypurine Reverse Hoogsteen Oligonucleotides. International Journal of Molecular Sciences, 24(1), 378. https://doi.org/10.3390/ijms24010378